
Visible light-mediated intramolecular C–H arylation of diazonium salts of N-(2-aminoaryl)benzoimines: a facile synthesis of 6-arylphenanthridines†
Palani
Natarajan
*,
Naveen
Kumar
and
Manjeet
Sharma
Department of Chemistry & Centre for Advanced Studies in Chemistry, Panjab University, Chandigarh - 160 014, India. E-mail: pnataraj@pu.ac.in
First published on 2nd August 2016
Abstract
Utilizing [Ru(bpy)3]Cl2 as a photoredox catalyst and a blue LED as an irradiation source, moderate to good yields of 6-arylphenanthridines were obtained from aryl diazonium salts in situ formed from the reactions of N-(2-aminoaryl)benzoimines and tert-butyl nitrite (tBuONO). Although few visible light-mediated protocols are available for the synthesis of 6-arylphenanthridines, this work provides a new method to access them readily from inexpensive and environmentally benign starting materials.
Introduction
The phenanthridine skeletons1 are very important due to their pharmaceutical and biological properties including antibacterial, antifungal, antiseptic, antitumor, antiviral, cytotoxic, DNA intercalation and glutamic dehydrogenase inhibitory properties.1b Thus, the development of new methods to access phenanthridine and its derivatives is of continuing interest to industry and academia.2 The classical Pictet–Hubert condensation,3 Morgan–Walls cyclization,4 Diels–Alder cycloadditions,5 radical-mediated reactions,6 metal-catalyzed cross couplings,7 microwave-assisted reactions,8 cascade/tandem reactions9 and so on10 are some well-established synthetic protocols for phenanthridine syntheses. During the past few years, visible light-mediated syntheses have also emerged as viable strategies for the preparation of various organic compounds including phenanthridine and its derivatives.11 For example, Yu et al.12 reported that 6-alkylated phenanthridines could be synthesized by somophilic isocyanide insertion reactions, cf. Scheme 1a. Gu et al.13 reported a metal-free approach for the exclusive preparation of 6-arylphenanthridines from 2-isocyanobiphenyls and arylsulfonyl chlorides act as a radical source under visible light-irradiation conditions (Scheme 1b). Zhou et al.14 also reported a metal-free protocol for the synthesis of 6-substituted phenanthridines from 2-isocyanobiphenyls and various radicals. In this case, the authors generated functional radicals from hydrazines by visible light-irradiation (Scheme 1c). In 2014, Yu et al.15 again reported a new methodology for the synthesis of 6-substituted phenanthridines via intramolecular C–H arylation of iminyl-radicals produced from acyl oximes in the presence of visible light photoredox catalysis and irradiation, cf. Scheme 1d. They also disclosed a visible-light-mediated preparation of 6-trifluoromethyl phenanthridines (Scheme 1e) using the Umemoto reagent as a CF3 radical precursor.16 Very recently, the same group once again reported a strategy for the synthesis of 6-mono- and difluoromethylated phenanthridines from 2-isocyanobiphenyls and ethyl bromofluoroacetate/ethyl bromodifluoroacetate (Scheme 1f and g).17 Of course, each of these methods has its own applications, but remains associated with some disadvantages such as low to moderate yields of products, use of toxic, expensive and unstable precursors, etc.18 As a consequence, extension of a visible light-induced method for the synthesis of phenanthridines utilizing readily available and environmentally benign starting compounds is very desirable and remains a challenging area for exploration. | ||
| Scheme 1 Visible light-mediated synthesis of 6-substituted phenanthridines reported in the literature (a–g) and this work (h). | ||
Aryl diazonium salts19 have been employed as a source of aryl radicals under visible light-photocatalyzed conditions to synthesize arylboronates, arylsulfides, benzothiophenes, biaryls, biheteroaryls, phenanthrenes, α-phenylketones, stilbenes, trifluoroaryls as well as other compounds.20 Moreover, the availability, stability and safety of diazonium salts are improved by an in situ strategy without being isolated.21 In these cases, the amino group (–NH2) formally acts as a leaving group. To our surprise, there is still no report on the synthesis of 6-arylphenanthridines from aryl diazonium salts or anilines under visible light-photocatalyzed conditions.
Herein, we describe the visible light-mediated intramolecular C–H arylation of aryl diazonium salts to give 6-arylphenanthridines (PHE1–13, Scheme 1h) in good to excellent yields. Moreover, a tentative mechanism is described for the reaction, vide infra. It is worth noting that the various aryl diazonium salts used in this study were produced in situ from inexpensive and ecofriendly starting materials, i.e. N-(2-aminoaryl)benzoimines (IME1–13, Scheme 1h) and tert-butyl nitrite (tBuONO).
Results and discussion
At the outset of this investigation, a solution of N-(2-aminophenyl)benzoimine (IME1, 1.0 mmol.), tBuONO (1.0 mmol) and 1 mol% of Eosin Y in anhydrous acetonitrile under argon was irradiated with a 5 W blue LED lamp at room temperature. After about 24 h, we were pleased to find a desired 6-phenylphenanthridine (PHE1) in 26% yield (Table 1, entry 1). We then explored the influence of solvents on the formation of PHE1. Of the solvents tested (Table 1, entries 2–8), dry DMSO gave a moderate yield (Table 1, entry 7). After this, the amounts of tBuONO were inquired. 1.3 equiv. of tBuONO provided a satisfactory yield (64%, Table 1, entry 10). Further optimizations revealed that this reaction proceeded more efficiently (93%, Table 1, entry 13) in the presence of 1 mol% of [Ru(bpy)3]Cl2 (bpy = 2,2′-bipyridine), however other visible light-photoredox catalysts Eosin B and Rose bengal failed to produce satisfactory yields (Table 1, entries 14 and 15). We believe that the superior reduction capacity in the excited state of [Ru(bpy)3]Cl2 may be the reason for the higher yield of PHE1 (Table 1, entry 13).11 Subsequently, the influence of the quantity of [Ru(bpy)3]Cl2 was investigated. With an increase in the catalyst amount from 1 mol% to 3 mol% and 5 mol%, the reaction time obviously shortened with 92–94% yield of PHE1 (Table 1, entries 16 and 17). A further increase in the catalyst amount showed a profound effect neither on the reaction time nor on the yield of the desired product (Table 1, entry 18). However, no product was observed in the absence of either [Ru(bpy)3]Cl2 or tBuONO or the irradiation source (Table 1, entries 19–21). Thus, the optimized conditions for this transformation (Scheme 1h) were 1.0 equiv. of N-(2-aminoaryl)benzoimines (IME), 1.3 equiv. of tBuONO and 1 mol% of [Ru(bpy)3]Cl2 in anhydrous DMSO at room temperature for 6 h, cf. ESI.†|
|
||||
|---|---|---|---|---|
| Entry | Solventb | Catalystc | Time (h) | Yieldd (%) |
| a Unless stated otherwise all reactions were performed in a Schlenk tube with 1.0 equiv. of IME1, 1.0–2.0 equiv. of tBuONO and 1 mol% of photoredox catalyst in dry solvent under an inert atmosphere with a 5 W blue LED source. b Solvents were rigorously purified following the methods described in ref. 22. c All catalysts, in high quality, were purchased from the commercial source and used as such. d Isolated yields. e 1.1 equiv. of tBuONO were used. f 1.3 equiv. of tBuONO were used. g 1.5 equiv. of tBuONO were used. h 2.0 equiv. of tBuONO were used. i 3.0 mol% of [Ru(bpy)3]Cl2 were used. j 5.0 mol% of [Ru(bpy)3]Cl2 were used. k 10 mol% of [Ru(bpy)3]Cl2 were used. l No irradiation source was used. m No tBuONO was used. NR: no reaction. | ||||
| 1 | CH3CN | Eosin Y | 24 | 26 |
| 2 | CH2Cl2 | Eosin Y | 24 | <10 |
| 3 | C2H4Cl2 | Eosin Y | 24 | <10 |
| 4 | Hexane | Eosin Y | 24 | <5 |
| 5 | Heptane | Eosin Y | 24 | <5 |
| 6 | Glyme | Eosin Y | 24 | 18 |
| 7 | DMSO | Eosin Y | 24 | 43 |
| 8 | DMF | Eosin Y | 24 | 36 |
| 9 | DMSO | Eosin Y | 24 | 51e |
| 10 | DMSO | Eosin Y | 24 | 64f |
| 11 | DMSO | Eosin Y | 24 | 66g |
| 12 | DMSO | Eosin Y | 24 | 65h |
| 13 | DMSO | [Ru(bpy)3]Cl2 | 6 | 93 |
| 14 | DMSO | Eosin B | 24 | 32 |
| 15 | DMSO | Rose bengal | 24 | 30 |
| 16 | DMSO | [Ru(bpy)3]Cl2 | 4 | 92i |
| 17 | DMSO | [Ru(bpy)3]Cl2 | 2 | 94j |
| 18 | DMSO | [Ru(bpy)3]Cl2 | 2 | 93k |
| 19 | DMSO | None | 24 | NR |
| 20 | DMSO | [Ru(bpy)3]Cl2 | 24 | NRl |
| 21 | DMSO | [Ru(bpy)3]Cl2 | 24 | NRm |

