
Approach to the synthesis of the C1–C11 and C14–C18 portion of Leucascandrolide A†‡
T. J.
Hunter§
a,
J.
Zheng§
b and
G. A.
O'Doherty
*b
aMilliporeSigma, 645 Science Drive, Madison, Wisconsin 53711, USA
bDepartment of Chemistry and Chemical Biology, Northeastern University, Boston, Massachusetts 02115, USA. E-mail: g.odoherty@neu.edu
First published on 12th July 2016
Abstract
An asymmetric synthesis of the C1 to C11 and C14 to C18 fragments of the macrocyclic portion of the antibiotic Leucascandrolide A was achieved in 21 total steps from an achiral dienoate. The key 4-hydroxy-2,5-pyran portion of the natural product was established by oxy-Michael cyclization of a 5,7,9,11-tetraol intermediate, which in turn was established by an iterative asymmetric-hydration of dienoates. Alternative strategies for establishing the polyol stereochemistry were explored.
Introduction
As part of humankind's continuous search for unique biologically active structures from the deep blue sea, Leucascandrolide A (1) was discovered in the Coral Sea off the northeastern coast of New Caledonia.1 In 1996, Pietra reported the isolation of the macrocyclic lactone from the calcareous sponge Leucascandra caveolata. A combination of 2D-NMR and Mosher ester analysis was used to assign the absolute and relative stereochemistry of the macrocyclic natural product. In addition to its novel structure, interest in Leucascandrolide A was further peaked by discovery that it possessed potent anticancer and antifungal activity, with IC50 value against the human KB (70 nM) and P388 (350 nM) cell lines and activity against Candida albicans.1 As is often the problem associated with marine natural products isolated from sponges, as opposed to the bacteria that most likely produce it, Leucascandrolide A no longer appears to be available from the same sponge.2 These factors have inspired synthetic organic chemist to pursue the total synthesis of Leucascandrolide A (Fig. 1).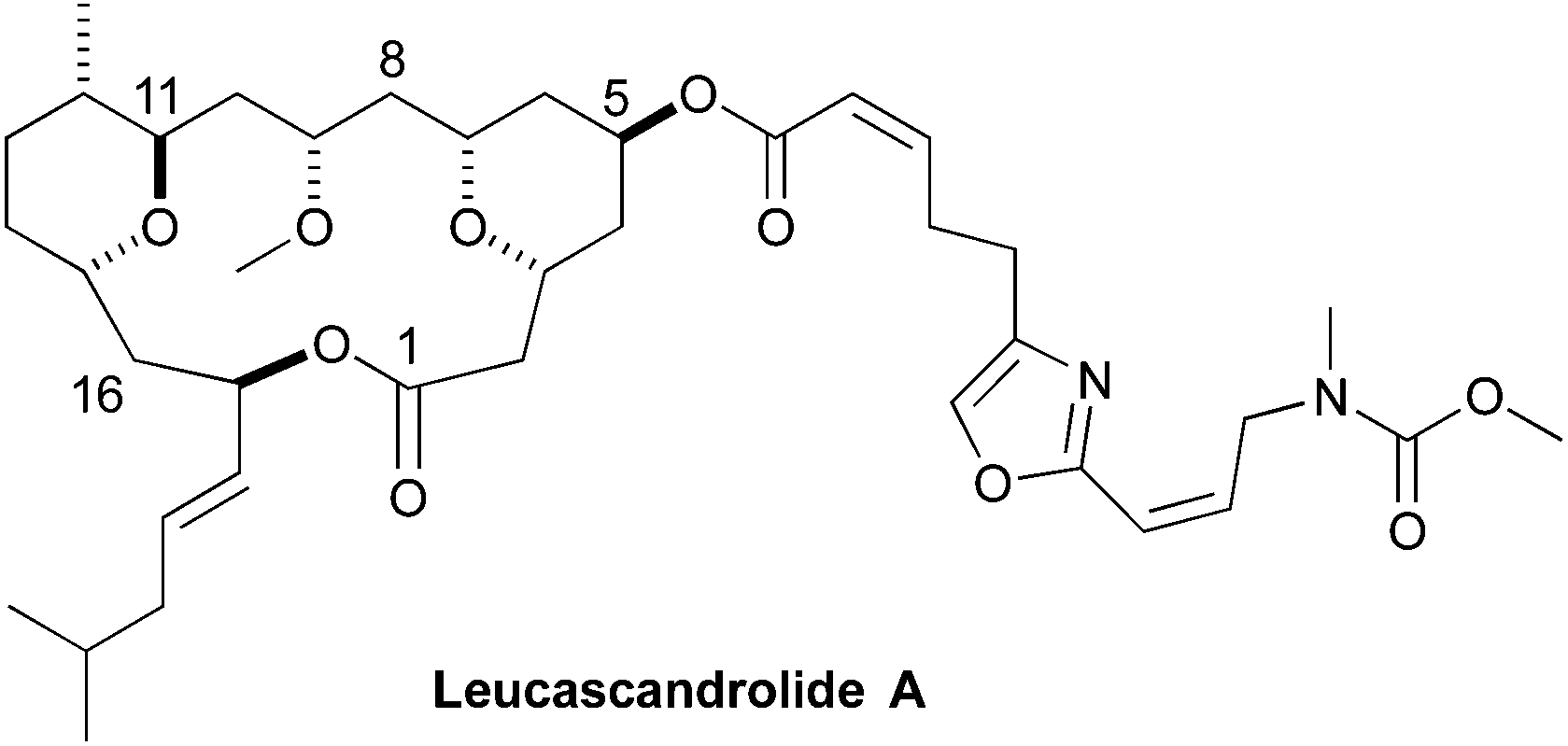 | ||
| Fig. 1 Structure of Leucascandrolide A. | ||
A mere four years after its isolation, the first total synthesis of Leucascandrolide A was completed by Leighton.3 Since the Leighton synthesis there have been 10 other total4 and 11 formal syntheses.5 More recently, Kozmin was able to show that Leucascandrolide A inhibits oxidative phosphorylation via interaction with cytochrome bc1 complex.6 As part of a larger program aimed at the synthesis and SAR-study of anticancer agents that act by inhibition of ion transport, we became interested in the synthesis of Leucascandrolide A.
Retrosynthetically, we envisioned that the Leucascandrolide A macrocycle (1) could be derived by ring closing cross metathesis and reductive cyclization of diene 2 (Scheme 1). Compound 2 in turn could be derived from an anti-selective crotylation of aldehyde 3. Aldehyde 3 could be prepared by an esterification of alcohol 4 and acid 5. Carboxylic acid 5 could be derived from protected tetraol 6, which could result from a diastereoselective allylation/cross metathesis of protected trihydroxy ester 7.7 Previously we have shown that esters such as 7 can be derived by the iterative asymmetric hydration of dienoates like 8.8 The β-hydroxy enone 4 was also envisioned as ultimately arising from an asymmetric hydration of a dienoate. Herein we describe our latest efforts aimed at the synthesis of the C1 to C11 and C14 to C18 containing fragment of Leucascandrolide A, aldehyde 3.
