
Self-healing hydrogels triggered by amino acids†
Nicola
Zanna
*,
Andrea
Merlettini
and
Claudia
Tomasini
*
Dipartimento di Chimica Ciamician, Alma Mater Studiorum Università di Bologna, Via Selmi, 2, 40126 Bologna, Italy. E-mail: claudia.tomasini@unibo.it; nicola.zanna2@unibo.it
First published on 28th September 2016
Abstract
Nine amino acids with different chemical properties have been chosen to promote the formation of hydrogels based on the bolamphiphilic gelator A: three basic amino acids (arginine, histidine and lysine), one acidic amino acid (aspartic acid), two neutral aliphatic amino acids (alanine and serine) and three neutral aromatic amino acids (phenylalanine, tyrosine and tryptophan). Although hydrogels are obtained under any conditions, strong and thermoreversible hydrogels are formed by the addition of Arg to the bolamphiphilic gelator. These hydrogels have physiological pH and self-healing properties and may be used for regenerative medicine applications.
Introduction
Hydrogels are very promising materials for both regenerative medicine and drug delivery applications,1–3 since they are soft materials with a high water content,4–7 similar to extracellular matrices.8,9The improvement of the properties of the most studied gelators is still a challenge,10–12 as both natural and synthetic polymers still suffer from significant drawbacks. The impossibility to tune the gelation and mechanical properties of natural biopolymers and the low biocompatibility and biodegradation of synthetic polymers recently opened new routes in the search for the perfect gelator.13–15 The main concern about these applications is the gels non-toxicity, so polymeric crosslinked materials have been avoided and replaced by small molecules, called low molecular weight gelators (LMWGs)6,16 as they promote the gelation process without interfering with the metabolic activities.
The last two decades have witnessed an upsurge of research activities in the area of LMWGs, as they may be used as supports for 3D cell culture,17,18 for drug delivery systems,19–21 for wound dressing,22–24 in the food industry25 or to prepare photoconductive xerogels.26
Although computational models, based on experimental data, have been reported very recently,27 gelators are serendipitously obtained, as their rational design and synthesis is still a major challenge. A wide variety of amino acids may be used to tune the material properties for a given application. Usually the gelator is a small peptide that may be either protected with Fmoc or other aromatic groups, or totally deprotected.28–30
Finally optimization of the gel properties is an important task to be accomplished.31 The gelation trigger choice is also very important as it should be biocompatible and induce the formation of strong, elastic and transparent gels. Several methods have been recently developed such as temperature variation,24 ultrasound sonication,32,33 enzymatic cleavage,34,35 salts’ addition,36–38 pH change,39–41 dissolution in solvent mixtures,8 light irradiation,42 and use of cross-linkers that are often toxic (i.e. glutaraldehyde) or very expensive (i.e. genipin).43 Herein, we want to show an inexpensive method to promote water gelation using amino acids as gelation triggers.
