
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intermolecular oxidative dehydrogenative 3,3′-coupling of benzo[b]furans and benzo[b]thiophenes promoted by DDQ/H+: total synthesis of shandougenine B†‡
T.
Wirtanen
*a,
M.
Muuronen
a,
J.
Hurmalainen
b,
H. M.
Tuononen
b,
M.
Nieger
a and
J.
Helaja
*a
aDepartment of Chemistry, A.I. Virtasen aukio 1, University of Helsinki, 00014, Finland. E-mail: juho.helaja@helsinki.fi
bDepartment of Chemistry, Nanoscience Center, University of Jyväskylä, P.O. Box 35, FI-40014, Jyväskylä, Finland
First published on 27th September 2016
Abstract
With an excess of a strong acid, 2,3-dichloro-5,6-dicyano-1,4-quinone (DDQ) is shown to promote metal-free intermolecular oxidative dehydrogenative (ODH) 3,3′-coupling of 2-aryl-benzo[b]furans and 2-aryl-benzo[b]thiophenes up to 92% yield as demonstrated with 9 substrates. Based on the analysis of oxidation potentials and molecular orbitals combined with EPR, NMR and UV-Vis observations, the studied reaction is initiated by a DDQ-substrate charge transfer complex and presumably proceeds via oxidation of the substrate into an electrophilic radical cation that further reacts with another molecule of a neutral substrate. The coupling reactivity can easily be predicted from the oxidation potential of the substrate and the morphology of its frontier molecular orbitals. The intermolecular ODH coupling reaction allowed a concise total synthesis of the natural product shandougenine B.
Introduction
Quinones are important oxidizers in both biological and synthetic chemical conversions.1 For example, the high oxidative power of 2,3-dichloro-5,6-dicyano-1,4-quinone (DDQ) makes it a frequently used oxidant in various chemical conversions. Broadly categorizing, there are three commonly accepted mechanistic pathways associated with oxidation: hydride abstraction, hydrogen atom transfer and single electron transfer (SET).2It has previously been shown that quinone promoted heteroatomic oxidations associated with α-deprotonation (Scheme 1a) or direct hydride transfer (Scheme 1b) lead to the formation of good electrophiles (e.g. iminium, oxonium or allylic cations) that can be coupled with various nucleophiles (Nu).3,4 Similarly, an electrophile can be generated by one electron oxidation of a neutral substrate into a radical cation that can subsequently react with different nucleophiles such as water or aryl (Scheme 1c).3,5,6a The last mechanistic pathway operates in many quinone mediated intramolecular oxidative couplings of aryls, but reports on analogous intermolecular couplings are quite rare.3,6a–c Common to these reactions is that quinones are used in the presence of a strong acid promotor (e.g. TFA, MsOH, H2SO4, TfOH, HClO4 or HFSO3), which has been shown to increase the oxidative power of the quinone considerably.6
 | ||
| Scheme 1 Quinone mediated metal-free approaches for intermolecular oxidative dehydrogenative (ODH) couplings. | ||
Numerous synthetic methods have been used to construct the 3,3′-bibenzo[b]furan and 3,3′-bibenzo[b]thiophene skeletons intermolecularly.7–13 This structural motif is found in natural products,14 diphosphine metal ligands,7a,8a,b diphosphine oxide organocatalysts15 and in organic chiral electrodes.16 Nevertheless, many of the modern high yielding coupling methods require the use of prefunctionalized starting materials, stoichiometric or substoichiometric amounts of transition metals or air sensitive or pyrophoric reagents (Scheme 2a). However, benzo[b]furans and benzo[b]thiophenes can be 3,3′-coupled from unfunctionalized monomers directly by the Brønsted acid mediated Friedel–Crafts reaction to dihydro-3,3′-benzofused intermediates in variable yields (Scheme 2b).10,17 The McMurry coupling of 3-oxobenzofurans has also been shown to offer a viable route to dihydrogenated intermediates (Scheme 2b).7 These intermediates can then be dehydrogenated either thermally (Friedel–Crafts) or with a stoichiometric amount of an oxidant such as DDQ (McMurry).