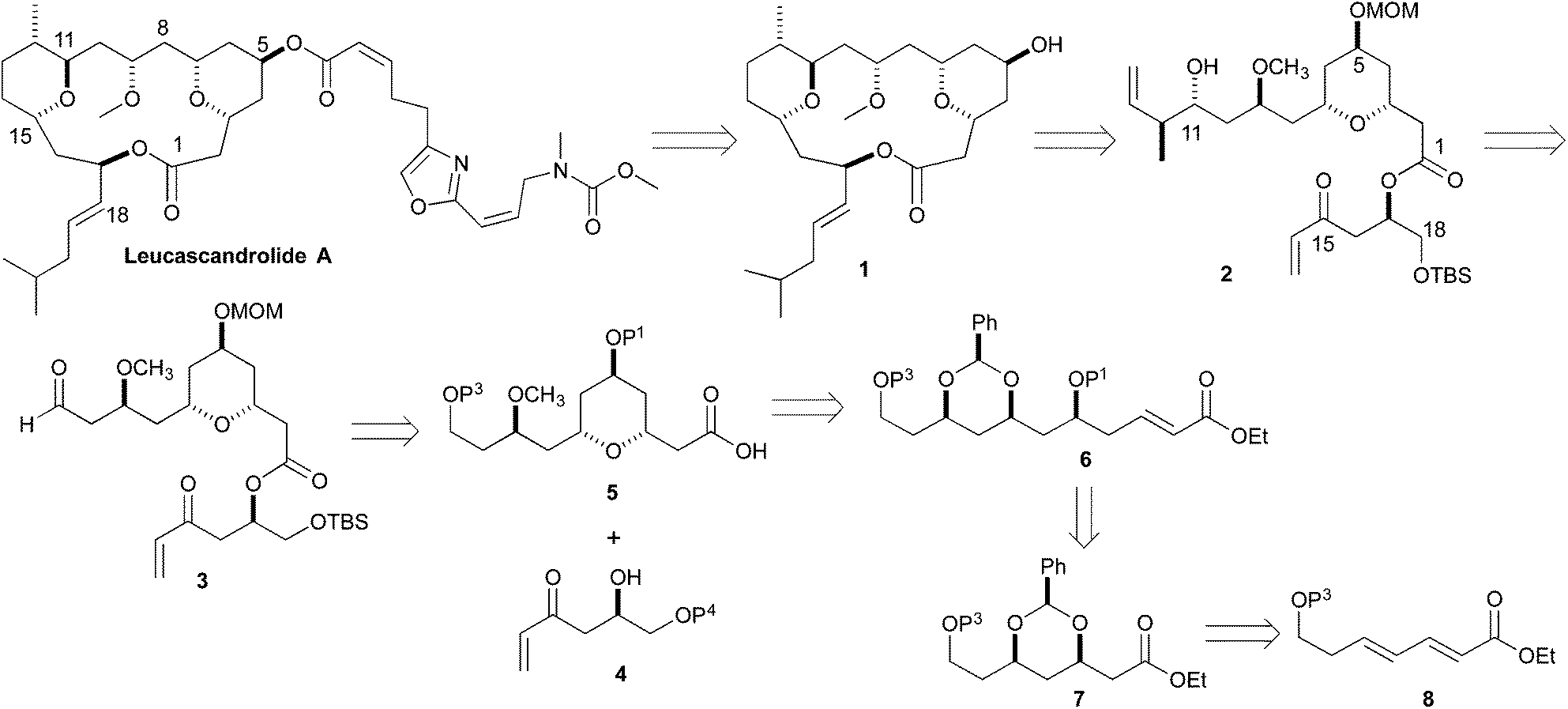 | ||
| Scheme 1 Retrosynthesis of Leucascandrolide A. | ||
Results and discussion
Our synthetic efforts aimed at Leucascandrolide A began with aldehyde 9 (Scheme 2), which we previously had shown could be made from the DibalH reduction of esters like 7.9 Exposure of aldehyde 9 to the vinylogous Horner–Wadsworth–Emmons reagent 10 and LiOH as base gave the desired dienoate 11 (68%),10 which could be converted into the desired syn/anti-1,3,5-triol by our three step asymmetric hydration protocol. Thus, treating the E,E-dienoate 11 to the typical Sharpless asymmetric dihydroxylation conditions (5% OsO4, 6% (DHQ)2PHAL, K3FeCN6, MeSO2NH2), provided diol 12 in good yield (71%) and as a single diastereomer.11 Exposing diol 12 to a pyridine solution of triphosgene converted it into a cyclic carbonate 13 in good yield (79%). When cyclic carbonate 13 was exposed to our Pd-reduction conditions (HCO2H/Et3N, 1% Pd2(dba)3·CHCl3/5% PPh3) it cleanly reacted to give the 5-hydroxy-1-enoate anti-14a in an excellent yield (79%).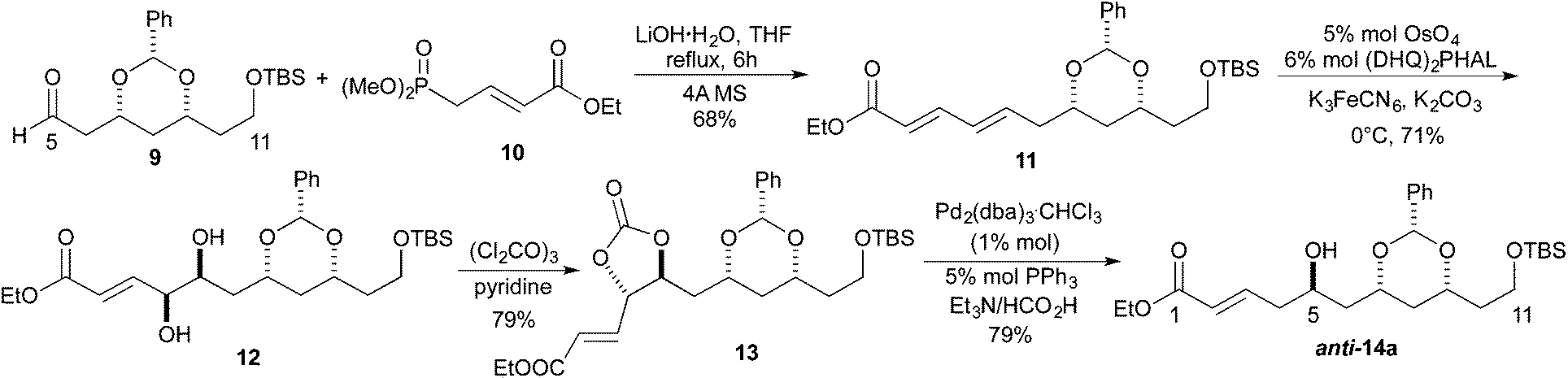 | ||
| Scheme 2 Os/Pd approach to protected triol. | ||
Following a nearly identical procedure, we previously have shown that anti-14b (Table 1), with a C11 PMB-group, can be prepared from a dienoate like 8 (P3 = PMB).8 Similarly, the C5 diastereomer syn-14b, was also prepared from the same dienoate, by simply replacing the ligand in the Sharpless dihydroxylation to (DHQD)2PHAL.