To test this method, we used the already reported bolamphiphilic pseudopeptide HO-D-Oxd-L-Phe-CO(CH2)7CO-L-Phe-D-Oxd-OH A which possesses two L-Phe-D-Oxd [Phe = phenylalanine; Oxd = (4R,5S)-4-methyl-5-carboxyl-oxazolidin-2-one] dipeptide units coupled with an azelaic acid unit as a gelator (Fig. 1).33
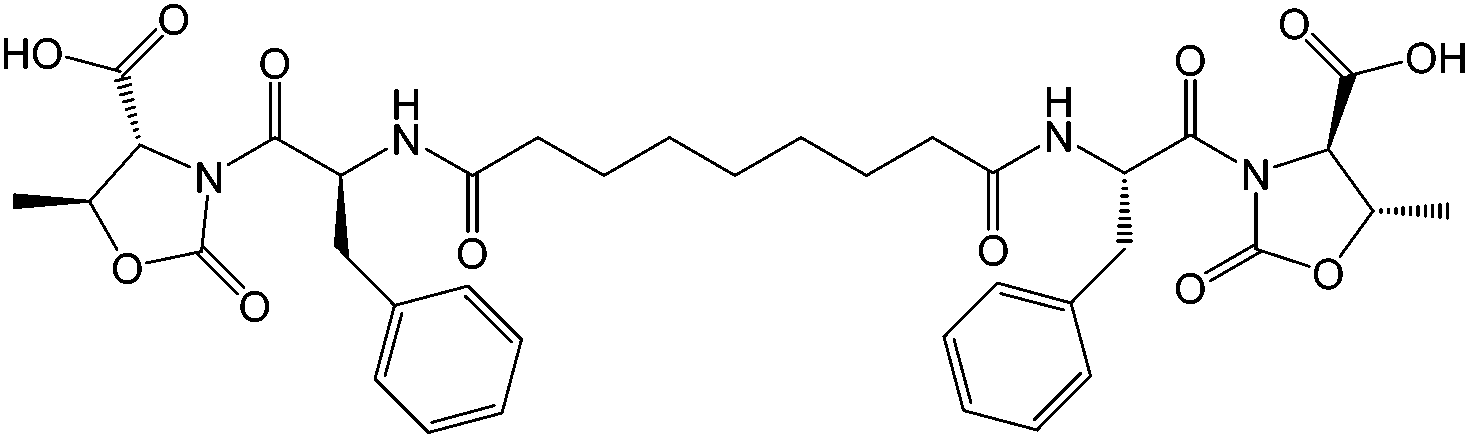 | ||
| Fig. 1 Chemical structure of the gelator A. | ||
The L-Phe-D-Oxd moiety is a privileged scaffold for the formation of supramolecular materials and gels.44 The constraint imposed by the trans conformation of the two carbonyls of the Oxd moiety, together with the presence of the Phe aromatic ring, allows intramolecular interactions that lead to the formation of fibers which, in the presence of water, can self-assemble to yield a gel.
The gel formation induced by A has been studied in the past using ultrasound irradiation as a gelation trigger.29 Under these conditions, A showed a good propensity to form a gel using mixtures of solvents (methanol/H2O and ethanol/H2O), but any attempt to form gels in pure water failed.
In this work we demonstrate that the gelator A, together with the selected amino acids, may promote the gelation of pure water at physiological pH, as the amino acids behave as non-covalent cross-linkers. The use of amino acids as biocompatible triggers has never been described before and the different properties of the tested amino acids affect their ability to induce the hydrogel formation.
Results and discussion
The bolamphiphilic gelator A was prepared in a multigram scale starting from Phe, Thr (Thr = threonine) and azelaic acid.29 The final purification of the fully protected bolamphiphilic derivative was performed by ultrasound irradiation with acetonitrile followed by filtration. The benzyl deprotection by hydrogenolysis yields pure A, which may be used to form hydrogels.Nine amino acids have been chosen to promote the formation of hydrogels based on their different chemical properties: three basic amino acids (arginine, histidine and lysine), one acidic amino acid (aspartic acid), two neutral aliphatic amino acids (alanine and serine) and three neutral aromatic amino acids (phenylalanine, tyrosine and tryptophan), to compare the effects of the different weak interactions. The hydrogels have been prepared using the gelator A both in 1% and in 2% w/w concentrations, adding either 1 or 2 equivalents of each amino acid. All the mixtures were stirred for about 5 minutes and then left to stand in the test tube for a couple of hours (for more details, see the ESI†).
For comparison, we prepared two more hydrogels with the gelator A both in 1% and in 2% w/w concentrations, using pH variation induced by addition of GdL as a gelation trigger (for more details, see the ESI†).40 This method has been recently reported and leads to the formation of strong and transparent gels, due to a slow pH variation induced by GdL hydrolysis.45
In all cases a hydrogel is obtained, although only a few gels are transparent. Both 1/1 and 1/2 gelator A/amino acid ratios produce good hydrogels, but generally the hydrogels obtained with 1/1 ratios look more transparent and homogeneous.