 | ||
| Scheme 2 Various synthetic routes to 3,3′-bibenzo[b]furan and 3,3′-bibenzo[b]thiophene skeletons. | ||
Results and discussion
We began the development of our method with non-optimized reaction conditions, 1 eq. of DDQ, 5 eq. of MsOH and 1 eq. of 1a in DCM (0.05 M) at RT, which yielded 2,2′-bis(phenyl)-3,3′-bibenzo[b]furan 2a in 61% yield in 1 h.18b In preliminary studies, we found that an acid additive was necessary for the progress of the reaction. Therefore, we tested the effect of several acids on the reaction and found that, out of these, only MsOH worked satisfactorily (Table 1, entries 1–3, see the ESI‡ for kinetic data). Later we noticed that the behaviour of TFA was both concentration and substrate dependent. At higher concentrations TFA could promote the reaction of 1a to 2a, whereas in more dilute solutions it promoted the coupling of the more electron rich substrate 1c to 2c.19 Next, we screened solvents and found that DCE worked better than DCM or CHCl3, and that MeNO2 gave a low yield (Table 1, entries 3–6). We then employed a two level full factorial design for three factors to optimize the reaction conditions.20 Entry 3 was chosen as the centre point and molarity, temperature and the amount of DDQ were set as variables (Table 1, entries 3 and 7–14). One corner of the screened reaction space (+1, −1, −1) gave a very high (99%) yield (entry 11) and no further optimization was deemed necessary.21|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent (molarity) | T (°C) | Time (h) | Acid additive (3 eq.) | Amount of DDQ (eq.) | NMR yielda (%) |
| a Determined with 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. b Isolated yield. O/N = overnight. | ||||||
| 1 | DCM (0.05) | RT | O/N | TFA | 1 | n.r. |
| 2 | DCM (0.05) | RT | O/N | Sc(OTf)3 | 1 | n.r. |
| 3 | DCM (0.05) | RT | O/N | MsOH | 1 | 75 |
| 4 | CHCl3 (0.05) | RT | 1.5 | MsOH | 1 | 56 |
| 5 | DCE (0.05) | RT | 1.5 | MsOH | 1 | 88 |
| 6 | MeNO2 (0.05) | RT | 1.5 | MsOH | 1 | 11 |
| 7 | DCE (0.01) | 0 | 1.5 | MsOH | 0.5 | 0 |
| 8 | DCE (0.01) | 0 | 1.5 | MsOH | 1.5 | 0 |
| 9 | DCE (0.01) | 40 | 1.5 | MsOH | 0.5 | 0 |
| 10 | DCE (0.01) | 40 | 1.5 | MsOH | 1.5 | 25 |
| 11 | DCE (0.1) | 0 | 1.5 | MsOH | 0.5 | 99 (88) |
| 12 | DCE (0.1) | 0 | 1.5 | MsOH | 1.5 | 61 |
| 13 | DCE (0.1) | 40 | 1.5 | MsOH | 0.5 | 84 |
| 14 | DCE (0.1) | 40 | 1.5 | MsOH | 1.5 | 8 |

After determining the optimized reaction conditions, the scope of the reaction was studied next. In the case of 2-aryl-benzo[b]furans, we were able to obtain the corresponding 2,2′-bis(aryl)-3,3′-bibenzo[b]furans in good to excellent isolated yields (Scheme 3, 2a–c). A slightly lower yield (68%) of the coupling product 2d was obtained when the 2-aryl substituent was functionalized with an amide group at the para-position. However, the electron poor 2-(4-nitrophenyl)benzo[b]furan 1i could not be coupled even in the presence of a stronger acid TfOH or with different oxidants such as p-tetrachloro-o-quinone or ammonium persulfate (see below). To our pleasure, we could, however, obtain the corresponding 2,2′-bis(aryl)-3,3′-bibenzo[b]thiophenes, 2e–g, in equally high yields as the related bibenzo[b]furans 2a–c. Undesirably, the 3,3′-homocoupling product 2h, 2,2′-bis(2-thienyl)-3,3′-bibenzo[b]thiophene was obtained only in 14% yield. We believe that the reason for such a low yield is in the structure of 1h that enables reactivity at two sites, benzo[b]thiophene 3- and thiophene 5-positions, leading to oligomerization. The oxidative 3,3′-homocoupling of the electron rich substrate 1j would have given a direct route to the natural product kynapcin-24,14a but we could not obtain any product under the reaction conditions employed (see below).
 | ||
| Scheme 3 Reaction conditions and isolated yields for DDQ/H+ mediated homocoupling of benzofurans and benzothiofurans 1a–h to 2a–h. Substrates 1i and 1j failed to yield a product (see the text for details). | ||
Based on previous studies,6 it seemed reasonable that the reactions are initiated by a SET between the substrate and the DDQ/H+ and the oxidation potential of a particular substrate would be directly related to its reactivity. Consequently, we measured the relative oxidation potentials of compounds 1a–jvs. FcH/FcH+ couple, and compared the results with data from theoretical calculations (Table 2). It is clearly evident that both oxidation potentials normalized to 1c have strong systematic correlation (the absolute value is underestimated by ca. 0.31 V).23 Furthermore, both the calculated and the experimental oxidation potentials correlate well with the observed reactivity and provide a rationale as for why the homocoupling of 1i was unsuccessful. With an oxidation potential of 1.310 V, 1i is not oxidized by DDQ.