|
|
||||
|---|---|---|---|---|
| Entry | SM | R1 | R2 | Yield (syn-15) |
| Reaction conditions:a TBDPSCl (2 equiv.), imidazole (2.5 equiv.), DMF, 18 h.b Benzyl trichloroacetimidate (1.2 equiv.), TMSOTf, CH2Cl2.c BnBr (2 equiv.), NaH, TBAI, THF, 24 h.d MOMCl (4 equiv.), DMAP (3% equiv.), i-Pr2NEt, CH2Cl2, 0 °C.e NR = no reaction. | ||||
| 5d | syn- 14b | PMB | MOM | 52% (syn-15d) |
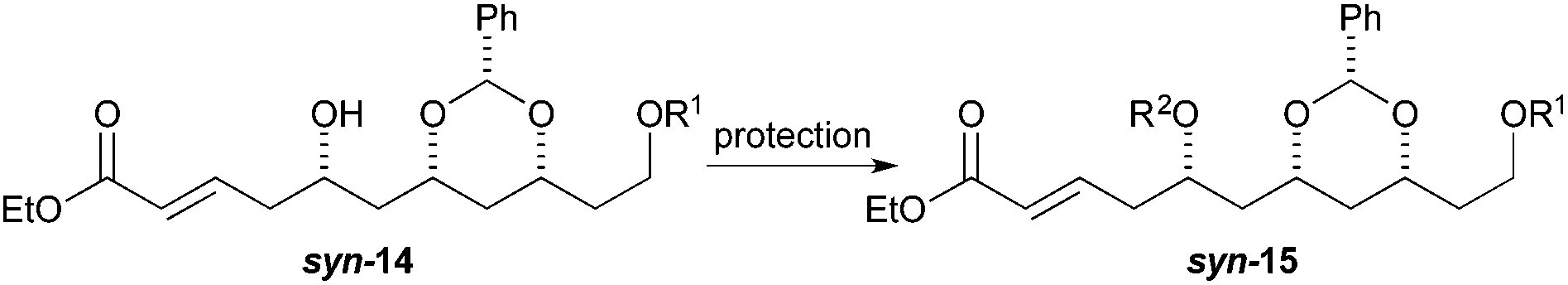
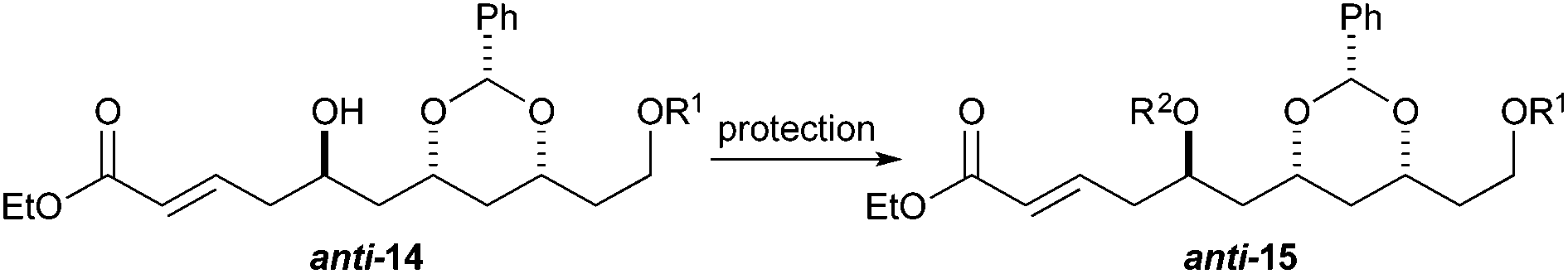
To our surprise, we found it difficult to protect the C5 alcohol of anti-14a with either a TBDPS-group or a benzyl-group (Table 1). The benzyl protection was similarly problematic, whether acidic or basic conditions were used. In contrast, we were able to react alcohol anti-14b with MOMCl to give MOM-ether anti-15d in a satisfactory yield (47%). To our delight, the C5 diastereomer syn-14b reacted under a similar procedure to give syn-15d in a similar yield (52%).
We surmised that the difficulty associated with the protection of the C5 hydroxyl group could be due to the acidity of the C4 position and the propensity of the substrate to undergo elimination to form dienoates 11. Thus we decided to explore an alternative procedure (Scheme 3) for the synthesis of enoates, like syn-15. Specifically, we decided to pursue a route where the protection step occurred before the ester group was introduced. This revised route was envisioned to occur through an allylation, protection and cross-metathesis sequence.