Both 1% and 2% w/w gelator concentrations lead to gel formation under any conditions, but the gels prepared with the gelator in 1% w/w concentration often results in them being quite fragile (for more details, see Table S1 and Fig. S1–S10†).
After the preliminary screening, we focused our attention on the promising hydrogels 1–6, that have been prepared with A in 2% w/w concentration and with five selected amino acids (1 equiv.) or GdL (2 equiv.) as a trigger, producing materials of a wide pH variety (Fig. 2).
 | ||
| Fig. 2 Photographs of hydrogels 1–6, all containing A (2% w/w concentration) and an amino acid (1 equiv.) or GdL (2 equiv.). From left to right: Arg, Hys, Asp, Ser, Phe, GdL. | ||
The first analysis useful to understand the strength of the hydrogel is the measurement of its melting point (Tgel), that is the temperature at which a glass ball suspended on the top of the gel starts to penetrate.46,47 The samples show very different behaviour after heating, as hydrogels 2, 5 and 6 give syneresis with water ejection, while hydrogels 1, 3 and 4 melt and are thermoreversible (Table 1).
| Hydrogel | Trigger (equiv.) | Final pH | Gel properties | T gel (°C) |
|---|---|---|---|---|
| 1 | Arg (1) | 8.0 | Transparent, thermoreversible | 98 |
| 2 | Hys (1) | 7.5 | Transparent, syneresis occurs | 100 |
| 3 | Asp (1) | 3.0 | Turbid, not thermoreversible | 63 |
| 4 | Ser (1) | 5.0 | Turbid, thermoreversible | 65 |
| 5 | Phe (1) | 3.5 | Turbid, syneresis occurs | 45 |
| 6 | GdL (2) | 4.0 | Opaque, syneresis occurs | 98 |
Gel 1 and 2, obtained respectively with Arg and Hys (left end of Fig. 2), look very transparent and homogeneous, as these amino acids induce a basic pH that help the dissolution of the acidic compound A. Both 1 or 2 have pH ≈ 7.5–8.0, a biocompatible pH that can never be obtained with GdL. These two peculiar properties make these hydrogels good candidates for applications in regenerative medicine as injectable stem cell delivery systems.43
In contrast, the addition of acid or neutral amino acids to the gelator A ends up in the formation of the acid gels 3–5, thus showing that the addition of these amino acids has no advantages compared with the already reported method using GdL (hydrogel 6).
Some more information on the nature of hydrogels 1–6 was obtained by SEM analysis of aerogels prepared by freeze-drying these samples (Fig. 3). The transparent and thermoreversible gels 1 and 2 (Fig. 3A and B) furnish aerogels characterized by dense fibrous networks, while gel 6, formed with GdL as a trigger (Fig. 3F), shows the formation of locally oriented long strips that cross on the large scale, thus forming a network. Fig. 3C–E show more complex patterns with a rough orientation, which are in agreement with the appearance of the gels 3–5 shown in Fig. 2.
 | ||
| Fig. 3 SEM images of the samples of xerogel obtained by freeze drying samples of hydrogel 1–6 prepared with A in 2% concentration and selected triggers: (A) Arg (1 equiv.), (B) Hys (1 equiv.), (C) Asp (1 equiv.), (D) Ser (1 equiv.), (E) Phe (1 equiv.), (F) GdL (2 equiv.). | ||
The X-ray powder diffraction analysis of samples 1–6 showed diffraction patterns at 0.48 nm that may be associated with a β-sheet structure (Fig. S24†). Aerogel 1 shows a peak at 1.0 nm that may be associated with molecular packing, while aerogels 5 and 6 show the typical pattern of a biological material with a strong peak at 1.6 nm, and weaker peaks at 1.0, 0.48 and 3.9 nm.
To check the presence of N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonds, IR spectra were recorded on aerogels 1–6 with the ATR technique (Fig. S25†). The presence of strong N–H stretching bands below 3400 cm−1 suggests the formation of N–H⋯OC hydrogen bonds.