With the optimized reaction conditions in hand (Table 1), we next explored the scope of the reaction with a series of N-(2-aminoaryl)benzoimines23 as shown in Table 2. Generally, the desired 6-arylphenanthridines were isolated in good to excellent yields (85–94%) irrespective of the position and electronic nature of substituents on the aniline moiety. For examples, the chloro and nitro groups were found to be compatible under these reaction conditions (Table 2, entries 6–9 and 12). Likewise, electron-donating substituents (methyl and methoxy groups) provided excellent yield of the products (Table 2, entries 2–5 and 11). Moreover, sterically hindered substrates, i.e.IME10, IME11, IME12 and IME13, also worked well in the present system affording the corresponding 6-arylphenanthridines in good yields (Table 2, entries 10–13). Thus, a wide range of electronically and structurally diverse 6-arylphenanthridines can be synthesized efficiently with the present methodology (ESI†).
|
|
|||
|---|---|---|---|
| Entry | Substrate (N-(2-aminoaryl)benzoimines) | Product (6-arylphenanthridines) | Yieldb,c (%) |
| a Unless stated otherwise all reactions were performed in a Schlenk tube with 1.0 equiv. of IME, 1.3 equiv. of tBuONO and 1 mol% of [Ru(bpy)3]Cl2 in dry DMSO at room temperature in argon with the irradiation of a 5 W blue LED for 6 h. b Isolated yield. c All products were characterized by IR and NMR spectroscopy (ESI) in comparison with known compounds from literature data. | |||
| 1 |