| Substrate | Exp.a (Exp.n.)b/V | Calc. (Calc.n.)b/V | Exp. − Calc. (Exp.n. − Calc.n.) b/V |
|---|---|---|---|
| a Measurements were carried out in DCM with 0.05 M NBu4PF6 as the supporting electrolyte. b Values in parenthesis have been normalized to 1c to see relative potentials. | |||
| 1a | 1.070 (0.280) | 0.749 (0.305) | 0.321 (−0.03) |
| 1b | 0.925 (0.135) | 0.627 (0.183) | 0.298 (−0.05) |
| 1c | 0.790 (0.000) | 0.444 (0.000) | 0.346 (0.00) |
| 1d | 0.810 (0.020) | 0.557 (0.113) | 0.253 (−0.09) |
| 1e | 1.055 (0.265) | 0.782 (0.338) | 0.273 (−0.07) |
| 1f | 1.000 (0.210) | 0.689 (0.245) | 0.311 (−0.04) |
| 1g | 0.845 (0.055) | 0.527 (0.083) | 0.318 (−0.03) |
| 1h | 0.910 (0.120) | 0.595 (0.151) | 0.315 (−0.03) |
| 1i | 1.310 (0.520) | 1.060 (0.616) | 0.250 (−0.10) |
| 1j | 1.100 (0.310) | 0.685 (0.241) | 0.415 (0.07) |
The oxidation potentials do not, however, explain why 1j fails to homocouple even though it has an oxidation potential comparable to 1a and 1f. If we consider that dimerization involves the energetically closest MOs,24 we can then pay our attention to the morphology of the SOMO of the radical cation and the HOMO of the neutral substrate. For a successful 3,3′-homocoupling, a high contribution from the C3-carbon to the nucleophile's HOMO and the electrophile's SOMO is imperative. As shown in Fig. 1, this is certainly true for 1a but not for 1j. Thus, it is clear that, in order to predict reactivity, both the oxidation potential of the substrate and the morphology of its frontier orbitals need to be considered.
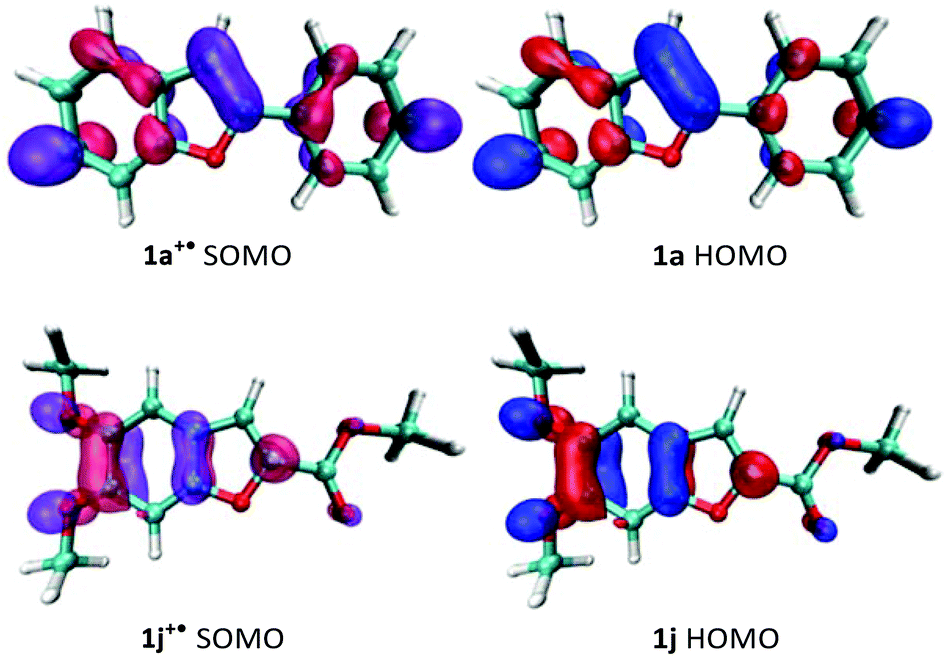 | ||
| Fig. 1 Frontier molecular orbitals of 1a and 1j (HOMOs) and the respective cation radicals (SOMOs). | ||
Finally, to demonstrate the applicability of our approach, we endeavoured to synthesise shandougenine B. Prior to the synthesis, we predicted the oxidative homocoupling to be favourable based on its low oxidation potential and a high contribution from the C3-carbon to the frontier MOs (see the ESI‡). Substrate 1k was prepared with the reported literature procedure25 and the homocoupling reaction promoted with 6 eq. of TFA and 0.5 eq. of DDQ (Scheme 4). To our delight, the dimerization of 1k proceeded smoothly and the corresponding dimer could be isolated in 80% yield. After subsequent removal of the benzyl groups, the antioxidant shandougenine B was obtained in 73% yield (NMR) under standard conditions. Interestingly, the biological origin of shandougenine B has been hypothesised to be oxidative coupling of the monomer Sophora as it is also present in the roots of Sophora tonkinensis.14b Our successful synthesis of shandougenine B via an oxidative route certainly supports this hypothesis.
