
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultigram chromatography-free synthesis of octa(ethylene glycol) p-toluenesulfonate†
Adam M.
Wawro
a,
Takahiro
Muraoka
bc,
Maho
Kato
b and
Kazushi
Kinbara
*b
aInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai 980-8577, Japan
bSchool of Life Science and Technology, Tokyo Institute of Technology, 4259 Nagatsuta-cho, Midori-ku, Yokohama 226-8501, Japan. E-mail: kkinbara@bio.titech.ac.jp; Web: http://www.kinbara.bio.titech.ac.jp
cPRESTO, Japan Science and Technology Agency, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 6th September 2016
Abstract
Here we report a detailed synthetic procedure for monodisperse octa(ethylene glycol) p-toluenesulfonate, a member of the heterobifunctional oligo(ethylene glycol) derivatives family. The method offers completely chromatography-free workup and purification, resulting in higher yields and a lower cost than any of the alternative strategies. We also introduce several upgrades, simplifying the method even more, and discuss the chromatography-free design limitations.
 Adam M. Wawro | Adam M. Wawro, born in 1988, received his MSc degree in Chemical Technology from the Warsaw University of Technology in Poland. Currently he is pursuing his PhD as a MEXT Scholar at Tohoku University in Japan under the supervision of Professor Kazushi Kinbara. |
 Takahiro Muraoka | Dr Takahiro Muraoka is an assistant professor at the Tokyo Institute of Technology since 2015. He obtained his Ph.D. in 2007 from the University of Tokyo under the supervision of Professor Takuzo Aida. After working as a JSPS postdoctoral researcher in Professor Samuel I. Stupp's group at Northwestern University, he was promoted to assistant professor at Tohoku University in 2008. Since 2013, he has been concurrently serving as a PRESTO researcher at the Japan Science and Technology Agency. His present research focuses on functional synthetic molecules inspired by proteins on the basis of monodisperse PEG and amphiphilic molecular design. |
 Maho Kato | Maho Kato was born in 1993 in Chiba, Japan. She completed her undergraduate course from the Tokyo Institute of Technology. Now she continues her research as an MSc graduate student in the Kinbara Group. |
 Kazushi Kinbara | Dr Kazushi Kinbara obtained a Ph.D. in Organic Chemistry in 1996 under the direction of Professor Kazuhiko Saigo from the University of Tokyo and then began an academic career there. In 2001, he moved to Professor Takuzo Aida's group at the School of Engineering, University of Tokyo, as a lecturer and associate professor. In 2008, he was promoted to professor at the Institute of Multidisciplinary Research for Advanced Materials, Tohoku University. In 2015, he moved to the Graduate School of Bioscience and Biotechnology, Tokyo Institute of Technology. |
Introduction
Poly(ethylene glycol)s (PEGs) are frequently used in chemistry, most notably for protein cross-linking and modification, surface functionalization, and as flexible and hydrophilic building blocks of functional compounds. Although traditionally PEGs have been produced by polymerization, yielding statistically distributed mixtures of individual polymer molecules, the recent demand for more uniform PEGs has stimulated studies on well-defined, single-molecule polymers. These PEGs, known as ‘discrete’, ‘unimolecular’ or ‘monodisperse’, consist of a single chemical species with a specific molecular weight. Monodisperse PEGs are obtained by classical step-by-step chemical synthesis, usually from inexpensive short (n ≤ 4) PEG oligomers, rather than by polymerization. Due to their discrete structure, monodisperse PEGs require the same purification and analytical regime as small-molecule compounds.Among PEG derivatives, the highest versatility can be attributed to heterobifunctional PEGs. Due to their asymmetric structure, the two chain termini can be functionalized to show orthogonal reactivity. Such asymmetric oligomeric PEGs are used for the surface modification, protein cross-linking and PEGylation of small drug molecules. Unfortunately, heterobifunctional PEGs are the most challenging derivatives from the synthetic point of view. Most of the known methods give low overall yields and are either hampered by numerous chromatographic separations or require the use of expensive reagents. On top of that, commercially available asymmetrically substituted PEGs usually cost above US $500 per gram.
Previous methods
Methods of asymmetric PEG derivatives’ synthesis can be divided into two types. The first group involves firstly the synthesis of unsubstituted PEGs of the desired length, followed by non-selective monofunctionalization. Although the initial part is relatively simple, the final step gives moderate yields, since a symmetric bis-substituted product is obtained concurrently. Excess of PEG can shift the product ratio towards the monosubstituted derivative, but in this case recovery and purification of the unreacted, precious starting material is highly desired. The other group of methods focuses on the direct synthesis of heterobifunctional PEGs. The asymmetric molecular structure is introduced at one of the early steps and then the PEG chain is extended until the desired length is obtained. This approach is generally more convenient and most of the recent reports utilize it.There are four main strategies belonging to the latter group. Loiseau et al.1 and later French et al.2 proposed the use of two orthogonally reactive protective groups. By selective manipulation of the terminal functional groups, shorter PEG fragments are assembled into longer chains. The protective groups can be removed at any stage and the product can be further functionalized. This approach generates a small amount of by-products, but requires chromatographic purification after nearly every step.
Li et al.3 designed a benzyl-type fluorinated tag attached to the grown PEG chain. This feature allowed the use of chromatography based on fluorine interactions, simplifying the workup and purification process. However, purity of the products was not discussed.
Székely et al.4,5 employed a trifunctional benzyl-type core on which three PEG chains are grown simultaneously. PEG arms of the resulting star-shaped molecule are elongated and eventually the core is cleaved. This approach requires reverse-phase column chromatography with a relatively small amount of the stationary phase.
Finally, Zhang et al.6,7 proposed a new type of building block. Instead of using asymmetric PEG tosylates, they synthesized macrocyclic PEG sulfates. These sulfates are proasymmetric, i.e. upon ring-opening reaction an asymmetrically substituted derivative is obtained. This method requires fewer chromatographic separations, but employs toxic CCl4, transition metal catalyst and a large volume of the solvent for the cyclization process.
We have recently reported a new synthetic strategy for the synthesis of monodisperse oligo(ethylene glycol) p-toluenesulfonates.8 The toluenesulfonates are heterobifunctional and easily derivatizable to give a wide range of orthogonally reactive functional PEGs. Our method has significant advantages over the previously known ones: requires no chromatography, uses only simple and inexpensive reagents and offers the highest overall yields, with respect to the corresponding asymmetric PEGs obtained by other methods. These advantages have been achieved by focusing on three particular aspects: rational arrangement of the individual processes of a multistep procedure, carefully developed workup conditions and taking advantage of unique PEG physical properties, namely substituent-dependent partitioning between the organic and aqueous layers. In this paper we report an improved procedure, featuring a shorter reaction time and no use of hydrogen gas.