|

|
93 |
| 2 |
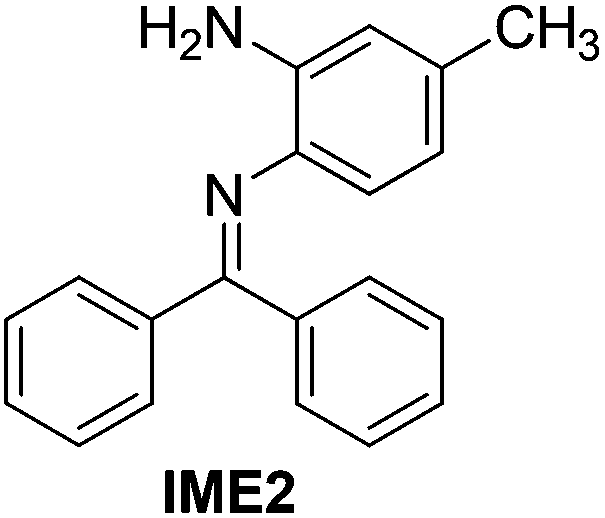
|
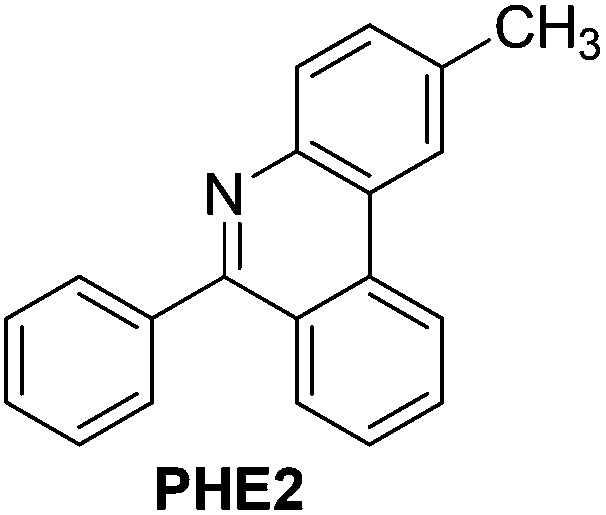
|
94 |
| 3 |