 | ||
| Scheme 3 Alternative synthesis of protected triol. | ||
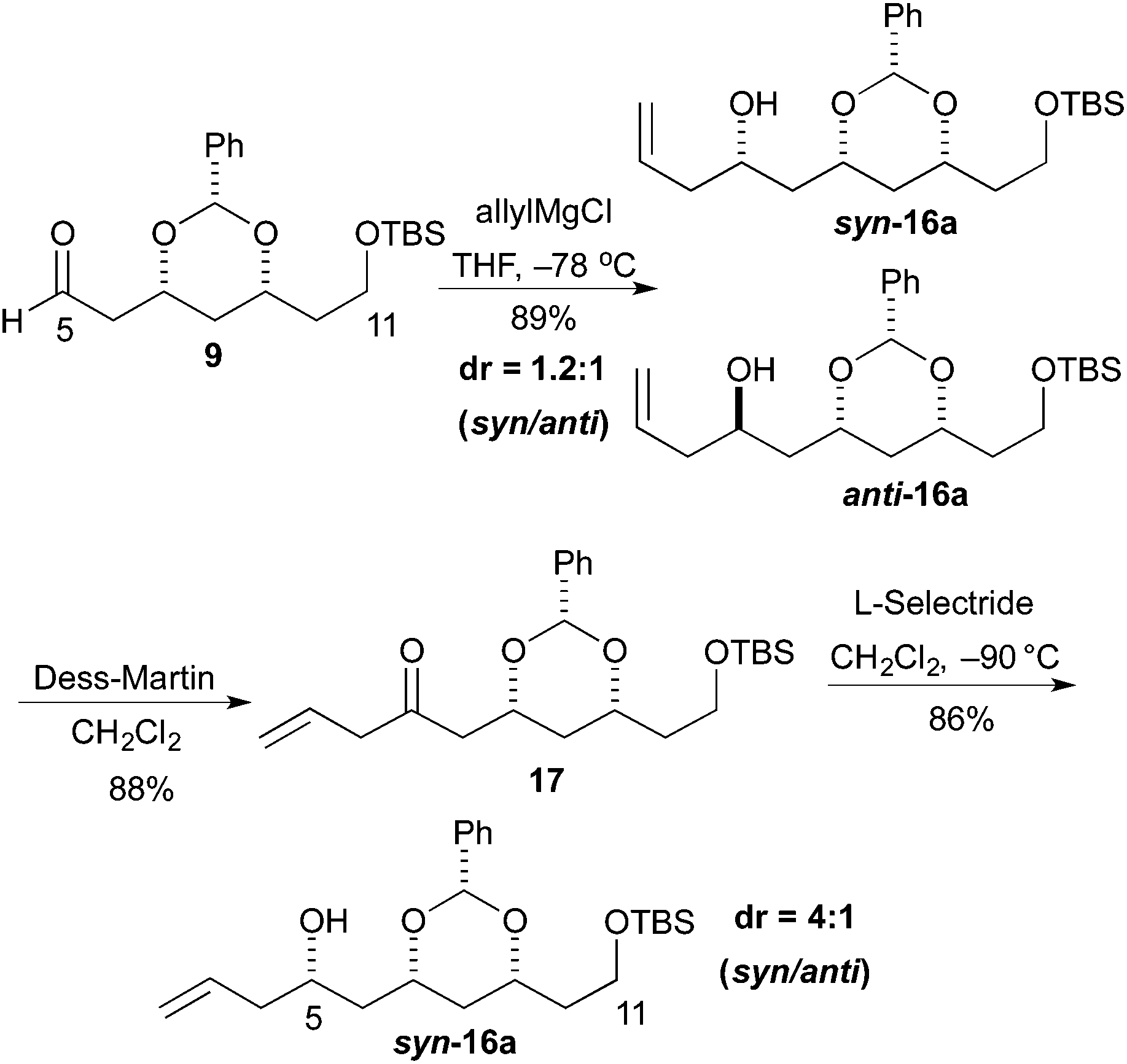
The revised route began with the unselective allylation of aldehyde 9 with an allylic Grignard reagent (AllylMgCl). The Grignard addition occurred to give a mixture of diastereomeric homo-allylic alcohols syn-16a and anti-16a (1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), which could be separated by silica gel chromatography. Fortunately, the diastereoselectivity for the formation of syn-16a could be improved by an oxidation/reduction sequence (Scheme 3).
:1), which could be separated by silica gel chromatography. Fortunately, the diastereoselectivity for the formation of syn-16a could be improved by an oxidation/reduction sequence (Scheme 3).
Thus, the mixture of alcohols syn/anti-16a was treated with the Dess–Martin reagent to give β,γ-unsaturated enone 17 in good yield (88%). Then reduction of enone 17 with L-selectride occurred to give the syn-16a as the major isomer. When the reaction was performed at −90 °C the maximal syn/anti ratio was achieved (>4:1).
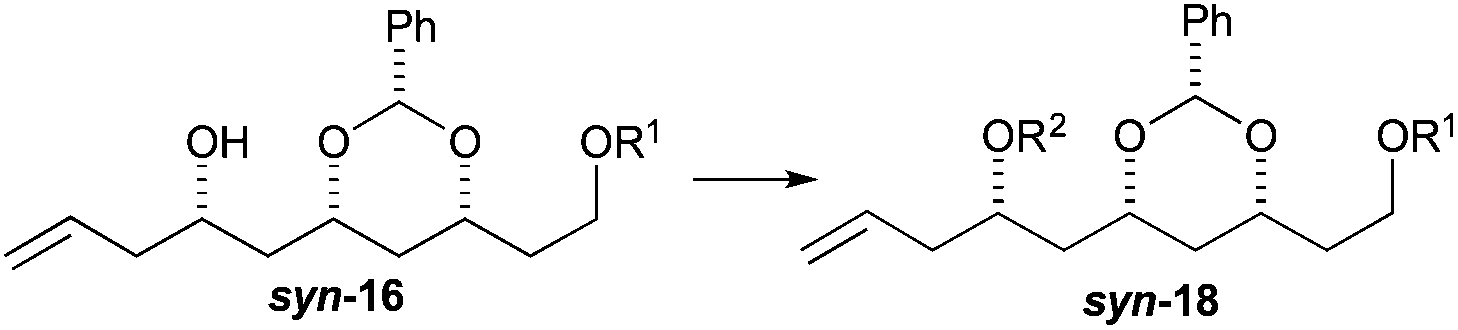
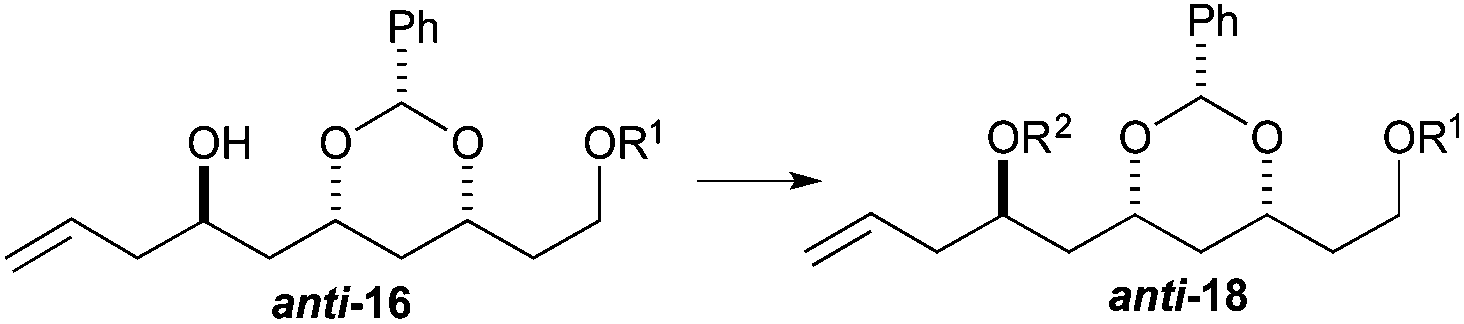
To our delight we were able to protect both homo-allylic alcohols syn-16a and anti-16a with both the TBDPS and Bn– ether groups, as well as the MOM-group. While the yields for the benzyl protection of either the syn- or anti-homoallylic alcohols were suboptimal, both TBDPS- and MOM-groups could be installed in excellent yields (78–87%), providing ample quantities syn-18a and syn-18d and anti-18a and anti-18d to pursue the cross metathesis protocol (Table 2).