Method description
Design
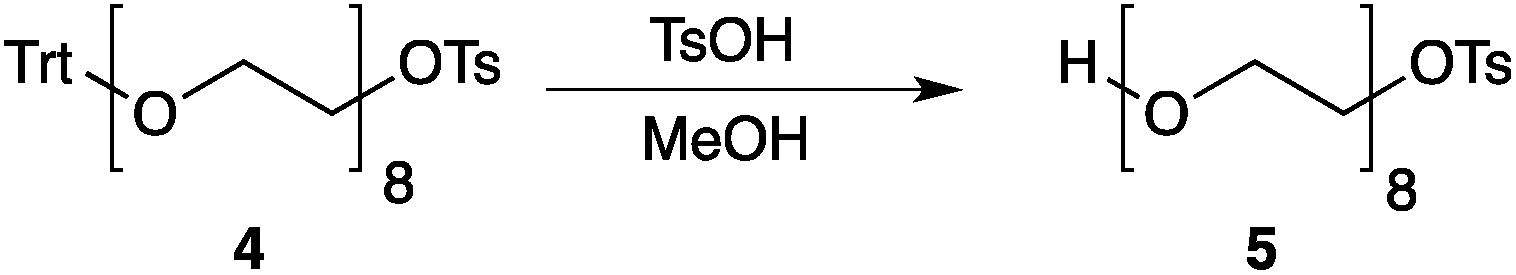
Our method consists of five main stages (Fig. 1). Initially, an excess of PEG4 is protected with the trityl group, leading to PEG4 trityl ether (1) contaminated with a small amount of symmetric bis-trityl ether. The by-product is not removed during this stage, but remains in a small quantity until removed later. The second stage, step B, is the exhaustive tosylation of the remaining hydroxy group. Due to the improved procedure originally reported by Ouchi et al.9 the starting material is completely transformed into the tosylate product (2) and no chromatographic separation is required to remove by-products. Step C is the chain extension stage, performed in THF with sodium hydride as the base. As in the case of step A, some symmetric bis-trityl ethers are generated during this reaction. Similarly, the crude product (3) is not purified before being used in the following reaction. Step D is the tosylation reaction once again; the product of this reaction (4) contains two symmetric bis-trityl impurities that had been generated during the previous stages. The final stage, step E, is trityl deprotection. In addition to the tosylate main product (5), the crude reaction mixture contains deprotected symmetric PEGs, obtained from the bis-trityl ethers, which are minor contaminants. During the reaction workup the main product is isolated in the pure state by means of liquid–liquid extraction between an organic solvent and brine.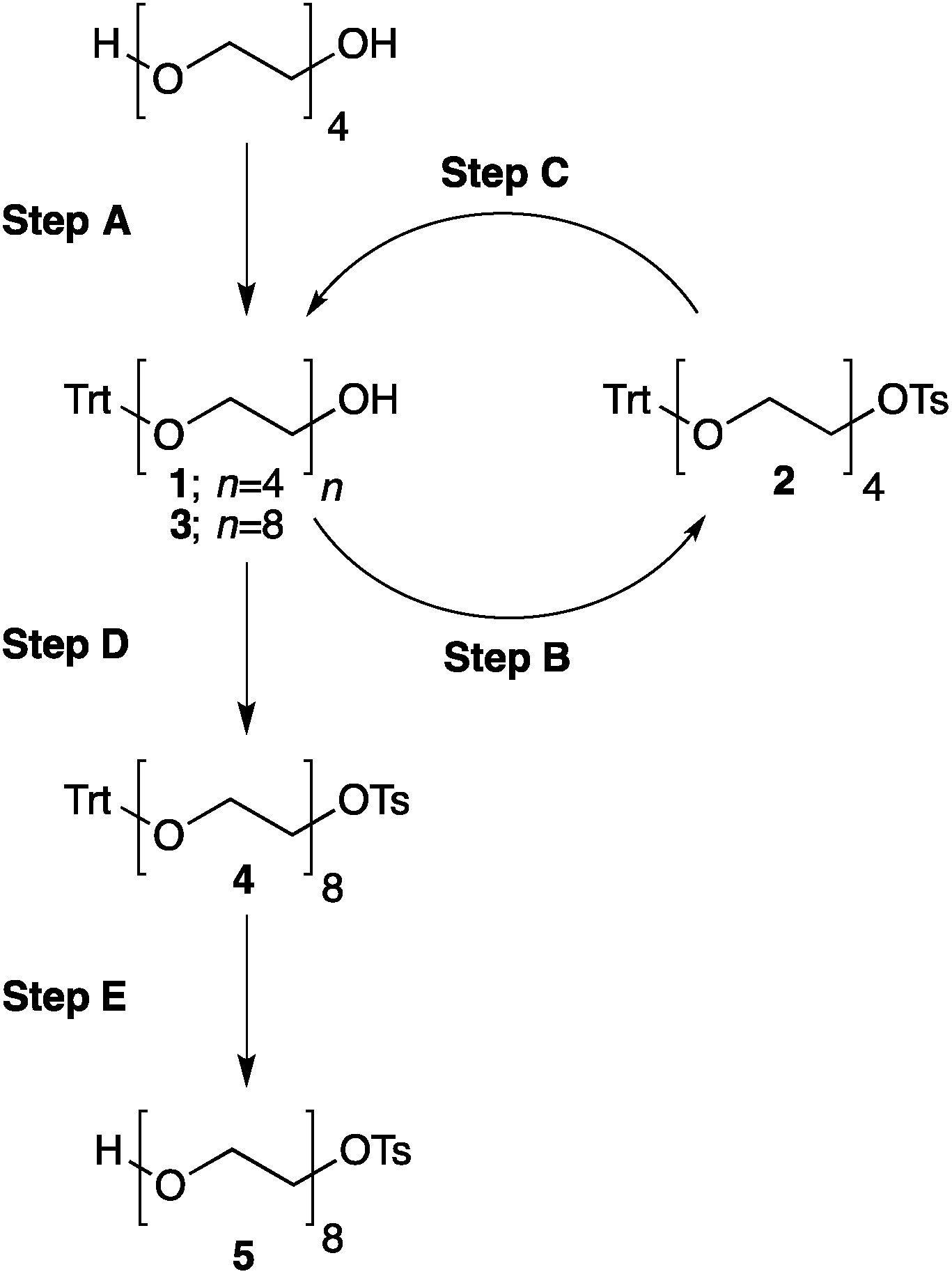 | ||
| Fig. 1 Chromatography-free PEG8 monotosylate synthesis scheme. Step A: TrtCl, Et3N; step B: TsCl, NaOH, H2O, THF; step C: PEG4, NaH, THF; step D: TsCl, NaOH, H2O, THF; step E: TsOH, MeOH. | ||
As seen from the above description, the intermediate products 2–4 are not isolated as pure compounds. Nevertheless, the final product 5 is obtained in high purity. Although the intermediates technically could be purified by simple chromatographic separations, these processes would not profit the quality of compound 5. In fact, chromatographic purifications on silica would lower the total yield, increase significantly the reaction costs and required time, as well as potentially lead to the acid-triggered undesired cleavage of trityl groups. In the Procedure and characterization section we indicated that the intermediates were used without additional purification, but we also provided details of voluntary chromatographic purification, as well as analytical characteristics of the pure compounds. As the ESI† we provided 1H NMR spectra of crude intermediates and 1H and 13C NMR spectra of the corresponding pure compounds.
Although here we described only the synthesis of PEG8 tosylates, the methodology described here applies not only to that particular oligomer, but also to shorter and longer oligomers. It can also be used to prepare PEG4 tosylates, simply by omitting steps B and C. The PEG chain length can be precisely controlled by using building blocks other than PEG4: ethylene glycol, PEG2 and PEG3. Longer PEGs can be obtained by the iterative repeating of steps B and C before terminating the synthesis with steps D and E. In our previous work we reported the synthesis of longer oligomers – PEG12 and PEG16 tosylates.8
The original procedure had some potentially troublesome steps. In particular, the long reaction time of the chain elongation reactions (step C) was a serious limiting factor. In addition, the final hydrogenation (step E) required a pressurised reactor, not widely available in all laboratories. In order to make the methodology more widely accessible and practical, here we introduced some improvements and technical changes.
Step A, the trityl group introduction, is now performed with triethylamine instead of pyridine. Triethylamine hydrochloride is insoluble in the reaction mixture and can be easily filtered off, isolated and used e.g. for amine recovery. In contrast, pyridine hydrochloride was well soluble in PEG4 and could not be isolated easily.
Step C, the Williamson ether synthesis reaction, is now performed at a higher temperature, under reflux (75–80 °C), compared to 40 °C previously. This allowed for a more precise temperature control and substantially shorter reaction time (6 h vs. 4 days). Importantly, this modification does not reduce the overall reaction yield or product purity.
Step E, the trityl group cleavage, is carried out by acid-catalysed solvolysis in methanol, instead of catalytic hydrogenation. This change simplified the process in many ways; no more pressurized steel hydrogen reactor and pyrophoric palladium carbon catalyst are required and the reaction time is greatly reduced.