|

|
89 |
| 4 |

|

|
94 |
| 5 |
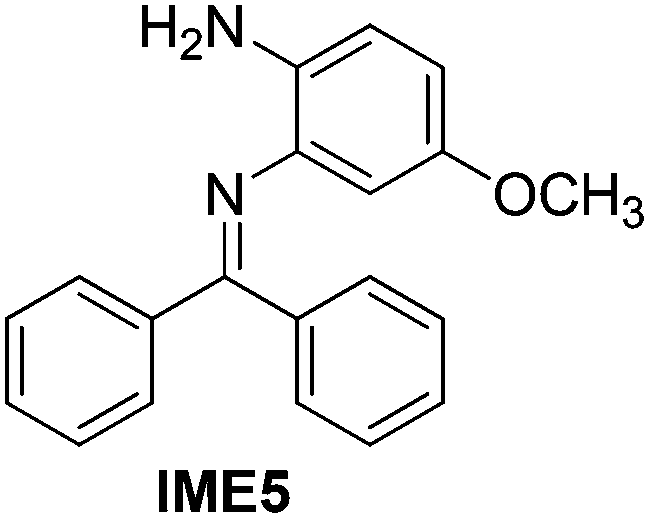
|
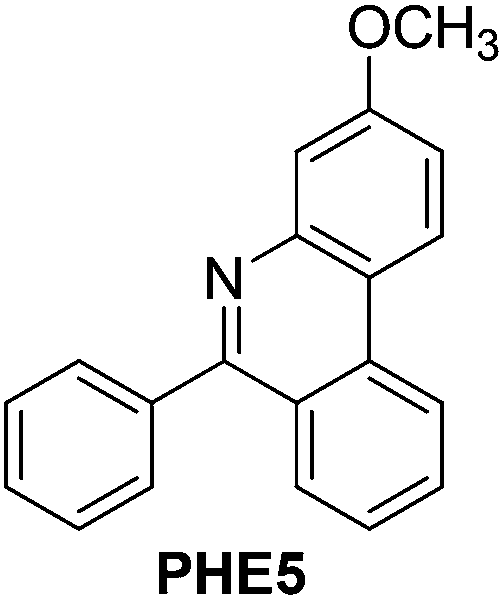
|
86 |
| 6 |
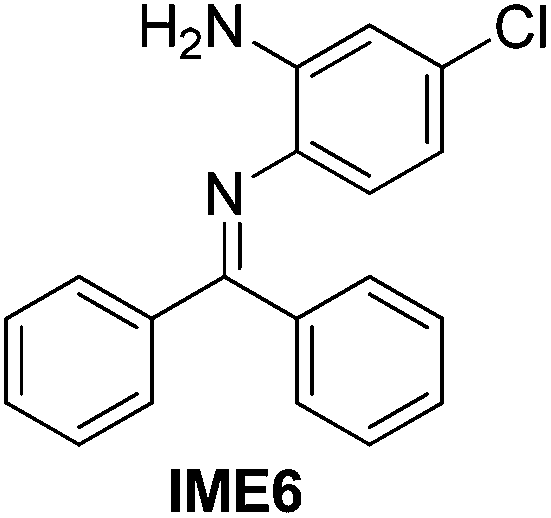
|