C hydrogen bonds, IR spectra were recorded on aerogels 1–6 with the ATR technique (Fig. S25†). The presence of strong N–H stretching bands below 3400 cm−1 suggests the formation of N–H⋯OC hydrogen bonds.
Possibly the amino acids are triggers for the hydrogel formation, as they behave as non-covalent cross-linkers, thus forming strong networks that could be more effective with basic amino acids such as Hys and Arg. Fig. 4 shows a schematic representation of what could happen by mixing molecule A and Arg: the basicity of Arg allows a better dissolution of A, then electrostatic interactions between amino acid groups of the bidentate A can form a supramolecular chain, thus mimicking a polymer. The additional presence of the aromatic rings could allow π–π stacking interactions, creating a well-structured 3D network.
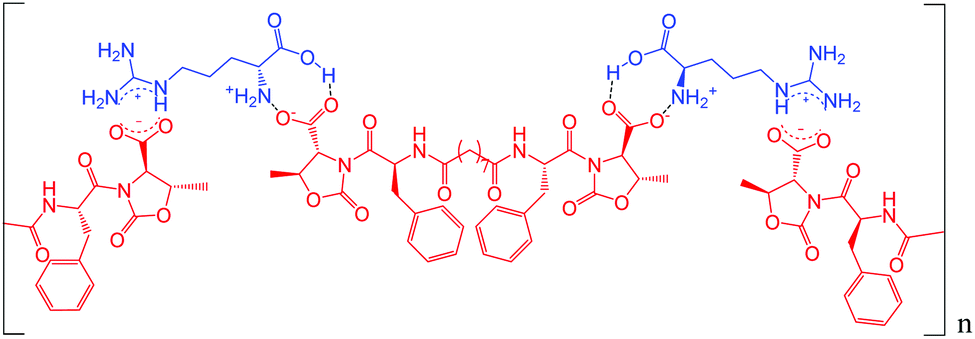 | ||
| Fig. 4 Hypothetical interactions formed by molecule A and Arg. | ||
Rheological analysis has been carried out to evaluate the viscoelastic properties of hydrogels 1–6 in terms of storage and loss moduli (G′ and G′′ respectively) (Table 2, Fig. S26†). All the obtained hydrogels are characterized by a storage modulus approximately an order of magnitude higher than the loss component, indicating their “solid-like” attitude. Frequency sweep analysis (Fig. S26†) pointed out that for all the obtained hydrogels both G′ and G′′ were almost independent of the frequency in the range from 0.1 to 100 rad s−1 (with G′ always greater than G′′) confirming the “solid-like” rheological behaviour.
| Hydrogel | Trigger (equiv.) | G′ (Pa) | G′′ (Pa) |
|---|---|---|---|
| 1 | Arg (1) | 160![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
18000 |
| 2 | Hys (1) | 135000 |
11000 |
| 3 | Asp (1) | 1500 | 100 |
| 4 | Ser (1) | 6000 | 350 |
| 5 | Phe (1) | 300 | 30 |
| 6 | GdL (2) | 40000 |
4500 |
Rheological studies nicely correlate with the previous Tgel analysis. Hydrogels 1 and 2 display the highest storage moduli, even one order of magnitude higher compared to hydrogel 6 (obtained using GdL), thus demonstrating that Arg and Hys create a stronger molecular network compared to GdL. On the other hand, hydrogels 3, 4 and 5 displayed storage moduli one order of magnitude lower than the GdL hydrogel 6, due to the lack of useful bidentate electrostatic interactions.
Finally, we tested if hydrogel 1 is provided with self-healing properties, that may be defined as the ability to autonomously reconstruct the bonding interactions after damage, like biological tissues,48–52 by a step strain experiment (Fig. 5). Multiple cycles composed of three steps were applied to the gel. During the first step, the sample was subjected to a strain value within the LVE region and was characterized by G′ values greater than G′′. When the applied strain was increased above the crossover point, the sample behavior switched from gel-like to sol-like, with G′′ values greater than G′. Finally, the sample was left at a fixed strain within the LVE range to check the recovery of the gel-like behavior.