Limitations
With regard to plausible limitations, we identified two main issues. Due to the synthetic design, the obtainable PEG length is limited by the point at which the PEG tosylate no longer prefers the organic layer during the liquid–liquid extraction at step E. We investigated this issue using a mixture of longer polydisperse PEG tosylates, within a molecular weight range of 600–1500 Da (Fig. 2).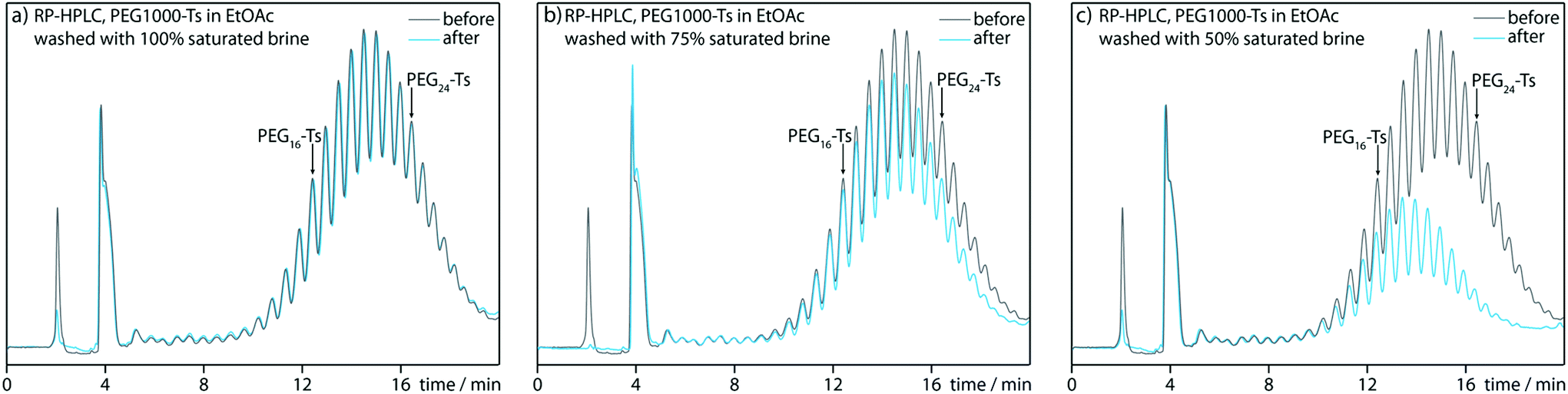 | ||
Fig. 2 Extraction of PEG tosylates from ethyl acetate solution with brine at various ionic strengths. A 2% solution of PEG1000-Ts in EtOAc was extracted with an equal volume of 100% saturated brine (a), 75% saturated brine (b) and 50% saturated brine (c). Gray curves represent the composition of the organic layer before extraction; blue curves represent the composition of the organic layer after extraction. Analysis performed with Cosmosil 5C18-AR-II column, 4.6 × 150 mm at 40 °C, eluted with 50–65% (v/v) MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O linear gradient during 20 min, with a constant flow rate of 0.8 mL min−1. :H2O linear gradient during 20 min, with a constant flow rate of 0.8 mL min−1. | ||
HPLC analysis indicated that the extraction of PEG tosylate solution in ethyl acetate with 100% saturated brine hardly affected the composition of the organic layer; even the PEG tosylates longer than PEG24-Ts were not transferred to the aqueous layer (Fig. 2a). Lower brine concentrations improve the extracting properties of the aqueous phase (Fig. 2b and c). Because even saturated brine can wash away unsubstituted PEGs efficiently, thus from this point of view our synthetic methodology can be potentially expanded towards PEG24-Ts synthesis.
The second limitation is the decreasing purity of longer oligomers. The more chain extension cycles are repeated, the more PEG(n−1) and PEG(n+1) contaminations are observed in the final product. We clearly experienced this effect in the previous paper.8 The oligomer purity decreased from almost 99% for PEG8-Ts to 97% for PEG16-Ts, as determined by HPLC. If longer PEGs were synthesized, the purity would further decrease, and the obtained products may not comply with high-level purity standards.
Procedure and characterization
General methods
1H and 13C NMR spectra were recorded in CDCl3 at 400 MHz and 101 MHz, respectively, with a Bruker Avance III 400 spectrometer. MALDI-TOF MS analyses were performed with a Bruker Autoflex Speed spectrometer in the positive mode. The samples were prepared on a ground steel target plate with an α-cyano-4-hydroxycinnamic acid (CHCA) matrix.HPLC analyses were performed with a JASCO LC-2000 Plus system equipped with a Nacalai Cosmosil 5C18-AR-II, 4.6 × 150 mm column at 40 °C. The UV detector wavelength was set at 276 nm.
A special note for students and junior researchers: when performing the experiments according to the chromatography-free procedure below, it is crucial to follow the basic rules of organic synthesis. In particular: (a) when a liquid is transferred from one to another container (flask, dropping funnel, separating funnel, etc.), the previous container should be rinsed two or three times with small volumes of the same solvent to ensure transferring of as much of the solution as possible; (b) ground glass joints should be kept clean and completely free of the reactants; (c) during the reaction course all the reactants should be in constant contact – none of them should be deposited on the flask wall, connector surface, etc.
1. A 500 mL, two-necked, round-bottomed flask equipped with a glass stopper is charged with tetra(ethylene glycol) (100 mL, 112 g, 577 mmol, 6.4 eq.)‡ and toluene (100 mL).
2. The flask is mounted on a rotary evaporator cooled with an ethylene glycol–water mixture (temp. −15 °C) and equipped with a heating water bath. Azeotropic evaporation of residual water (40 °C bath temperature, 0.15 bar initial pressure to 0.05 bar final pressure) is performed until ca. 70 mL of the liquid is collected.
3. The side neck is equipped with a glass hose barb adapter connected to an argon line and the central neck is equipped with a loosely attached polypropylene stopper. The flask is flushed with argon, equipped with an oval-shaped PTFE-coated magnetic stir bar and triethylamine (16 mL, 11.6 g, 115 mmol, 1.3 eq.) is added in one portion by using a glass syringe.
4. Trityl chloride (25.0 g, 89.7 mmol, 1 eq.) is added over 5 min through a glass funnel through the main neck against the argon gas flow. After the addition is completed, the flask wall is rinsed with a small volume of toluene. It is important that all trityl chloride is in contact with the reaction mixture.
5. The central neck is equipped with a glass stopper and the side connector is exchanged into a drying glass tube filled with anhydrous calcium chloride. The reaction is exothermic and the temperature of the mixture rises to 35–40 °C during the first 10 minutes. The reaction mixture turns from a colorless liquid to a yellow-orange suspension. Every 15 minutes the flask is gently agitated to disperse the solid accumulating on the flask wall. The stirring speed should be set so that the mixture is not splashing above the liquid surface level.
6. After 3 h of stirring at room temperature (20–23 °C), the mixture is diluted with ethyl acetate (100 mL) and stirred for additional 10 min. Then the mixture is vacuum-filtered through a Büchner or Kiriyama funnel (d = 6 cm) into a two-neck round-bottomed flask and the filtered solid is rinsed with ethyl acetate (2 × 20 mL).
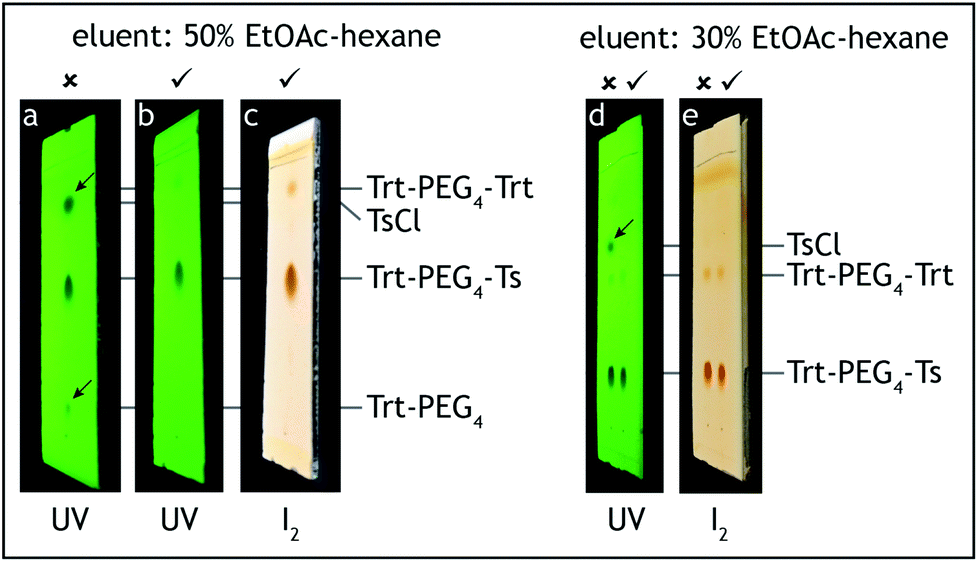
7. The filtrate is transferred to a 500 mL separation funnel and washed with water (2 × 150 mL, 3 × 50 mL), saturated aqueous solution of ammonium chloride (2 × 50 mL) and brine (50 mL).