 | ||
| Scheme 4 Synthesis of shandougenine B with DDQ-mediated oxidative homocoupling as the key strategic step. | ||
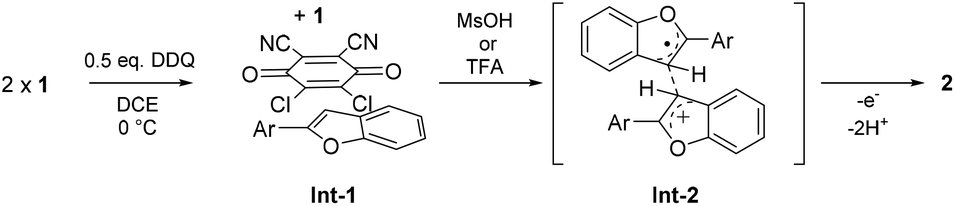
In the proposed mechanism (Scheme 5), DDQ initially forms a charge transfer (CT) complex with the substrate (Int-1). The formation of a CT complex is seen visually by a colour change and confirmed by UV-vis spectroscopy for 1a–c (Fig. 2 top). Furthermore, the structure of a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex of 1c and DDQ was determined by single crystal X-ray diffraction (Fig. 2 bottom, see the ESI‡ for details).26 The layered structure of the complex between 1c and DDQ shows a short (3.14 Å) distance between the electron donor and the acceptor, which is a prerequisite for efficient electron transfer.27
:1 complex of 1c and DDQ was determined by single crystal X-ray diffraction (Fig. 2 bottom, see the ESI‡ for details).26 The layered structure of the complex between 1c and DDQ shows a short (3.14 Å) distance between the electron donor and the acceptor, which is a prerequisite for efficient electron transfer.27
 | ||
| Scheme 5 Proposed reaction mechanism for the intermolecular oxidative dehydrogenative 3,3′-coupling of 2-aryl-benzo[b]furans and 2-aryl-benzo[b]thiophenes. | ||
 | ||
| Fig. 2 (Top) UV-vis spectra of DCE solutions of 1a–c and DDQ in a 2:1 ratio. (Bottom) Side view of the stacked packing of DDQ and 1c in the crystal structure of a 1:2 adduct between DDQ and 1c. | ||
In our mechanistic scenario, the actual coupling reaction is initiated by a SET from 1 to DDQ, which is, in turn, triggered and promoted by addition of an acid that increases the oxidative power of DDQ.6 The formed electrophilic radical cation 1+˙ can then react with a nucleophilic neutral substrate 1 to form a radical cation intermediate Int-2, from which product 2 is obtained after elimination of two protons and a second SET. Alternatively, 1+˙ could deprotonate and couple with neutral 1 to form a neutral radical intermediate, leading to 2via elimination of one proton and a second SET. At this point, the radical cation mechanism seems more likely as it is analogous to the one reported for MoCl5-mediated dehydrogenative coupling of arenes.28 Monitoring the reaction with NMR and EPR spectroscopy implies that the purported mechanism is conceivable as the disappearance of the NMR signals is accompanied by an appearance of an EPR signal upon addition of the acid (see the ESI‡). Moreover, our preliminary calculations show that the Int-2 structure is a local minimum on the potential energy surface when 1+˙ and 1 are coupled together via their C3-carbon atoms (see the ESI‡). Detailed computational investigations of the reaction mechanism are currently underway.
Conclusions
We have developed a metal free DDQ/H+ mediated 3,3′-homocoupling reaction of 2-aryl-benzo[b]furans and 2-aryl-benzo[b]thiophenes. The feasibility of the reaction was found to correlate with the oxidation potential of the substrate and the morphology of its frontier molecular orbitals. The usefulness of this approach was demonstrated in a priori examination of the monomer Sophora that was successfully dimerized with the developed synthetic method, resulting in the first total synthesis of a natural product shandougenine B.Experimental
Computational methods
All calculations were performed with the Turbomole 7.0 program package.29 The solvent environment was taken into account with the COSMO approach with a dielectric constant of 10.125 for 1,2-dichloroethane.30 The geometries were optimized using the TPSS functional31 augmented with D3 dispersion correction.32 Ahlrichs’ def2-TZVP basis sets were used for all atoms as implemented in Turbomole.33 A modified integration grid M4 was employed along with the MARI-J approximation augmented with a suitable auxiliary basis set.34,35 Vibrational frequencies were calculated numerically for all stationary points and were found to verify their correspondence to true minima (zero imaginary frequencies) and to obtain Gibbs free energies. Absolute oxidation potentials were calculated using a standard protocol (E° = ΔG298.15 K/nF; where n = 1 and F = 96.48 kcal mol−1 V−1) and were further normalized to the FcH/FcH+ couple in DCE by subtracting 4.927 eV from all calculated values.23 The use of FcH/FcH+ couple as a reference for data employing nonaqueous solvents is recommended by IUPAC.36 Molecular orbitals were visualized with the program VMD.24bGeneral remarks
Unless otherwise specified, all materials were purchased from commercial sources and used as received without further purification. Known benzo[b]thiophenes were prepared with CuI/TMEDA catalysed annulation of 2-bromo alkynylbenzenes.37 Known benzo[b]furans were synthesized from 2-iodophenol in a cascade Sonogashira/5-endo-dig cyclisation reaction similar to that described in the literature.381H, and 13C{1H} NMR spectra were recorded using a Varian Mercury 300 or Varian Inova 500 spectrometer at 27 °C. 1H spectra were referenced to tetramethylsilane (TMS, 0.0 ppm) or to residual solvent peaks (CDCl3 7.26 ppm; d6-DMSO 2.50 ppm, CD3OD 3.31 ppm and CD2Cl2 5.32 ppm). 13C{1H} NMR spectra were referenced to residual solvent peaks (CDCl3 77.16 ppm; d6-DMSO 39.52 ppm and CD3OD 49.00 ppm). HRMS data were acquired with a JEOL JMS-700 instrument using EI ionization mode. UV-vis spectra were recorded on a Varian Cary 50 spectrometer at 27 °C. Cyclic voltammetry was performed with a Gamry Reference 600 potentiostat using a scan rate of 150 mV s−1. Solid samples of 1a–j (1–2 mg) were dissolved into a DCM solution containing 0.05 M of NBu4PF6 as the supporting electrolyte. Potentials were scanned at RT in a single compartment cell fitted with Pt electrodes and referenced to FcH/FcH+ couple. Details of single crystal X-ray structure determinations are given in the ESI.‡
General procedure for homocouplings
Starting material (1 eq.) and DDQ (0.5 eq.) were mixed and dissolved in DCE (0.05 or 0.1 M). The formation of a CT-complex was observed from a colour change and the mixture was cooled to 0 °C and MsOH or TFA (3 or 6 eq.) was added to the mixture. After a given amount of time, the reaction mixture was filtered through a pad of basic Al2O3 and washed with chloroform. Purification of the crude product after evaporation of solvents was performed by flash chromatography (SiO2).:1 → 40:1 (hexanes:EtOAc) afforded 2a in 88% yield (44.7 mg, 0.116 mmol). Spectral data is in accordance with published data.18b1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 7.9 Hz, 4H), 7.62 (d, J = 8.3 Hz, 2H), 7.33 (t, J = 7.6 Hz, 2H), 7.26–7.21 (m, 6H), 7.16–7.08 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 154.5, 152.1, 130.6, 129.6, 128.7, 128.7, 126.4, 125.1, 123.2, 120.9, 111.4, 107.8.