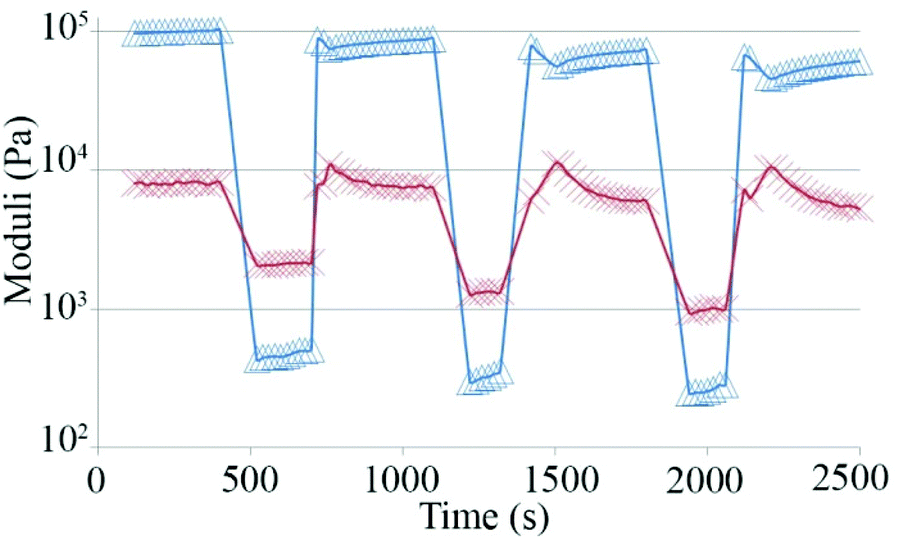 | ||
| Fig. 5 Values of storage moduli (Δ) and loss moduli (X) during a step strain experiment performed on hydrogel 1. | ||
Fig. 5 demonstrates that the sample is characterized by a great capability to regain the gel-like behavior after the application of strain well above its LVE region.
This rheological analysis prompted us to check the previously observed self-healing properties at the macroscopic level. So we prepared two blocks of hydrogel 1 (0.5 mL each) and let them stand overnight (Fig. 6). For demonstration purposes, we used some rhodamine dye (10−5 M concentration) to make one bright pink block. Then we cut each block into three bits. When different gel blocks were placed in direct contact along the cut surfaces without any external stimuli, they recombined within 20 min. We were able to construct a self-standing approx. 4.0 cm long bridge using alternating dye-doped and undoped gel blocks that could be stored for several days.
 | ||
| Fig. 6 (Top) The four images show the sequence followed to prepare the hydrogel bridge (length ≈ 4.0 cm). (Bottom) The four images demonstrate that the hydrogel has self-healing properties, the bridge is self-standing and may be easily handled, lifted up and stored. The right-end image was taken after one week. | ||
Conclusions
We prepared a small library of hydrogels based on the bolamphiphilic gelator HO-D-Oxd-L-Phe-CO(CH2)7CO-L-Phe-D-Oxd-OH A using nine amino acids as a trigger. After a general screening, we chose two basic amino acids (Arg and Hys), one acidic amino acid (Asp) and two neutral amino acids (Ser and Phe) and we compared their ability to promote the hydrogel formation in the presence of the gelator A. GdL was also tested as a trigger. Hydrogels have been obtained under various conditions, so the chemical and rheological properties of some selected samples have been analysed.The most promising hydrogel 1, obtained with A and Arg in a 1:1 ratio, is strong, elastic and thermoreversible. Moreover it has physiological pH and self-healing properties, thus it is a good candidate for regenerative medicine applications.