8. At this point all of tetra(ethylene glycol) and triethylamine should be washed away. The absence of these compounds can be confirmed by TLC analysis on silica gel with 100% ethyl acetate as the eluent and visualization by iodine. Tetra(ethylene glycol)trityl ether has Rf = 0.5, triethylamine and tetra(ethylene glycol) have Rf = 0 (Fig. 3).
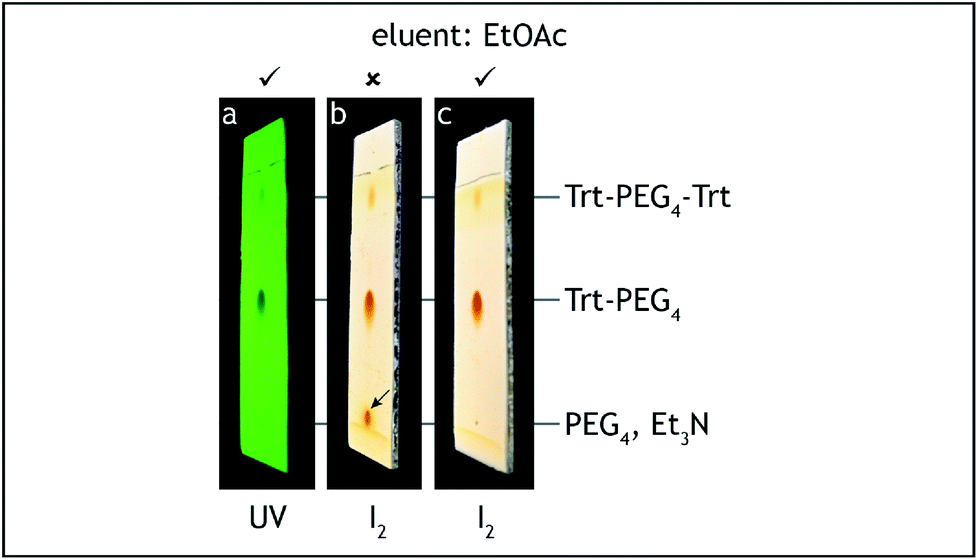 | ||
| Fig. 3 TLC analysis, step A. The reaction product (Trt-PEG4) is contaminated only with symmetric Trt-PEG4-Trt (a, c) and should not contain Et3N nor unreacted PEG4 (b). | ||
9. The organic layer is transferred to a 300 mL conical flask and dried over anhydrous sodium sulfate (2–3 g) over 1 h.
10. The mixture is then filtered through a cotton plug put into a glass funnel stem into a 500 mL round bottom flask. The solvent is removed with a rotary evaporator (40 °C bath temperature, 0.20 bar initial pressure to 0.05 bar final pressure). An oval-shaped magnetic stir bar is put into the flask and the product is further dried while stirring under vacuum (oil vacuum pump, 1.5 mmHg) for 1 h to afford crude product 1 as a yellow oil (38.5–40.5 g, ca. 78 mmol of pure compound 1), used in the following step without further purification.
The as-prepared 1 is contaminated with ca. 7 mol% of symmetric tetra(ethylene glycol) bis-trityl ether and traces of THF and ethyl acetate. None of these compounds affects the following steps. If required pure, compound 1 can be purified by column chromatography on silica gel with 70% ethyl acetate–hexane as the eluent. The pure compound shows the following properties: MALDI-TOF (positive mode, m/z): calcd for C27H32NaO5 [M + Na]+ 459.2147; found 459.2126. 1H NMR (400 MHz, CDCl3, δ): 7.46 (d, 6H), 7.34–7.18 (m, 9H), 3.72–3.64 (m, 12H), 3.58 (t, 2H), 3.24 (t, 2H), 2.47 (t, 1H, OH). 13C NMR (101 MHz, CDCl3, δ): 144.4, 128.9, 127.9, 127.1, 86.8, 72.7, 71.0, 70.9, 70.7, 63.6, 62.0.
1. Compound 1 (38.5–40.5 g, ca. 78 mmol of pure 1) put into a 500 mL round-bottomed flask equipped with an oval-shaped PTFE-coated magnetic stir bar is dissolved in THF (120 mL).
2. The mixture is stirred and cooled for 30 minutes on a water-ice bath. Sodium hydroxide (12.5 g, 313 mmol, 4 eq.) is put into a 100 mL beaker, dissolved in distilled water (40 mL) and left to cool down below 30 °C. Then it is added in one portion to the 500 mL flask through a glass funnel.
3. The resulting heterogeneous yellow mixture is stirred at 0 °C for 30 minutes. The flask is equipped with a 100 mL dropping funnel with a PTFE stopcock and fitted with a loosely attached polypropylene stopper.
4. p-Toluenesulfonyl chloride (17.0 g, 89.2 mmol, 1.14 eq.) is put into a 100 mL beaker and dissolved in THF (40 mL). The solution is transferred to the dropping funnel through a glass funnel. The solution is added slowly (ca. 1–2 drops per second) to the stirred reaction mixture over 15 min.
5. The dropping funnel is removed and the flask neck is equipped with a loosely attached polypropylene stopper. The mixture is stirred at 0 °C for 5 h. The stirring speed is set to ensure efficient contact between phases, but the mixture should not be splashing around the flask. If it happened, the flask inner wall should be rinsed with a small volume of THF to provide constant contact of all the reactants. The reaction progress is monitored by TLC analysis of the organic (top) layer on silica gel with 50% ethyl acetate–hexane as the eluent and visualization with UV and iodine. The trityl ether starting material has Rf = 0.1, and the tosylate product has Rf = 0.55 (Fig. 4).
 | ||
| Fig. 4 TLC analysis, step B. The reaction starting material Trt-PEG4 should be consumed before removing the ice bath (a–c). Before the reaction termination, TsCl (showing strong absorption at 254 nm and poorly stained with iodine) should completely disappear (b, c, d, e). | ||
6. The ice bath is removed and the mixture is stirred at room temperature (20–25 °C) overnight (12–16 h). Hydrolysis of tosyl chloride can also be monitored by TLC analysis on silica gel with 30% ethyl acetate–hexane as the eluent and visualization with UV. The tosylate product has Rf = 0.2, the symmetric bis-trityl by-product has Rf = 0.6 and TsCl has Rf = 0.7 (Fig. 4).
7. Then the reaction mixture is diluted with water (30 mL) and methyl t-butyl ether (100 mL) and transferred to a 500 mL separating funnel. The aqueous layer is discarded and the organic layer is washed with water (50 mL) and brine (2 × 50 mL).
8. The organic layer is transferred to a 500 mL conical flask and dried over anhydrous sodium sulfate (5 g) over 1.5 h. The resulting clear yellowish liquid is filtered through a cotton plug put into a glass funnel stem into a 500 mL round-bottomed flask and the solution is concentrated with a rotary evaporator (40 °C bath temperature, 0.40 bar initial pressure to 0.05 bar final pressure). An oval-shaped magnetic stir bar is put into the flask and the product is further dried while stirring under vacuum (oil vacuum pump, 1.5 mmHg) for 1 h to afford product 2 as a yellowish gum (50.0–52.0 g, containing ca. 78 mmol of pure compound 2), used in the following step without further purification.