:1 → 40:1 (hexanes:EtOAc) afforded 2b in 92% yield (37.9 mg, 0.0914 mmol). Spectral data is in accordance with published data.18b1H NMR (500 MHz, CDCl3) δ 7.69 (d, J = 8.2 Hz, 4H), 7.63 (d, J = 8.3 Hz, 2H), 7.35–7.30 (m, 2H), 7.16–7.09 (m, 4H), 7.07 (d, J = 8.2 Hz, 4H), 2.30 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 154.4, 152.4, 138.7, 129.8, 129.4, 127.9, 126.3, 124.8, 123.1, 120.7, 111.2, 107.2, 21.5.
:1 (hexanes:EtOAc) afforded 2c in 92% yield (40.1 mg, 0.0898 mmol). Spectral data is in accordance with published data.18b1H NMR (500 MHz, CDCl3) 7.76–7.71 (m, 4H), 7.61 (d, J = 8.2 Hz, 2H), 7.34–7.29 (m, 2H), 7.16–7.09 (m, 4H), 6.82–6.77 (m, 4H), 3.75 (s, 6H). 13C NMR (126 MHz, CDCl3) δ 159.9, 154.3, 152.3, 129.9, 127.9, 124.5, 123.4, 123.0, 120.6, 114.2, 111.1, 106.2, 55.3.
:1 (DCM:MeOH) afforded 2d in 68% yield (9.7 mg, 0.0194 mmol). 1H NMR (500 MHz, DMSO) δ 10.03 (s, 2H), 7.74 (d, J = 8.3 Hz, 2H), 7.61 (d, J = 8.7 Hz, 4H), 7.53 (d, J = 8.7 Hz, 4H), 7.36 (t, J = 7.7 Hz, 2H), 7.14 (t, J = 7.5 Hz, 2H), 7.03 (d, J = 7.7 Hz, 2H), 2.01 (s, 6H). 13C NMR (126 MHz, DMSO) δ 168.5, 153.6, 151.5, 139.9, 128.8, 126.4, 125.0, 124.2, 123.3, 119.9, 118.9, 111.4, 105.9, 24.0. HRMS (EI+) calcd for [C32H24N2O4]+m/z: 500.1736, found: 500.1731.
:1 → 40:1 (hexanes:EtOAc) afforded 2e in 86% yield (26.1 mg, 0.0624 mmol). Spectral data is in accordance with published data.18b1H NMR (500 MHz, CDCl3) δ 7.89 (d, J = 8.0 Hz, 2H), 7.39–7.32 (m, 4H), 7.27–7.22 (m, 2H), 7.12–7.08 (m, 2H), 7.07–7.01 (m, 8H). 13C NMR (75 MHz, CDCl3) δ 142.5, 140.8, 139.0, 134.2, 128.5, 128.4, 127.8, 126.6, 124.8, 124.7, 123.8, 122.3.
:1 (hexanes) → 40:1 (hexanes:DCM) → 20:1 (hexanes:EtOAc) → 10:1 (hexanes:EtOAc) → 1:1 (hexanes:EtOAc) afforded 2f in 86% yield (26.1 mg, 0.0624 mmol). Spectral data is in accordance with published data.18b1H NMR (500 MHz, CDCl3) δ 7.90–7.87 (m, 2H), 7.35–7.29 (m, 4H), 7.24–7.19 (m, 2H), 7.08 (d, J = 8.2 Hz, 4H), 6.93–6.89 (m, 4H), 2.24 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 142.4, 141.0, 138.7, 137.8, 131.4, 129.2, 128.2, 126.2, 124.6, 123.6, 122.2, 21.3.