|
|
||||
|---|---|---|---|---|
| Entry | SM | R1 | R2 | Yield (anti-18) |
| Reaction conditions:a TBDPSCl (2 equiv.), imidazole (2.5 equiv.), DMF, 18 h.b Benzyl trichloroacetimidate (1.2 equiv.), TMSOTf, CH2Cl2.c BnBr (2 equiv.), NaH, TBAI, THF, 24 h.d MOMCl (4 equiv.), DMAP (3% equiv.), i-Pr2NEt, CH2Cl2, 0 °C. | ||||
| 5a | anti- 16a | TBS | TBDPS | 78% (anti-18a) |
| 6b | anti- 16b | PMB | Bn | 52% (anti-18c) |
| 7d | anti- 16b | PMB | MOM | 85% (anti-18d) |
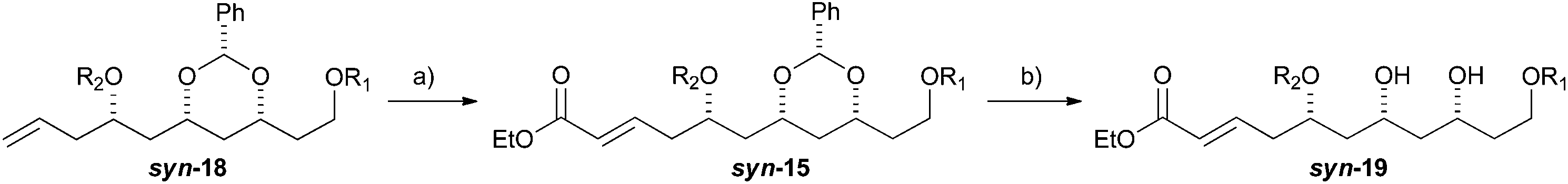
With an improved procedure for installing protecting groups at the C5 position in syn-/anti-18, we next investigated the cross metathesis of syn-18 and anti-18 to form syn-15 and anti-15 and the subsequent deprotection to form diols syn-19 and anti-19. Treatment of syn-18b–d with ethyl acrylate and Grubbs II catalyst (1.5%) cleanly provided the desired enoates syn-15b–d in excellent yields (90–96%). Unfortunately, the primary TBS-groups were not compatible with the benzylidene deprotection conditions, whereas the PMB-group survived. The deprotection of the benzylidene group was accomplished by heating the enoates syn-15c/d in 80% aqueous acetic acid (60 °C) to provide the syn-diols syn-19c/d (72% and 80%). Following a similar procedure, the anti-diastereomer anti-16d was also converted into anti-19d (Table 3).
|
|
|||||
|---|---|---|---|---|---|
| Entry | SM | R1 | R2 | Yield (syn-15) | Yield (syn-19) |
| 1 | syn- 18b | TBS | Bn | 96% (syn-15b) | NAc (syn-19b) |
| 2 | syn- 18c | PMB | Bn | 91% (syn-15c) | 72% (syn-19c) |
| 3 | syn- 18d | PMB | MOM | 90% (syn-15d) | 80% (syn-19d) |
|
|
|||||
|---|---|---|---|---|---|
| Entry | SM | R1 | R2 | Yield (anti-15) | Yield (anti-19) |
| Reaction conditions:a Ethyl acrylate (2.5 equiv.), Grubbs II catalyst (1.5% equiv.), CH2Cl2, reflux, 4 h.b 80% acetic acid/H2O, 60 °C, 4 h.c Not attempted. | |||||
| 4 | anti- 18a | TBS | TBDPS | 88% (anti-15a) | low (anti-19a) |
| 5 | anti- 18b | PMB | MOM | 92% (anti-15d) | 71% (anti-19d) |
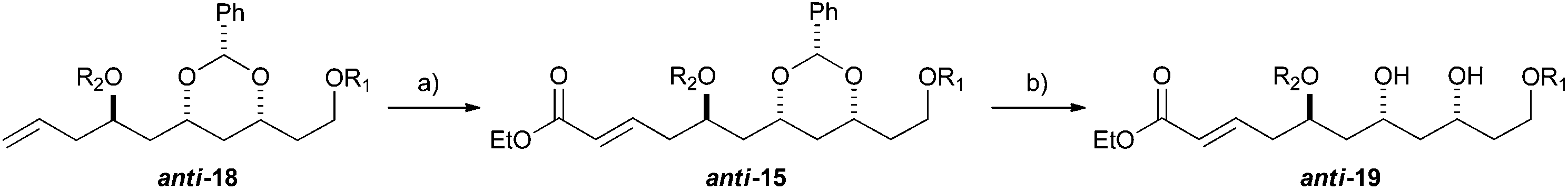
We next pursued the formation of the 2,6-cis-pyran ring. This was most easily accomplished under basic conditions. The addition of 1 equiv. of potassium t-butoxide to a −40 °C solution of syn-19c in THF, followed by warming to room temperature led to the formation of tetrahydropyran syn-20c (70%). Under similar condition syn-19d was converted into tetrahydropyran syn-20d (61%). These conditions produced tetrahydropyran syn-21c in 70% yield as a 6:1 ratio of cis and trans diastereomers, and tetrahydropyran syn-21d product in a 61% yield with a 7:1 ratio of cis to trans diastereomers. Formation of the methyl ether was accomplished using Me3OBF4 on syn-20c and syn-20d to produce the products syn-21c and syn-21d in excellent yields (75 and 97%, respectively) (Scheme 4). Turning to the synthesis of the C1 to C11 and the C14 to C18 portion of Leuscandrolide A, we undertook the synthesis of alcohol 24 from δ-hydroxy enoate 22. Previously we have shown that 22 can be prepared by an asymmetric hydration of the corresponding dienoate.8 The double bond of enoate 22 was oxidatively cleaved by a dihydroxylation (OsO4/NMO)/diol cleavage (NaIO4) sequence to form aldehyde 23. Addition of vinylmagnesium bromide to the aldehyde 23 and manganese dioxide oxidation of the allylic alcohol completed the synthesis of enone 24 (Scheme 5).