The as-prepared product 2 is contaminated with ca. 7 mol% of symmetric bis-trityl tetra(ethylene glycol) and traces of THF and MTBE. None of these compounds affects the following steps. If required pure, compound 2 can be purified by column chromatography on silica gel with 30% ethyl acetate–hexane as the eluent. The compound shows the following properties: MALDI-TOF (positive mode, m/z): calcd for C34H38NaO7S [M + Na]+ 613.2236; found 613.2252. 1H NMR (400 MHz, CDCl3, δ): 7.78 (d, 2H), 7.45 (d, 6H), 7.34–7.18 (m, 11H), 4.13 (t, 2H), 3.70–3.54 (m, 12H), 3.23 (t, 2H), 2.42 (s, 3H). 13C NMR (101 MHz, CDCl3, δ): 144.9, 144.4, 133.5, 130.0, 129.0, 128.2, 128.0, 127.1, 86.8, 71.0, 70.9, 69.4, 68.9, 63.6, 21.8.
1. A 300 mL, oven dried, round-bottomed flask is charged with tetra(ethylene glycol) (100 mL, 112 g, 577 mmol, 7.4 eq.) and toluene (70 mL).i
2. Azeotropic evaporation of residual water is performed with a rotary evaporator (40 °C bath temperature, 0.15 bar initial pressure to 0.05 bar final pressure) until ca. 50 mL of the liquid is collected. The dried reagent is stored under an argon atmosphere until it is added at point 7.
3. A 500 mL round-bottomed flask with 2 (50.0–51.5 g, containing ca. 78 mmol of pure 2) is purged with argon, dry THF (40 mL) is added through a glass syringe and the neck is equipped with a silicon stopper with a syringe needle to equalize the pressure in the flask. The flask is agitated until the yellow gum is dissolved. The reagent is stored under an argon atmosphere until it is added at point 8.
4. A 1000 mL, oven-dried, two-necked, round-bottomed flask is equipped with an oval-shaped PTFE-coated magnetic stir bar, a glass stopper and a three-way glass stopcock connected to a vacuum line and an argon line.
5. The flask is flushed twice with argon, the gas flow is continued through the side neck and sodium hydride (60% dispersion in paraffin, 4.8 g, 120 mmol, 1.5 eq.) is added through the main neck against the gas flow. The paraffin is washed out with dry hexane.§
6. The flask is then charged with dry THF (100 mL), added from a glass syringe against the argon flow. The central neck is equipped with a pressure-equalizing 200 mL dropping funnel with a loosely attached polypropylene stopper and the whole apparatus is purged with argon. With a constant gentle gas flow, the suspension is cooled on a water-ice bath and stirred for 15 minutes.
7. The mixture of tetra(ethylene glycol) and toluene is added to the dropping funnel through a glass funnel against the gas flow. The argon flow is stopped and the three-way stopcock is turned to an all-closed position. The tetra(ethylene glycol) is carefully added to the suspension of sodium hydride over 20 min. The emerging hydrogen gas is evacuated through the polypropylene stopper on the top of the dropping funnel. Upon the addition, the grey suspension turns into a yellow solution.
8. Then the argon gas flow is turned on once again in the two-necked flask and the solution of compound 2 is added to the dropping funnel through a glass funnel against the argon gas flow.
9. The solution of compound 2 is added to the reaction mixture over 5 minutes and the ice bath is removed.
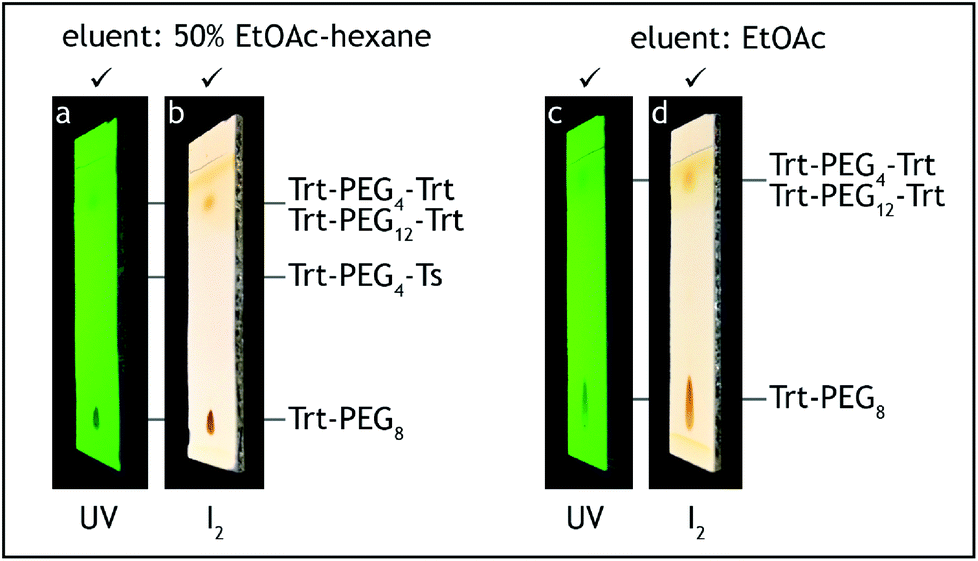
10. The dropping funnel is exchanged with a water-cooled Dimroth condenser fitted with a glass tube filled with anhydrous calcium chloride. The three-way stopcock is exchanged with a glass stopper and the reaction mixture is heated in an oil bath (external temperature 75–80 °C) under reflux for 6 hours (Fig. 5). The reaction progress was monitored by TLC analysis on silica gel with 50% ethyl acetate–hexane as the eluent and visualization with UV and iodine.¶ The tosylate starting material has Rf = 0.55, the product has Rf = 0.05 and shows significant tailing (Fig. 6).
 | ||
| Fig. 5 Reaction apparatus, step C. Addition of tetra(ethylene glycol) to the sodium hydride suspension through a dropping funnel (a). Once all reactants are added, the mixture is heated and stirred under reflux (b). | ||
 | ||
| Fig. 6 TLC analysis, step C. The starting material (Trt-PEG4-Ts) should be consumed before quenching the reaction (a–d). | ||
11. Then the oil bath is removed and the reaction mixture is left to cool below 40 °C. Brine (150 mL) and water (150 mL) are added and the mixture is transferred to a 1000 mL separation funnel.
12. The aqueous layer is discarded and the organic layer is washed with a water–brine mixture (1:1 v/v, 2 × 100 mL) and brine (50 mL). At this point all of tetra(ethylene glycol) should be washed away. The absence of this compound can be confirmed by TLC analysis on silica gel with 10% methanol–ethyl acetate as the eluent and visualization by iodine. The reaction product has Rf = 0.5, tetra(ethylene glycol) has Rf = 0.
13. The solution is transferred to a 500 mL conical flask and dried over anhydrous sodium sulfate (10 g) for 2 hours.
14. The solution is filtered through a glass funnel with a cotton plug put in the stem into a 500 mL round-bottomed flask. The solution is concentrated with a rotary evaporator (40 °C bath temperature, 0.30 bar initial pressure to 0.05 bar final pressure). An oval-shaped magnetic stir bar is put into the flask and the product is further dried while stirring under vacuum (oil vacuum pump, 1.5 mmHg) for 1 h to afford product 3 as a yellow oil (49.5–52.5 g, containing ca. 70 mmol of pure compound 3), used in the following step without additional purification.
The as-prepared product 3 is contaminated with ca. 7 mol% of bis-trityl tetra(ethylene glycol), ca. 5 mol% of bis-trityl dodeca(ethylene glycol) and traces of THF. None of these compounds affects the following steps. If required pure, compound 3 can be purified by column chromatography on silica gel with 2% methanol–ethyl acetate as the eluent. The compound shows the following properties: MALDI-TOF (positive mode, m/z): calcd for C35H48NaO9 [M + Na]+ 635.3196; found 635.2828. 1H NMR (400 MHz, CDCl3, δ): 7.46 (d, 6H), 7.28 (t, 6H), 7.21 (t, 3H), 3.72–3.56 (m, 30H), 3.23 (t, 2H). 13C NMR (101 MHz, CDCl3, δ): 144.2, 128.8, 127.8, 127.0, 86.6, 72.6, 70.9, 70.8, 70.7, 70.5, 63.4, 61.8.