:1 (hexanes:EtOAc) → 10:1 (hexanes:EtOAc) afforded 2g in 93% yield (44.3 mg, 0.0884 mmol). Spectral data is in accordance with published data.18b1H NMR (500 MHz, CDCl3) δ 7.88 (d, J = 8.0 Hz, 2H), 7.36–7.28 (m, 4H), 7.22 (t, J = 7.5 Hz, 2H), 7.08 (d, J = 8.8 Hz, 4H), 6.64 (d, J = 8.8 Hz, 4H), 3.71 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 159.4, 142.2, 141.0, 138.6, 129.6, 126.8, 125.7, 124.6, 124.5, 123.5, 122.2, 113.9, 55.3.
:1 → 40:1 afforded 2h in 15% yield (5.2 mg, 0.01237 mmol). 1H NMR (500 MHz, CDCl3) δ 7.87 (d, J = 8.1 Hz, 2H), 7.38–7.30 (m, 2H), 7.22–7.18 (m, 6H), 7.07 (d, J = 5.0 Hz, 2H), 6.86 (t, J = 4.1 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ 140.7, 138.3, 137.2, 135.9, 127.5, 127.1, 126.5, 125.3, 125.1, 123.2, 122.2, 77.2. HRMS (EI+) calcd for [C24H14S4]+m/z: 429.9978, found: 429.9984
Benzylated shandougenine B (2k)
Following the general procedure under dry conditions and under Ar with 16.1 mg (0.0357 mmol, 1 eq.) of 1k, 4.1 mg (0.0179 mmol, 0.5 eq.) of DDQ, 0.72 mL of DCE and 24.4 mg (0.214 mmol, 6 eq., 16.5 μl) of TFA. Purification of the crude product with 20:1 → 5:1 → 3:1 (hexanes:EtOAc) afforded 2k in 80% yield (12.9 mg, 0.01435 mmol). 1H NMR (300 MHz, CDCl3) δ 7.42–7.31 (m, 10H), 7.23–7.16 (m, 6H), 7.09–7.03 (m, 4H), 7.01–7.00 (m, 2H), 6.96 (d, J = 8.5 Hz, 2H), 6.67–6.66 (m, 2H), 6.31 (d, J = 2.3 Hz, 2H), 6.26 (dd, J = 8.5, 2.3 Hz, 2H), 5.96 (dd, J = 5.1, 1.3 Hz, 4H), 4.91 (s, 4H), 4.61 (s, 4H). 13C NMR (75 MHz, CDCl3) δ 160.2, 157.4, 150.3, 149.8, 145.9, 144.2, 136.9, 131.6, 128.8, 128.3, 128.2, 127.6, 126.9, 122.8, 113.9, 110.4, 105.9, 101.2, 100.9, 99.4, 93.3, 70.2, 69.9.
Shandougenine B
2k (6.5 mg, 0.00723 mmol) and 25 mg of 5% Pd/C (wetted ∼50% H2O) in 2.5 mL ethyl acetate were stirred vigorously for 10 h 30 min under a H2-balloon. After filtration through diatomaceous earth, evaporation of volatiles and redissolvation to CD3OD, shandougenine B was obtained in 73% yield (NMR). The analytical sample was purified with column chromatography using 10:1 (DCM:MeOH) as the eluent. Spectral data is in accordance with published data.14b1H NMR (500 MHz, CD3OD) δ 7.40 (d, J = 8.5 Hz, 2H), 6.99 (s, 2H), 6.40 (dd, J = 8.5, 2.4 Hz, 2H), 6.27 (s, 2H), 6.19 (d, J = 2.3 Hz, 2H), 5.85 (d, J = 7.2 Hz, 4H). 13C NMR (75 MHz, CD3OD) δ 160.8, 157.0, 151.5, 151.1, 147.2, 145.5, 131.8, 123.5, 111.6, 110.7, 108.8, 103.7, 102.4, 99.8, 93.9. HRMS (EI+) calcd for [C30H18O10]+m/z 538.0900 found: 538.0899.
Acknowledgements
TW is grateful to Finnish Cultural, Emil Aaltonen, Magnus Ehrnrooth and Otto Malm Foundations for funding. Financial support from the Academy of Finland [project no. 129062 (J. H.) and project no. 289172 (H. M. T.)] is acknowledged. The Finnish National Centre for Scientific Computing (CSC) is recognized for computational resources. M.Sc. Mikko Mäkelä and M.Sc. Otto Seppänen from University of Helsinki are acknowledged for their help in kinetic experiments.Notes and references
- S. Patai and Z. Rappoport, The Chemistry of the quinonoid compounds, Wiley, Chichester, 1988, vol. 2 Search PubMed.
- F. Wurche, W. Sicking, R. Sustmann, F.-G. Klärner and C. Rüchardt, Chem. – Eur. J., 2004, 10, 2707–2721 CrossRef CAS PubMed.
- Selected reviews: (a) M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2013, 52, 9900–9930 CrossRef CAS PubMed; (b) A. E. Wendlandt and S. S. Stahl, Angew. Chem., Int. Ed., 2015, 54, 14638–14658 CrossRef CAS PubMed; (c) R. Narayan, K. Matcha and A. P. Antonchick, Chem. – Eur. J., 2015, 21, 14678–14693 CrossRef CAS PubMed.