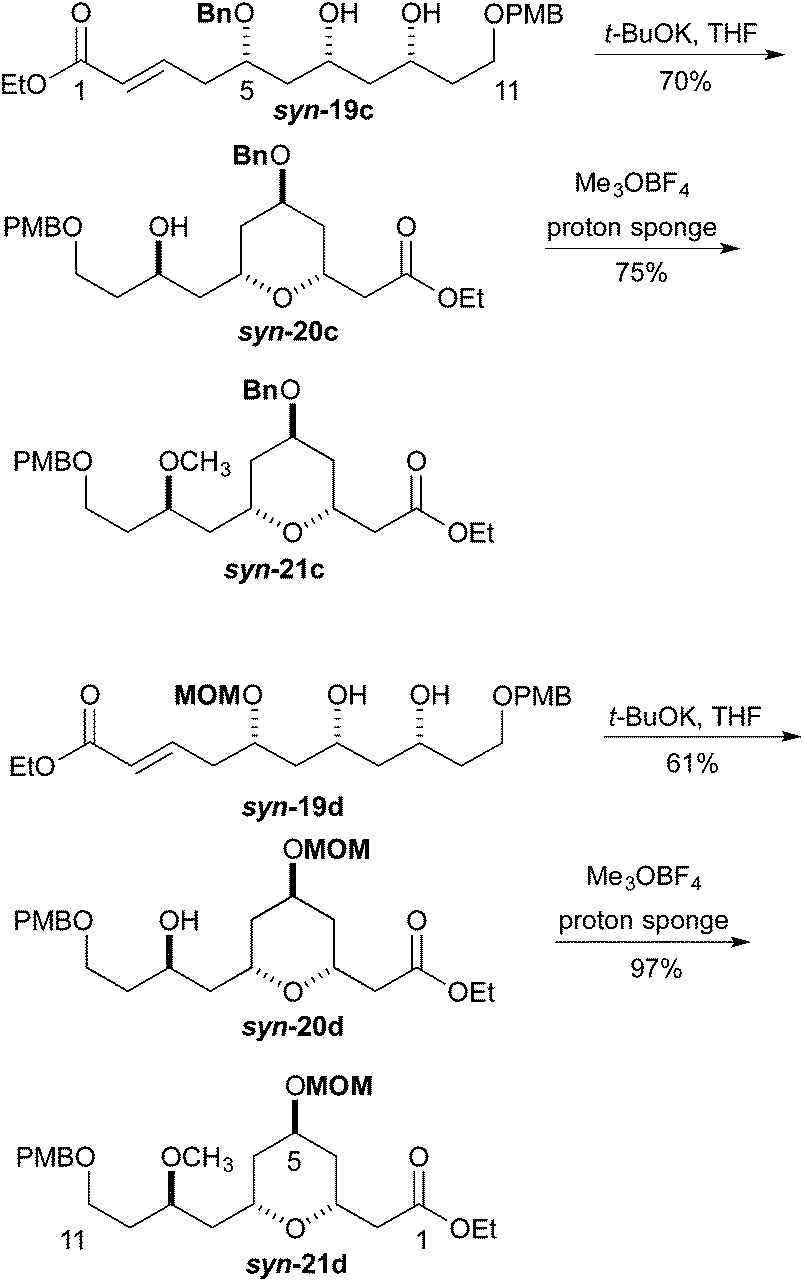 | ||
| Scheme 4 Synthesis of tetrahydropyran. | ||
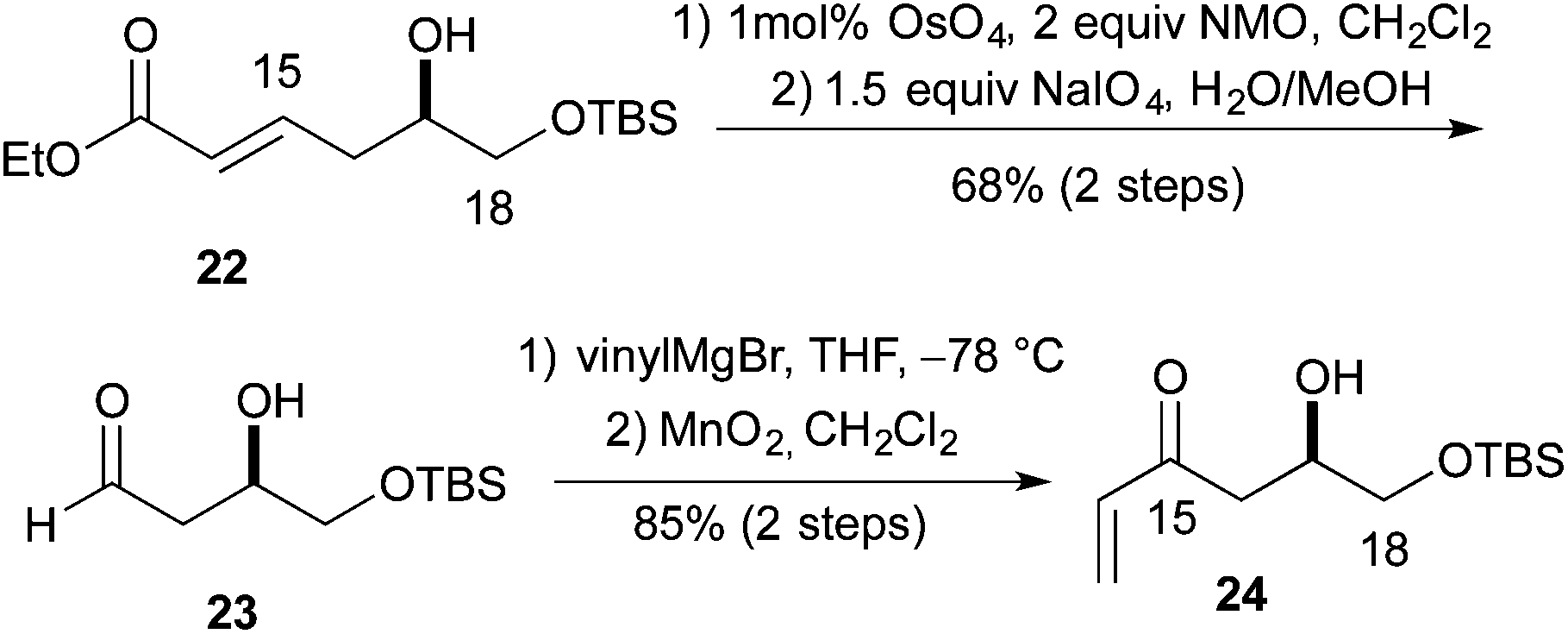 | ||
| Scheme 5 Synthesis of enone. | ||
To demonstrate the viability of the synthesis going forward, we chose to pursue the fragment coupling sequence with ester syn-21d. With the synthesis of enone 24 complete, the ester 26 was synthesized via a DCC coupling of the carboxylic acid of ester 25 and alcohol 24. The hydrolysis of ester syn-21d was accomplished by treatment with LiOH in THF and H2O. The conversion of the PMB-ether to the aldehyde began with a DDQ deprotection. Treatment of a dichloromethane solution of 26 with H2O and DDQ produced the primary alcohol 27 in 78% yield. Oxidation of the primary alcohol 27 to the aldehyde 3 was accomplished with Dess–Martin reagent (Scheme 6).