1. Product 3 (49.5–52.5 g, containing ca. 70 mmol of pure compound 3) put into a 500 mL round-bottomed flask equipped with an oval-shaped PTFE-coated magnetic stir bar is dissolved in THF (140 mL).
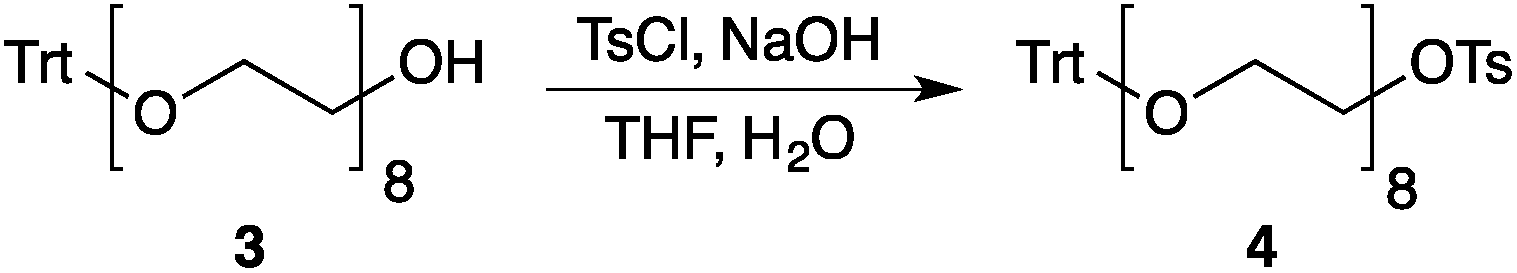
2. The mixture is stirred and cooled for 30 minutes on a water-ice bath. Sodium hydroxide (12.5 g, 313 mmol, 4.5 eq.) is put into a 100 mL beaker, dissolved in distilled water (40 mL) and left to cool down below 30 °C. Then it is added in one portion to the 500 mL flask through a glass funnel.
3. The heterogeneous yellow mixture is stirred at 0 °C for 30 minutes. The flask is equipped with a 100 mL dropping funnel with a PTFE stopcock and fitted with a loosely attached polypropylene stopper.
4. p-Toluenesulfonyl chloride (17.0 g, 89.2 mmol, 1.27 eq.) is put into a 100 mL beaker and dissolved in THF (40 mL). The solution is transferred to the dropping funnel through a glass funnel. The solution is added slowly (ca. 1–2 drops per second) to the stirred reaction mixture over 15 minutes.
5. The dropping funnel is removed and the flask neck is equipped with a loosely attached polypropylene stopper. The mixture is stirred at 0 °C for 1.5 h. The reaction progress was monitored by TLC analysis on silica gel with ethyl acetate as the eluent and visualization with UV and iodine. The starting material has Rf = 0.1, and the tosylate product has Rf = 0.6, both showing significant tailing. Just above the starting material one weak additional blunt tailing spot may appear with Rf = 0.15 (Fig. 7). This spot may remain even when the starting material is consumed and it is important not to confuse it with the starting material.||
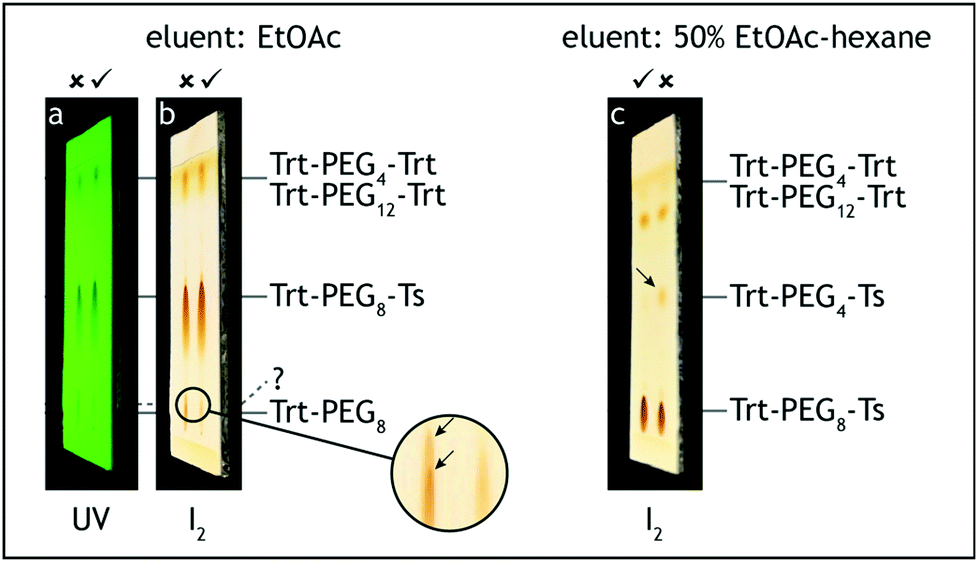 | ||
| Fig. 7 TLC analysis, step D. The reaction starting material (Trt-PEG8) should be consumed before removing the ice bath (a, b). A faint spot, located slightly above the starting material, may appear and is acceptable (b). Quality of the intermediate product may be roughly checked by confirming the lack of Trt-PEG4-Ts (c). | ||
The ice bath is removed and the mixture is stirred at room temperature (20–25 °C) overnight (12–16 h).
6. Then the reaction mixture is diluted with water (30 mL) and methyl t-butyl ether (100 mL) and transferred to a 500 mL separating funnel. The aqueous layer is discarded and the organic layer is washed with water (50 mL) and brine (2 × 50 mL).
7. The organic layer is transferred to a 500 mL conical flask and dried over anhydrous sodium sulfate (12 g) over 2 h.
8. The resulting clear yellowish liquid is filtered through a cotton plug put into a glass funnel stem into a 500 mL round-bottomed flask and the solution is concentrated with a rotary evaporator (40 °C bath temperature, 0.30 bar initial pressure to 0.05 bar final pressure). An oval-shaped magnetic stir bar is put into the flask and the product is further dried while stirring under vacuum (oil vacuum pump, 1.5 mmHg) for 1 h to afford product 4 as a yellow gum (60.0–63.5 g, containing ca. 70 mmol of pure compound 4), used in the following step without additional purification.
The as-prepared product 4 is contaminated with ca. 7 mol% of bis-trityl tetra(ethylene glycol), ca. 5 mol% of bis-trityl dodeca(ethylene glycol) and traces of THF and methyl t-butyl ether. None of these compounds affects the following steps. If required pure, compound 4 can be purified by column chromatography on silica gel with 70% ethyl acetate–hexane as the eluent. The compound shows the following properties: MALDI-TOF (positive mode, m/z): calcd for C42H54NaO11S [M + Na]+ 789.3285; found 789.3271. 1H NMR (400 MHz, CDCl3, δ): 7.79 (d, 2H), 7.46 (d, 6H), 7.36–7.18 (m, 11H), 4.15 (t, 2H), 3.72–3.54 (m, 28H), 3.23 (t, 2H), 2.43 (s, 3H). 13C NMR (101 MHz, CDCl3, δ): 144.9, 144.3, 133.4, 130.0, 128.9, 128.1, 127.9, 127.1, 86.7, 71.0, 70.9, 70.8, 70.7, 69.4, 68.9, 63.5, 21.8.