- Oxidative C–H functionalization reviews: (a) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed; (b) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138–12204 CrossRef CAS PubMed.
- Triplet state DDQ benzene oxidation: K. Ohkubo, A. Fujimoto and S. Fukuzumi, J. Am. Chem. Soc., 2014, 135, 5368–5371 CrossRef PubMed.
- DDQ Ered of 0.6 V (vs. SCE) is shifted up to ∼1.7 V (vs. SCE) with an acid additive (a)–(c); (a) L. Zhai, R. Shukla, S. H. Wadumethrige and R. Rathore, J. Org. Chem., 2010, 75, 4748–4760 CrossRef CAS PubMed; (b) L. Zhai, R. Shukla and R. Rathore, Org. Lett., 2009, 11, 3474–3477 CrossRef CAS PubMed; (c) I. M. Matheson, O. C. Musgrave and C. J. Webster, Chem. Commun., 1965, 278–279 RSC; (d) T. S. Navale, K. Thakur and R. Rathore, Org. Lett., 2011, 13, 1634–1637 CrossRef CAS PubMed; (e) S. Fukuzumi, K. Ishikawa, K. Hironaka and T. Toshio, J. Chem. Soc., Perkin Trans. 2, 1987, 751–760 RSC; (f) R. Rathore and J. K. Kochi, Acta Chem. Scand., 1998, 52, 114–130 CrossRef CAS; (g) K. L. Handoo and K. Gadru, Curr. Sci., 1986, 55, 920–922 CAS; (h) O. Hammerich and D. V. Parker, Acta Chem. Scand. Ser. B, 1982, 36, 63–64 CrossRef; (i) H. Y. Cho and L. T. Scott, Tetrahedron Lett., 2015, 56, 3458–3462 CrossRef CAS.
- McMurry coupling of ketones followed by an oxidation with DDQ: (a) N. G. Andersen, M. Parvez and B. A. Keay, Org. Lett., 2000, 2, 2817–2820 CrossRef CAS PubMed; (b) N. G. Andersen, M. Parvez, R. McDonald and B. A. Keay, Can. J. Chem., 2004, 82, 145–161 CrossRef CAS.
- Transition metal mediated homocoupling of 3-halogenbenzo[b](thio)furans: (a) T. Benincori, E. Brenna, F. Sannicolò, L. Trimarco, P. Antognazza, E. Cesarotti, F. Demartin and T. Pilati, J. Org. Chem., 1996, 61, 6244–6251 CrossRef CAS PubMed; (b) T. Benincori, E. Brenna, F. Sannicolò, L. Trimarco, P. Antognazza and E. Cesarotti, J. Chem. Soc., Chem. Commun., 1995, 685–686 RSC; (c) M. Wynberg and M. Cabell, J. Org. Chem., 1973, 38, 2814–2816 CrossRef; (d) T. Qi, Y. Guo, Y. Liu, H. Xi, H. Zhang, X. Gao, Y. Liu, K. Lu, C. Du, G. Yu and D. Zhu, Chem. Commun., 2008, 6227–6229 RSC; (e) Y. Fort, S. Becker and P. Caubère, Tetrahedron, 1994, 50, 11893–11902 CrossRef CAS; (f) Z. Wang, C. Zhao, D. Zhao, C. Li, J. Zhang and H. Wang, Tetrahedron, 2010, 66, 2168–2174 CrossRef CAS; (g) M. Mézlova, H. Petříčková, P. Maloň, V. Kozmík and J. Svoboda, Collect. Czech. Chem. Commun., 2003, 68, 1020–1038 CrossRef.
- Gold-catalyzed oxidative coupling of 2-arylbenzo[b]furans: M. G. Auzias, M. Neuburger and H. A. Wegner, Synlett, 2010, 2443–2448 CAS.
- Acid mediated Friedel–Crafts type coupling to form a dihydrogenated benzofused dimer followed by ODH: (a) benzo[b]thiofurans in 10% H3PO4: A. R. Katritzky and M. Balasubramanian, Energy Fuels, 1992, 6, 431–438 CrossRef CAS; (b) PPA-SiO2 mediated coupling of furanocoumarins: S. Koul, T. K. Razdan, C. S. Andotra, A. K. Kalla, S. Koul and S. C. Taneja, Synth. Commun., 2002, 32, 1529–1534 CrossRef CAS.
- BuLi/S8 mediated coupling of benzo[b]furans: T. Janosik, B. Stensland and J. Bergman, J. Org. Chem., 2002, 67, 6220–6223 CrossRef CAS PubMed.
- Palladium catalyzed coupling of 3-halogen and 3-stannate/boronic acid derivatives: (a) L.-Y. Yang, C.–F. Chang, Y.–C. Huang, Y.-J. Lee, C. C. Hu and T.-H. Tseng, Synthesis, 2009, 1175–1179 CAS; (b) O. Galangau, T. Nakashima, F. Maurel and T. Kawai, Chem. – Eur. J., 2015, 21, 8471–8482 CrossRef CAS PubMed; (c) K. Tanaka, H. Suzuki and H. Osuga, Tetrahedron Lett., 1997, 38, 457–460 CrossRef CAS.