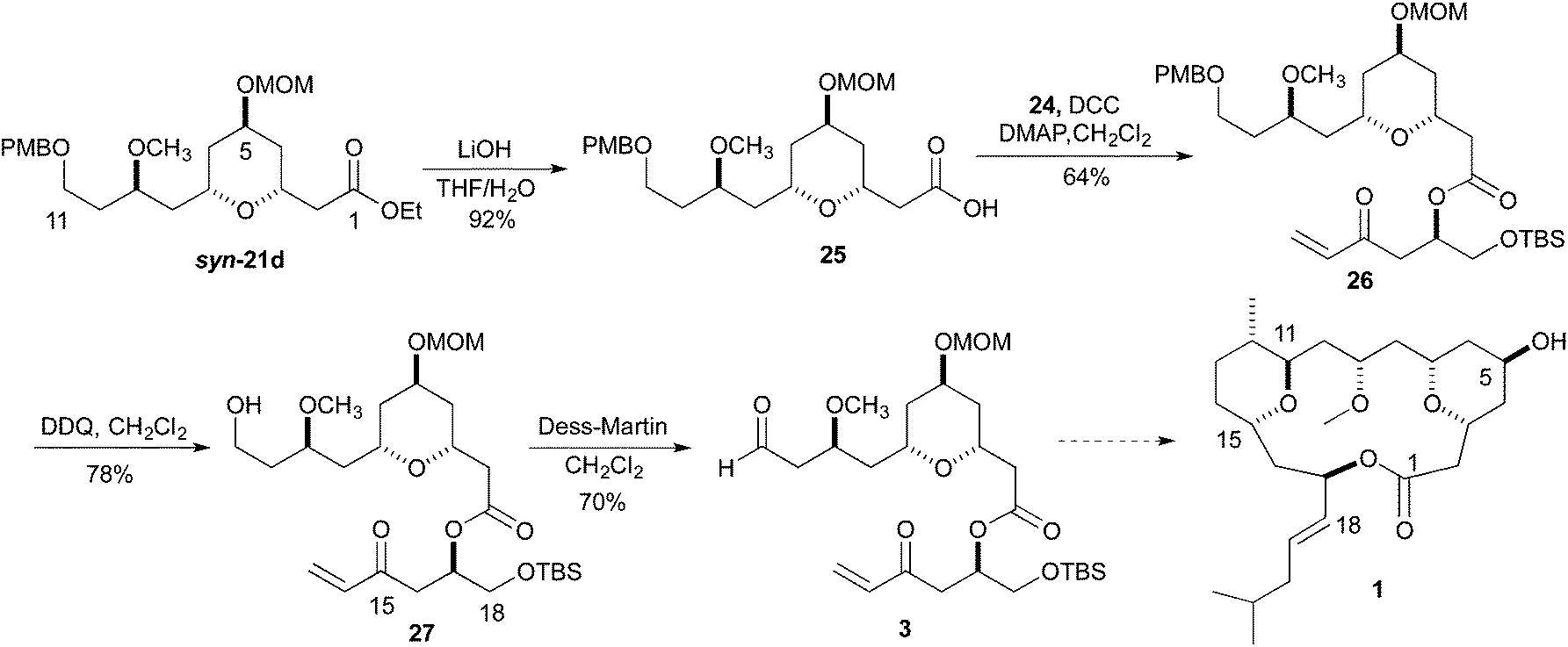 | ||
| Scheme 6 Completion of Leucascandrolide A fragment. | ||
Conclusions
In conclusion we have shown the viability of the asymmetric hydration of dienoate for the synthesis of key chiral building blocks for the synthesis of a potential Leuscandrolide A precursor 2. The further elaboration of this advanced intermediate is ongoing and will be reported in due course.Acknowledgements
We are grateful to NIH (GM090259) and NSF (CHE-1565788) for financial support of this research.Notes and references
- M. D'Ambrosio, A. Guerriero, C. Debitus and F. Pietra, Helv. Chim. Acta, 1996, 79, 51 CrossRef.
- M. D'Ambrosio, M. Tato, C. Debitus and F. Pietra, Helv. Chim. Acta, 1999, 82, 347 CrossRef.
- K. R. Hornberger, C. L. Hamblett and J. L. Leighton, J. Am. Chem. Soc., 2000, 122, 12894 CrossRef CAS.
- (a) I. Paterson and M. Tudge, Angew. Chem., Int. Ed., 2003, 42, 343 CrossRef CAS PubMed; (b) I. Paterson and M. Tudge, Tetrahedron, 2003, 59, 6833 CrossRef CAS; (c) Y. Wang, J. Janjic and S. A. Kozmin, J. Am. Chem. Soc., 2002, 124, 13670 CrossRef CAS PubMed; (d) Y. Wang, J. Janjic and S. A. Kozmin, Pure Appl. Chem., 2005, 77, 1161 CrossRef CAS; (e) A. Fettes and E. M. Carreira, Angew. Chem., Int. Ed., 2002, 41, 4098 CrossRef CAS; (f) A. Fettes and E. M. Carreira, J. Org. Chem., 2003, 68, 9274 CrossRef CAS PubMed; (g) P. Wipf and J. T. Reeves, Chem. Commun., 2002, 2066 RSC; (h) M. T. Crimmins and P. Siliphalvanh, Org. Lett., 2003, 5, 4641 CrossRef CAS PubMed; (i) D. R. Williams, S. Patnaik and S. V. Plummer, Org. Lett., 2003, 5, 5035 CrossRef CAS PubMed; (j) P. A. Evans and W. J. Andrews, Angew. Chem., Int. Ed., 2008, 47, 5426 CrossRef CAS PubMed; (k) H. H. Jung, J. R. Seiders and P. E. Floreancig, Angew. Chem., Int. Ed., 2007, 46, 8464 CrossRef CAS PubMed; (l) Q. Su, L. A. Dakin and J. S. Panek, J. Org. Chem., 2007, 72, 2 CrossRef CAS PubMed; (m) Q. Su and J. S. Panek, Angew. Chem., Int. Ed., 2005, 44, 1223 CrossRef CAS PubMed; (n) L. J. Van Orden, B. D. Patterson and S. D. Rychnovsky, J. Org. Chem., 2007, 72, 5784 CrossRef CAS PubMed.