1. Product 4 (60.0–63.5 g, containing ca. 70 mmol of pure compound 4) put into a 500 mL round-bottomed flask equipped with an oval-shaped PTFE-coated magnetic stir bar is suspended in methanol (150 mL).**
2. p-Toluenesulfonic acid monohydrate (0.5 g, 2.6 mmol, 0.04 eq.) is added and the mixture is stirred for 2.5 h at room temperature (20–25 °C). The reaction progress is monitored by TLC on silica gel with ethyl acetate as the eluent and visualization with UV and iodine. The trityl ether starting material has Rf = 0.6, the reaction product has Rf = 0.1 with significant tailing (Fig. 8a and b).
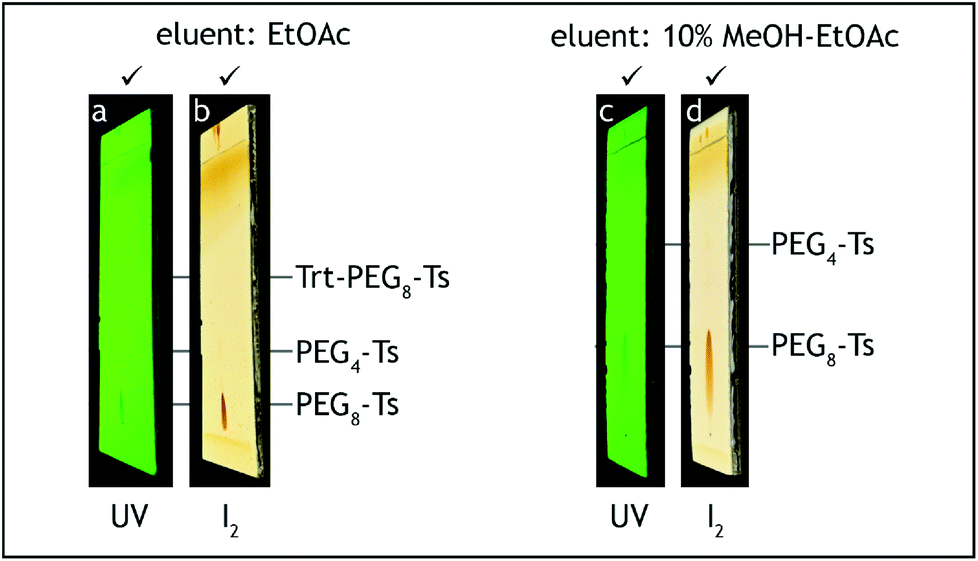 | ||
| Fig. 8 TLC analysis, step E. The starting material (Trt-PEG8-Ts) should be consumed before termination of the reaction (a, b). Absence of PEG4-Ts can be roughly checked before HPLC analysis (c, d). | ||
Upon reaction, the heterogeneous mixture turns into a yellow clear solution, occasionally followed by spontaneous precipitation of a white solid.
3. If the solid does not appear when the reaction is completed, precipitation is triggered by the slow addition of crushed ice (5–15 g).†† The ice is added until a significant amount of a coarse crystalline precipitate is observed.
4. Water (150 mL) is slowly added over 5 min and the mixture is cooled below 10 °C on an ice bath. Then it is filtered through a Büchner funnel (d = 6 cm), and the solid is rinsed with water (3 × 30 mL).
5. The filtrate is partially concentrated with a rotary evaporator (40 °C bath temperature, 0.25 bar initial pressure to 0.05 bar final pressure) until 100–120 mL of the liquid is collected.‡‡
6. The remaining mixture is transferred to a 1000 mL separation funnel, brine is added (100 mL) and the mixture is extracted with ethyl acetate (2 × 150 mL). If after two extractions a significant amount of the product remains in the aqueous layer, two or three additional extractions are performed (up to total 5 × 150 mL). The extraction process is monitored by checking the UV absorbance of both layers (e.g. with a fluorescent TLC plate). For more details consult the ESI.†
7. Combined organic layers are washed with a brine–water mixture (1:1 v/v, 2 × 100 mL), saturated aqueous solution of sodium hydrogen carbonate (50 mL), brine (2 × 50 mL), transferred to a 500 mL conical flask and dried over anhydrous sodium sulfate (10 g) for 2 h. After the aqueous washes the organic layer should contain no more tetra(ethylene glycol) and dodeca(ethylene glycol), by-products generated during steps A and C, respectively. The absence of these compounds can be confirmed by TLC analysis on silica gel with 10% methanol–ethyl acetate as the eluent and visualization by iodine. Tetra(ethylene glycol) and dodeca(ethylene glycol) have Rf = 0 (Fig. 8c and d).
8. Activated charcoal (0.7 g) is added to the flask, the mixture is agitated and left still for 15–30 minutes.
9. The mixture is filtered through fluted filter paper and the solvent is removed with a rotary evaporator (40 °C bath temperature, 0.20 bar initial pressure to 0.05 bar final pressure). An oval-shaped magnetic stir bar is put into the flask and the product is further dried while stirring under vacuum (oil vacuum pump, 1.5 mmHg) for 12 hours to afford compound 5 as a yellowish liquid (33.5–35.5 g, 64–68 mmol, 71–76% over 5 steps).§§
The compound shows the following properties: IR (neat, cm−1): 3469, 2870, 1598, 1451, 1351, 1292, 1249, 1189, 1176, 1095, 1018, 921, 817, 775. MALDI-TOF (positive mode, m/z): calcd for C23H40NaO11S [M + Na]+ 547.2189; found 547.2189. 1H NMR (400 MHz, CDCl3, δ): 7.79 (d, 2H), 7.34 (d, 2H), 4.16 (t, 2H), 3.78–3.58 (m, 30H), 2.57 (t, 1H, OH), 2.45 (s, 3H). 13C NMR (101 MHz, CDCl3, δ): 144.8, 133.6, 129.9, 128.1, 72.7, 70.9, 70.8, 70.7, 70.6, 69.4, 68.9, 61.9, 21.7. HPLC (oligomeric purity analysis, C18 column, particle size 5 μm, 4.6 × 150 mm, 1:1 MeOH–H2O, flow rate 0.8 mL min−1, temperature 40 °C, detection with UV absorption at λ = 276 nm) tR = 8.3 min.
Reagents and solvents
• Tetra(ethylene glycol) (Sigma (99%), CAS no. 112-60-7). The quality of this reagent is crucial for a high oligomer purity of the product.• Triethylamine (Sigma-Aldrich (99.5%), CAS no. 121-44-8)
• Trityl chloride (Aldrich (97%), CAS no. 76-83-5)
• p-Toluenesulfonyl chloride (Fluka (99%), CAS no. 98-59-9)
• Sodium hydroxide (Nacalai Tesque (97%), CAS no. 1310-73-2)
• Sodium hydride (TCI Chemicals (60% dispersion in paraffin), CAS no. 7646-69-7)
• p-Toluenesulfonic acid monohydrate (Nacalai Tesque (99%), CAS no. 6192-52-5)
• Tetrahydrofuran, dry (Kanto Chemicals (dehydrated, without stabilizer, 99.5%), CAS no. 109-99-9)
• Toluene (Nacalai Tesque (99%), CAS no. 108-88-3)
• Ethyl acetate (Nacalai Tesque (99%), CAS no. 141-78-6)
• Tetrahydrofuran (Nacalai Tesque (99.5%), CAS no. 109-99-9)
• Methyl t-butyl ether (Nacalai Tesque (99.5%), CAS no. 1634-04-4)
• Methanol (Nacalai Tesque (99%), CAS no. 67-56-1)
Reagent setup
Tetrahydrofuran, dry• Before using it was passed through a glass contour solvent purification system.
Troubleshooting
If the above procedure is performed correctly and precisely, the product should be obtained in the yield and purity as reported. However, certain mistakes during performing the procedure may cause a lower overall yield or purity. On the basis of our experience with this procedure, here we describe the causes and solutions, if they exist, of some possible issues.Step A
Problem: PEG4 is not completely removed after the workup.
Answer: Perform additional washes with brine and/or 1:1 water–brine.