Experimental
Materials
All chemicals and solvents were purchased from Sigma-Aldrich, VWR or Iris Biotech and used as received.Acetonitrile was distilled under an inert atmosphere before use. MilliQ water (Millipore, resistivity = 18.2 mΩ cm) was used throughout. For the synthetic details for the preparation of A and for chemical characterization, see the ESI.†
Conditions for the gel formation with amino acid trigger
A portion of compound A (10–20 mg, depending on the final concentration, ranging from 1% to 2% w/w) was placed in a test tube (diameter: 8 mm), then the selected amino acid (1 or 2 equiv.) was added. The mixture was stirred for about 10 minutes, until sample dissolution, then it was allowed to stand quiescently until gel formation that occurs over a period of time ranging between 5 and 60 minutes.Conditions for the gel formation with a pH trigger
A portion of compound A (10–20 mg, depending on the final concentration, ranging from 1% to 2% w/w) was placed in a test tube (diameter: 8 mm), then MilliQ water (≈0.95 mL) and aqueous 0.5 N NaOH (2 equiv.) were added and the mixture was stirred and sonicated in turn for about 30 minutes, until sample dissolution. Then glucono-δ-lactone (GdL: 2.2 equiv.) was added in one portion to the mixture. After a rapid mixing to allow the complete dissolution of GdL, the sample was allowed to stand quiescently until gel formation, which occurs over a number of hours.Conditions for Tgel determination
T gel was determined by heating some test tubes (diameter: 8 mm) containing the gel and a glass ball (diameter: 5 mm, weight: 165 mg) on the top of it. When the gel is formed, the ball is suspended atop. The Tgel is the temperature at which the ball starts to penetrate inside the gel. Some hydrogel samples melt, producing a clear solution, while in other cases the gelator shrinks and water is ejected, as syneresis occurs.Aerogel preparation
Some samples of hydrogels 1–6 were freeze dried using a BENCHTOP Freeze Dry System LABCONCO 7740030 with the following procedure: 0.5 mL of a water mixture containing the gelator 1 in 2% concentration and the selected amino acid or GdL was poured into an Eppendorf test tube at room temperature. After 16 hours, the sample was dipped in liquid nitrogen for 10 minutes, then it was freeze-dried for 24 hours in vacuo (0.2 mbar) at −50 °C.SEM analysis
Scanning electron micrographs of the samples were recorded using a Hitachi 6400 field emission gun scanning electron microscope.X-ray powder diffraction
The X-ray powder diffraction patterns were obtained using a Philips X'PertPro diffractometer. The diffraction patterns were collected using a voltage of 40 kV and a current of 40 mA. A diffraction region between 20° and 60° of 2θ was scanned.IR analysis
High quality infrared spectra (64 scans) were obtained with an ATR-FT-IR Bruker Alpha System spectrometer. All compounds were dried in vacuo and all sample preparations were performed under a nitrogen atmosphere.Rheology
Rheology experiments were carried out on an Anton Paar Rheometer MCR 102 using a parallel plate configuration (25 mm diameter). The experiments were performed at a constant temperature of 23 °C controlled by the integrated Peltier system and a Julabo AWC100 cooling system. To keep the sample hydrated a solvent trap was used (H-PTD200). Amplitude and frequency sweep analyses were performed with a fixed gap value of 0.5 mm on the gel samples prepared directly on the upper plate of the rheometer once the gelation reaction was complete. The samples were prepared the day before the analysis and left overnight at a controlled temperature of 20 °C to complete the gelation process (around 20 hours). Oscillatory amplitude sweep experiments (γ: 0.01–100%) were carried out in order to determine the linear viscoelastic (LVE) range at a fixed frequency of 1 rad s−1. Once the LVE of each hydrogel was established, frequency sweep tests were performed (ω: 0.1–100 rad s−1) at a constant strain within the LVE region of each sample.The step strain experiment was conducted on hydrogel 1 to demonstrate the self-healing behaviour of the material. The sample was subjected to consecutive deformation and recovery steps. The deformation step was performed by applying to the gel a constant strain of 10%, i.e. above the LVE region of the sample for a period of 5 minutes. The recovery step was performed by keeping the sample at a constant strain of 0.25%, i.e. within the LVE region, for 7 minutes. The cycles were performed 3 times at a fixed frequency of 1 rad s−1.