- (a) M. T. Crimmins, C. A. Carroll and B. W. King, Org. Lett., 2000, 2, 597 CrossRef CAS PubMed; (b) L. A. Dakin, N. F. Langille and J. S. Panek, J. Org. Chem., 2002, 67, 6812 CrossRef CAS PubMed; (c) L. A. Dakin and J. S. Panek, Org. Lett., 2003, 5, 3995 CrossRef CAS PubMed; (d) L. Ferrie, L. Boulard, F. Pradaux, S. Bouzbouz, S. Reymond, P. Capdevielle and J. Cossy, J. Org. Chem., 2008, 73, 1864 CrossRef CAS PubMed; (e) L. Ferrie, S. Reymond, P. Capdevielle and J. Cossy, Org. Lett., 2007, 9, 2461 CrossRef CAS PubMed; (f) D. J. Kopecky and S. D. Rychnovsky, J. Am. Chem. Soc., 2001, 123, 8420 CrossRef CAS PubMed; (g) S. A. Kozmin, Org. Lett., 2001, 3, 755 CrossRef CAS PubMed; (h) K. Lee, H. Kim and J. Hong, Org. Lett., 2011, 13, 2722 CrossRef CAS PubMed; (i) D. R. Williams, S. V. Plummer and S. Patnaik, Angew. Chem., Int. Ed., 2003, 42, 3934 CrossRef CAS PubMed; (j) D. R. Williams, S. V. Plummer and S. Patnaik, Tetrahedron, 2011, 67, 5083 CrossRef CAS PubMed; (k) P. Wipf and T. H. Graham, J. Org. Chem., 2001, 66, 3242 CrossRef CAS PubMed; (l) J. S. Yadav, M. R. Pattanayak, P. P. Das and D. K. Mohapatra, Org. Lett., 2011, 13, 1710 CrossRef CAS PubMed.
- O. A. Ulanovskaya, J. Janjic, M. Suzuki, S. S. Sabharwal, P. T. Schumacker, S. J. Kron and S. A. Kozmin, Nat. Chem. Biol., 2008, 4, 418 CrossRef CAS PubMed.
- (a) C. M. Smith and G. A. O'Doherty, Org. Lett., 2003, 5, 1959 CrossRef CAS PubMed; (b) M. Li and G. A. O'Doherty, Org. Lett., 2006, 8, 6087 CrossRef CAS PubMed; (c) H. Guo, M. S. Mortensen and G. A. O'Doherty, Org. Lett., 2008, 10, 3149 CrossRef CAS PubMed.
- (a) T. J. Hunter and G. A. O'Doherty, Org. Lett., 2001, 3, 2777 CrossRef CAS PubMed; (b) T. J. Hunter, Y. Wang, J. Zheng and G. A. O'Doherty, Synthesis, 2016, 1700 CAS; (c) S. D. Garaas, T. J. Hunter and G. A. O'Doherty, J. Org. Chem., 2002, 67, 2682 CrossRef CAS PubMed; (d) M. Li and G. A. O'Doherty, Org. Lett., 2006, 8, 3987 CrossRef CAS PubMed; (e) Y. Wang, Y. Xing, Q. Zhang and G. A. O'Doherty, Chem. Commun., 2011, 47, 8493 RSC.
- (a) Y. Wang and G. A. O'Doherty, J. Am. Chem. Soc., 2013, 135, 9334–9337 CrossRef CAS PubMed; (b) T. J. Hunter and G. A. O'Doherty, Org. Lett., 2001, 3, 1049–1052 CrossRef CAS PubMed.
- (a) R. S. Burden and L. Crombie, J. Chem. Soc. C, 1969, 2477 RSC; (b) F. Bohlmann and C. Zdero, Chem. Ber., 1973, 106, 3779 CrossRef CAS; (c) W. R. Roush, J. Am. Chem. Soc., 1980, 102, 1390 CrossRef CAS; (d) D. A. Evans and D. M. Fitch, J. Org. Chem., 1997, 62, 454 CrossRef CAS PubMed; (e) G. H. Posner, J. K. Lee, M. C. White, R. H. Hutchings, H. Dai, J. L. Kachinski, P. Dolan and T. W. Kensler, J. Org. Chem., 1997, 62, 3299 CrossRef CAS PubMed; (f) A. G. M. Barrett, D. Hamprecht, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1997, 119, 8608 CrossRef CAS; (g) J. M. Takacs, M. R. Jaber, F. Clement and C. Walters, J. Org. Chem., 1998, 63, 6757 CrossRef CAS; (h) M. Nazare and H. Waldmann, Chem. – Eur. J., 2001, 7, 3363 CrossRef CAS PubMed; (i) M. Sun, Y. Deng, E. Batyreva, W. Sha and R. G. Salomon, J. Org. Chem., 2002, 67, 3575 CrossRef CAS PubMed; (j) T. Tsuzuki, K. Tanaka, S. Kuwahara and T. Miyazawa, Lipids, 2005, 40, 147 CrossRef CAS PubMed; (k) R. Barth and J. Mulzer, Angew. Chem., Int. Ed., 2007, 46, 5791 CrossRef CAS PubMed; (l) J. T. Markiewicz, D. J. Schauer, J. Lofstedt, S. J. Corden, O. Wiest and P. Helquist, J. Org. Chem., 2010, 75, 2061 CrossRef CAS PubMed.
- (a) H. Berker, M. A. Soler and K. B. Sharpless, Tetrahedron, 1995, 51, 1345 CrossRef; (b) Y. Zhang and G. A. O'Doherty, Tetrahedron, 2005, 61, 6337–6351 CrossRef CAS. For synthetic application of this, see: (c) T. J. Hunter and G. A. O'Doherty, Org. Lett., 2002, 4, 4447 CrossRef CAS PubMed; (d) Y. Xing and G. A. O'Doherty, Org. Lett., 2009, 11, 1107 CrossRef CAS PubMed; (e) Md. M. Ahmed, M. S. Mortensen and G. A. O'Doherty, J. Org. Chem., 2006, 71, 7741 CrossRef CAS PubMed; (f) Md. M. Ahmed and G. A. O'Doherty, Tetrahedron Lett., 2005, 46, 3015–3019 CrossRef CAS; (g) Md. M. Ahmed, B. P. Berry, T. J. Hunter, D. J. Tomcik and G. A. O'Doherty, Org. Lett., 2005, 7, 745 CrossRef CAS PubMed.
Footnotes |
| † This manuscript is dedicated to Barry M. Trost on the occasion of this 75th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00284f |
| § Co-first authors. |
| This journal is © the Partner Organisations 2016 |