- Homocoupling of boronic acids: S. A. R. Mulla, S. S. Chavan, M. Y. Pathan, S. M. Inamdar and T. M. Y. Shaihk, RSC Adv., 2015, 5, 24675–24680 RSC.
- (a) K.-S. Song and I. Raskin, J. Nat. Prod., 2002, 65, 76–78 CrossRef CAS PubMed; (b) G. Luo, Y. Yang, M. Zhou, Q. Ye, Y. Liu, J. Gu, G. Zhang and Y. Luo, Fitoterapia, 2014, 99, 21–27 CrossRef CAS PubMed.
- V. Simonini, M. Benaglia and T. Benincori, Adv. Synth. Catal., 2008, 350, 561–564 CrossRef CAS.
- S. Arnaboldi, T. Benincori, R. Cirilli, W. Kutner, M. Magni, P. R. Mussini, K. Noworyta and F. Sannicolò, Chem. Sci., 2015, 6, 1706–1711 RSC.
- Originally, the idea of acid catalyzed dimerization of benzo-fused heterocycles is presented in skatole 2,2′-dimerization in HCl diethylether: F. Smith and A. E. Walter, J. Chem. Soc., 1961, 940–943 RSC.
- (a) J. E. Perea-Buceta, T. Wirtanen, O.-V. Laukkanen, M. K. Mäkelä, M. Nieger, M. Melchionna, N. Huittinen, J. A. Lopez-Sanchez and J. Helaja, Angew. Chem., Int. Ed., 2013, 52, 11835–11839 CrossRef CAS PubMed; (b) T. Wirtanen, M. K. Mäkelä, J. Sarfraz, P. Ihalainen, S. Hietala, M. Melchionna and J. Helaja, Adv. Synth. Catal., 2015, 357, 3718–3726 CrossRef CAS.
- In DCM, TFA fails to promote the catalysis of 1a to 2a at 0.15 M, while the coupling proceeds effectively in 0.3 M TFA in DCE. In the case of 1c, the TFA concentration of 0.2 M in DCM is enough to promote the coupling to 2c.
- R. Carlson and J. E. Carlson, Design and Optimization in Organic Synthesis, Elsevier, 2005, ISBN: 0-444-51527-5 Search PubMed.
- It can be concluded that the molarity was the most important parameter among those tested.
- M. Namazian, C. Y. Lin and M. L. Coote, J. Chem. Theory Comput., 2010, 6, 2721–2725 CrossRef CAS PubMed.
- The systematic underestimation of oxidation potentials with pure DFT functionals is well-known: M.-H. Baik and R. A. Friesner, J. Phys. Chem. A, 2002, 106, 7407–7412 CrossRef CAS.
- (a) For example, the energy difference between the SOMO of radical 1a+˙ and the HOMO and LUMO of neutral 1a is −1.41 and −4.49 eV, respectively; ; (b) Orbitals visualized using VMD software with 0.06 cutoff: W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- B. A. McKittrick, R. T. Scannell and R. Stevenson, J. Chem. Soc., Perkin Trans. 1, 1982, 3017–3020 RSC.
- From the mixture of DDQ and 1c in DCM–hexane, we were able to grow a 1:2 DDQ:1c crystal suitable for X-ray characterization.
- Charge-transfer complex has been shown to be a real intermediate in hydride transfer from the Michler's hydride to DDQ: K. M. Zaman, S. Yamamoto, N. Nishimura, J. Maruta and S. Fukuzumi, J. Am. Chem. Soc., 1994, 116, 12099–12100 CrossRef CAS.
- The radical cation mechanism is analogous to the one found in mechanistic study of MoCl5-mediated dehydrogenative coupling of arenes: J. Leppin, M. Schubert, S. R. Waldvogel and K. Heinze, Chem. – Eur. J., 2015, 21, 4229–4232 CrossRef CAS PubMed.
- TURBOMOLE V7.0 2015, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- A. Schafer, A. Klamt, D. Sattel, J. C. W. Lohrenz and F. Eckert, Phys. Chem. Chem. Phys., 2000, 2, 2187–2193 RSC.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. Sierka, A. Hogekamp and R. Ahlrichs, J. Chem. Phys., 2003, 118, 9136–9148 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- G. Gritzner and J. Kuta, Pure Appl. Chem., 1984, 56, 461–466 CrossRef.
- L.-L. Sun, C.-L. Deng, R.-Y. Tang and X.-G. Zhang, J. Org. Chem., 2011, 76, 7546–7550 CrossRef CAS PubMed.
- N. G. Kundu, M. Pal, J. S. Mahanty and S. K. Dasgupta, J. Chem. Soc., Chem. Commun., 1992, 41–42 RSC.
Footnotes |
| † This paper is dedicated to Professor Ari M. P. Koskinen on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Crystallographic data, xyz-coordinates, cyclic voltammetry data and EPR, 1H and 13C NMR spectra (PDF). And X-ray crystallographic data for DDQ:1c (1:2) complex (CIF). CCDC 1477482 (1c_DDQ). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00331a |
| This journal is © the Partner Organisations 2016 |