Step B
Problem: 1 is not consumed after 5 h stirring at 0 °C.Answer: If the spot of 1 at TLC is very weak and tosyl chloride remains in the mixture, proceed to the next point (stirring at RT). If the unreacted amount of 1 is large and tosyl chloride remains in the mixture, probably the reaction mixture stirring was insufficiently effective. Improve the organic–water phase contact by more efficient stirring and keep at 0 °C until the reaction is completed. If the unreacted amount of 1 is large and no tosyl chloride remains, the reaction mixture was not properly cooled or tosyl chloride addition was too fast. Provide sufficient cooling; add more tosyl chloride solution dropwise and the corresponding amount of sodium hydroxide solution.
Problem: Excess of tosyl chloride is not hydrolyzed after overnight stirring at room temperature.
Answer: Keep stirring at room temperature until the hydrolysis is completed. Caution: do not confuse the TLC spot of TsCl with the one of Trt-PEG4-Trt!
Step C
Problem: 2 is not consumed after 6 h stirring at 75–80 °C.Answer: Keep stirring at the same temperature until 2 is completely consumed.
Step D
Problem: 1 is not consumed after 5 h stirring at 0 °C.Answer: See Troubleshooting for step B.
Problem: Excess of tosyl chloride is not hydrolyzed after overnight stirring at room temperature.
Answer: See Troubleshooting for step B.
Step E
Problem: Although until step D the obtained amounts of products were correct, the yield after step E is low.Answer: Compound 5 was not fully extracted from the aqueous phase. Inspect the aqueous layer and perform additional extractions as necessary.
Problem: The final product contains methyl triphenylmethyl ether.
Answer: Precipitation of the crystalline solid during the workup was not complete. Dissolve the product in cold water, filter and repeat extractions as described in the procedure.
Problem: The product contains too large an amount of PEG4-Ts.
Answer: There are two possibilities, both related to step C: the Williamson reaction was not completed and/or excess of PEG4 was not washed away completely after the reaction. We are not aware of any reasonable method of PEG4-Ts removal from the final product, apart from chromatography.
Problem: The product contains too large an amount of PEG7-Ts and/or PEG9-Ts.
Answer: The purity of PEG4 used for the synthesis was too low. Use a higher grade (99% or above) of PEG4.
Conclusions
Here we presented a detailed procedure of the chromatography-free synthesis of octa(ethylene glycol) monotosylates. The main advantages of this method include high yield, scalability, use of simple reactants and low cost. The method can also be applied to the synthesis of other PEG tosylates, from PEG4-Ts to PEG24-Ts. PEG derivatives within this molecular weight range are the most common for protein crosslinking and drug–antibody conjugation. Our highly practical protocol for monodisperse PEGn-Ts synthesis shall significantly improve the availability of heterobifunctional monodisperse PEG-based reagents for bioconjugations and materials science.Author contribution
The original procedure was carried out by A. M. W. (three times at the reported scale) and repeated by M. K. (once at half scale).Acknowledgements
This work was supported by the Ministry of Education, Culture, Sports, Science and Technology in Japan (MEXT), Grants-in-Aid for Scientific Research on Innovative Areas “Spying minority in biological phenomena” (no. 3306, 23115003, KK) and Scientific Research C (26410170, TM), JST PRESTO “Molecular technology and creation of new functions”, and the Management Expenses Grants for National Universities Corporations.References
- F. A. Loiseau, K. K. Hii and A. M. Hill, J. Org. Chem., 2004, 69, 639–647 CrossRef CAS PubMed.
- A. C. French, A. L. Thompson and B. G. Davis, Angew. Chem., Int. Ed., 2009, 48, 1248–1252 CrossRef CAS PubMed.
- Y. Li, Q. Guo, X. Li, H. Zhang, F. Yu, W. Yu, G. Xia, M. Fu, Z. Yang and Z.-X. Jiang, Tetrahedron Lett., 2014, 55, 2110–2113 CrossRef CAS.
- G. Székely, M. Schaepertoens, P. R. J. Gaffney and A. G. Livingston, Polym. Chem., 2014, 5, 694–697 RSC.
- G. Székely, M. Schaepertoens, P. R. J. Gaffney and A. G. Livingston, Chem. – Eur. J., 2014, 20, 10038–10051 CrossRef PubMed.
- H. Zhang, X. Li, Q. Shi, Y. Li, G. Xia, L. Chen, Z. Yang and Z.-X. Jiang, Angew. Chem., Int. Ed., 2015, 54, 3763–3767 CrossRef CAS PubMed.
- Y. Li, X. Qiu and Z.-X. Jiang, Org. Process Res. Dev., 2015, 19, 800–805 CrossRef CAS.
- A. M. Wawro, T. Muraoka and K. Kinbara, Polym. Chem., 2016, 7, 2389–2394 RSC.
- M. Ouchi, Y. Inoue, Y. Liu, S. Nagamune, S. Nakamura, K. Wada and T. Hakushi, Bull. Chem. Soc. Jpn., 1990, 63, 1260–1262 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra; qNMR experiment; HPLC oligomer purity analysis; report from the experiment reproduced by M. K.; extraction process details. See DOI: 10.1039/c6qo00398b |
| ‡ Excess (6.4 eq.) of tetra(ethylene glycol) is used in order to promote the formation of monotrityl ether over the bis-trityl product. Decreasing the ratio will reduce the amount of tetra(ethylene glycol) used, but also decrease the overall process yield. For instance, under conditions described in this procedure after step A product 1 is contaminated by ca. 7 mol% of the symmetric by-product. Since formation of the by-product required twice as much trityl chloride as for the main product, only ca. 87% of the total trityl chloride was transformed into the desired compound. |
| § Dry hexane (40 mL) is added from a glass syringe against the gas flow. The slurry is stirred for 3 min and then is left still for 3 min. The supernatant is collected carefully with a 3 mL glass pipette, which was flushed inside with argon gas in advance. The washing with hexane is repeated once more and the remaining hexane is removed by connecting the flask through the three-way stopcock to the vacuum line. |
| ¶ Before TLC analysis, the sample taken from the reaction mixture should be processed. A small volume of the mixture is partitioned between equal volumes of water and ethyl acetate. The organic (top) layer is analyzed by TLC. |
| || At this point one can verify if the reaction mixture workups after steps B and C were done properly. If the TLC analysis is performed with 50% ethyl acetate–hexane as the eluent and visualized by iodine, two spots should be observed; the reaction product with Rf = 0.1 and bis-trityl tetra(ethylene glycol), the by-product from step A, with Rf = 0.85. If the reaction during steps B or C is not completed, or tetra(ethylene glycol) is not washed away completely during the reaction workup at step C, a weak spot of tetra(ethylene glycol)trityl ether p-toluenesulfonate is observed with Rf = 0.5 (Fig. 7c). |
| ** At this concentration the mixture has a miscibility gap. Initially, after adding 50 mL of methanol, the mixture is homogeneous and upon addition of remaining 100 mL of methanol it separated into two liquid phases. |
| †† If the ice is added too fast, instead of a white crystalline solid a yellow oil may appear. This event is rare, but highly undesired (see the Troubleshooting section). |
| ‡‡ Generally, the larger the volume of methanol evaporated now, the better. If the amount removed is too low, repeated extraction may be necessary at point 6 or, in an extreme case, a water–ethyl acetate–methanol mixture forms one phase. |
| §§ Although the starting material 4 was significantly contaminated with PEG-class impurities, the product is obtained in high purity. Because elemental analysis is purposeless for analysis of a sample contaminated with compounds with a homologous chemical formula, we developed a two-step purity assay. First, the amount of total PEG tosylates in the sample was quantified by quantitative NMR (99.3%) with dimethyl terephthalate as a reference. Then, oligomeric purity, i.e. distribution of homologous oligomers in the mixture, was determined by reverse-phase HPLC. Compound 5 was the major (98.4%, tR = 8.3 min) component of the total PEG tosylates, with traces of tetra(ethylene glycol) p-toluenesulfonate (0.4%, tR = 5.8 min), hepta(ethylene glycol) p-toluenesulfonate (0.8%, tR = 7.7 min) and nona(ethylene glycol) p-toluenesulfonate (0.4%, tR = 9.1 min). In total, the purity of the product 5 was determined as 97.7%. |
| This journal is © the Partner Organisations 2016 |