Notes and references
- R. Ravichandran, M. Griffith and J. Phopase, J. Mater. Chem. B, 2014, 2, 8466–8478 RSC
.
- C. Madeira, A. Santhagunam, J. B. Salgueiro and J. M. S. Cabral, Trends Biotechnol., 2015, 33, 35–42 CrossRef CAS PubMed
- K. Shroff, E. L. Rexeisen, M. A. Arunagirinathan and E. Kokkoli, Soft Matter, 2010, 6, 5064–5072 RSC
- D. J. Adams and P. D. Topham, Soft Matter, 2010, 6, 3707–3721 RSC
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed
- A. Dasgupta, J. H. Mondal and D. Das, RSC Adv., 2013, 3, 9117–9149 RSC
- W. Liyanage, K. Vats, A. Rajbhandary, D. S. W. Benoit and B. L. Nilsson, Chem. Commun., 2015, 51, 11260–11263 RSC
- X. Zhang, M. R. Battig, N. Chen, E. R. Gaddes, K. L. Duncan and Y. Wang, Biomacromolecules, 2016, 17, 778–787 CrossRef CAS PubMed
- D. K. Smith, Nat. Chem., 2010, 2, 162–163 CrossRef CAS PubMed
- M. A. Greenfield, J. R. Hoffman, M. O. De La Cruz and S. I. Stupp, Langmuir, 2010, 26, 3641–3647 CrossRef CAS PubMed
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed
- H. Sato, E. Nogami, T. Yajima and A. Yamagishi, RSC Adv., 2014, 4, 1659–1665 RSC
- K. L. Morris, L. Chen, A. Rodger, D. J. Adams and L. C. Serpell, Soft Matter, 2015, 11, 1174–1181 RSC
- C. Tomasini and N. Castellucci, Chem. Soc. Rev., 2013, 156–172 RSC
- C. Cheng, M. C. Tang, C. S. Wu, T. Simon and F. H. Ko, ACS Appl. Mater. Interfaces, 2015, 7, 19306–19315 CAS
- J. L. Vanderhooft, M. Alcoutlabi, J. J. Magda and G. D. Prestwich, Macromol. Biosci., 2009, 9, 20–28 CrossRef CAS PubMed
- N. Sahiner and X. Jia, Turk. J. Chem., 2008, 32, 397–409 CAS
- C. Gong, T. Qi, X. Wei, Y. Qu, Q. Wu, F. Luo and Z. Qian, Curr. Med. Chem., 2013, 20, 79–94 CrossRef CAS PubMed
- M. Reza Saboktakin and R. M. Tabatabaei, Int. J. Biol. Macromol., 2015, 75, 426–436 CrossRef PubMed
- A. B. Lugão, S. O. Rogero and S. M. Malmonge, Radiat. Phys. Chem., 2002, 63, 543–546 CrossRef
- E. A. Kamoun, X. Chen, M. S. Mohy Eldin and E. R. S. Kenawy, Arabian J. Chem., 2015, 8, 1–14 CrossRef CAS
- C. Pratoomsoot, H. Tanioka, K. Hori, S. Kawasaki, S. Kinoshita, P. J. Tighe, H. Dua, K. M. Shakesheff and F. R. A. J. Rose, Biomaterials, 2008, 29, 272–281 CrossRef CAS PubMed
- S. R. Jadhav, H. Hwang, Q. Huang and G. John, J. Agric. Food Chem., 2013, 61, 12005–12011 CrossRef CAS PubMed
- E. R. Draper, J. R. Lee, M. Wallace, F. Jäckel, A. J. Cowan and D. J. Adams, Chem. Sci., 2016, 7, 6499–6505 RSC
- J. Gupta, D. J. Adams and N. G. Berry, Chem. Sci., 2016, 4713–4719 RSC
- N. Castellucci, G. Sartor, N. Calonghi, C. Parolin, G. Falini and C. Tomasini, Beilstein J. Org. Chem., 2013, 9, 417–424 CrossRef CAS PubMed
- L. Milli, N. Castellucci and C. Tomasini, Eur. J. Org. Chem., 2014, 5954–5961 CrossRef CAS
- N. Zanna, A. Merlettini, G. Tatulli, L. Milli, M. L. Focarete and C. Tomasini, Langmuir, 2015, 31, 12240–12250 CrossRef CAS PubMed
- P. Curcio, F. Allix, G. Pickaert and B. Jamart-Grégoire, Chem. – Eur. J., 2011, 17, 13603–13612 CrossRef CAS PubMed
- A. Pramanik, A. Paikar and D. Haldar, RSC Adv., 2015, 5, 53886–53892 RSC
- N. Castellucci, G. Angelici, G. Falini, M. Monari and C. Tomasini, Eur. J. Org. Chem., 2011, 3082–3088 CrossRef CAS
- G. Fichman, T. Guterman, L. Adler-abramovich and E. Gazit, CrystEngComm, 2015, 17, 8105–8112 RSC
- Z. Yang, G. Liang and B. Xu, Acc. Chem. Res., 2008, 41, 315–326 CrossRef CAS PubMed
- T. Otsuka, T. Maeda and A. Hotta, J. Phys. Chem. B, 2014, 118, 11537–11545 CrossRef CAS PubMed
- L. Chen, T. O. McDonald and D. J. Adams, RSC Adv., 2013, 3, 8714 RSC
- N. Castellucci, G. Falini, G. Angelici and C. Tomasini, Amino Acids, 2011, 41, 609–620 CrossRef CAS PubMed
- D. J. Adams, L. M. Mullen, M. Berta, L. Chen and W. J. Frith, Soft Matter, 2010, 6, 1971–1980 RSC
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC
- S. Basak, J. Nanda and A. Banerjee, Chem. Commun., 2014, 50, 2356–2359 RSC
- J. Raeburn, T. O. McDonald and D. J. Adams, Chem. Commun., 2012, 48, 9355–9357 RSC
- S. P. Zustiak, Y. Wei and J. B. Leach, Tissue Eng., Part B, 2013, 19, 160–171 CrossRef CAS PubMed
- G. Angelici, G. Falini, H.-J. Hofmann, D. Huster, M. Monari and C. Tomasini, Angew. Chem., Int. Ed., 2008, 47, 8075–8078 CrossRef CAS PubMed
- S. Sutton, N. L. Campbell, A. I. Cooper, M. Kirkland, W. J. Frith and D. J. Adams, Langmuir, 2009, 25, 10285–10291 CrossRef CAS PubMed
- A. Takahashi, M. Sakai and T. Kato, Polym. J., 1980, 12, 335–341 CrossRef CAS
- M. Yamanaka and H. Fujii, J. Org. Chem., 2009, 74, 5390–5394 CrossRef CAS PubMed
- S. Zhang, M. a. Greenfield, A. Mata, L. C. Palmer, R. Bitton, J. R. Mantei, C. Aparicio, M. O. de la Cruz and S. I. Stupp, Nat. Mater., 2010, 9, 594–601 CrossRef CAS PubMed
- J. Nanda, A. Biswas and A. Banerjee, Soft Matter, 2013, 9, 4198–4208 RSC
- S. K. Maji, D. Haldar, A. Banerjee and A. Banerjee, Tetrahedron, 2002, 58, 8695–8702 CrossRef CAS
- G. Deng, C. Tang, F. Li, H. Jiang and Y. Chen, Macromolecules, 2010, 43, 1191–1194 CrossRef CAS
- S. Roy, A. Baral and A. Banerjee, Chemistry, 2013, 19, 14950–14957 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and supporting tables and figures. See DOI: 10.1039/c6qo00476h |
| This journal is © the Partner Organisations 2016 |