|
92 |
| 7 |

|
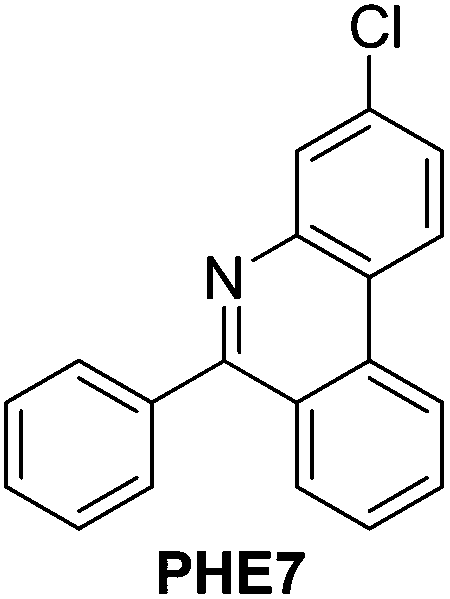
|
91 |
| 8 |

|

|
87 |
| 9 |

|

|
89 |
| 10 |

|

|
85 |
| 11 |

|

|
89 |
| 12 |

|

|
91 |
| 13 |

|

|
86 |
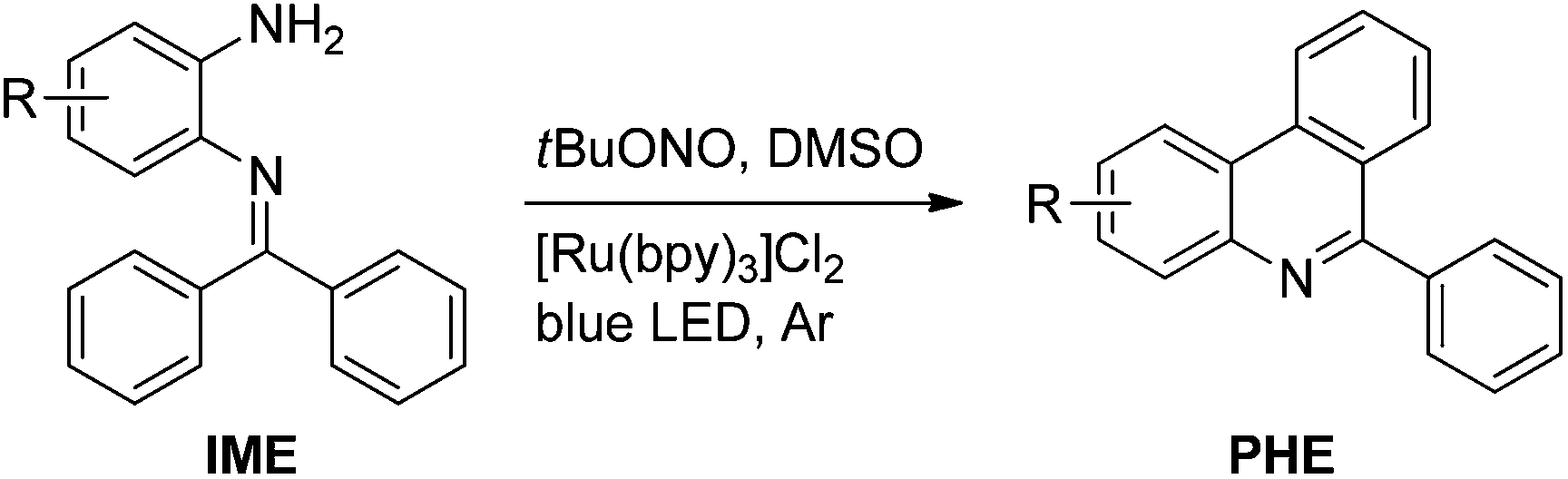
A plausible reaction mechanism for the preparation of 6-arylphenanthridines from N-(2-aminoaryl)benzoimines is outlined in Scheme 2 on the basis of blank experiments and an earlier proposed mechanism.11,24 It seems that reactions proceed through a visible light-induced single electron transfer mechanism as no product was observed without the photocatalyst or visible light-irradiation (Table 1, entries 19 and 20). Thus, the irradiation of [Ru(bpy)3]Cl2 with visible light generates the excited *[Ru(bpy)3]Cl2 species. A single electron transfer from the excited *[Ru(bpy)3]Cl2 to the diazonium salts, formed in situ from the reactions of N-(2-aminoaryl)benzoimine and tBuONO, affords [Ru(bpy)3]3+ and the N-(o-phenyl radical)benzoimine (I, Scheme 2) by extrusion of N2. Radical I undergoes intramolecular C–H aromatic coupling to form a cyclohexadienyl-type radical (II), which was then oxidized (path a) by [Ru(bpy)3]3+ to form a cationic intermediate III and regenerate [Ru(bpy)3]Cl2 or collision with diazonium salts to produce radical I through path b. Ultimately, deprotonation of III by tBuO− yields the desired 6-arylphenanthridines, cf. Scheme 2.
 | ||
| Scheme 2 A plausible mechanism for the visible light-induced preparation of 6-arylphenanthridines from N-(2-aminoaryl)benzoimines. SET, single electron transfer. | ||
In summary, we have successfully developed a novel method for the synthesis of 6-arylphenanthridines from aryl diazonium salts, in situ formed from the reactions of N-(2-aminoaryl)benzoimines and tert-butyl nitrite, under visible light-photocatalyzed conditions. Mild reaction conditions, inexpensive reagents and non-polluting byproducts are few advantages of this method while compared to other visible light-mediated protocols reported yet for the synthesis of 6-arylphenanthridines. Currently, studies on the DNA intercalation and antibacterial properties of selected 6-arylphenanthridines are underway in our laboratories.
Acknowledgements
The authors are pleased to acknowledge the financial support from the Council of Scientific and Industrial Research (CSIR), New Delhi, vide project no.: 02(0246)/15/EMR-II. N. K. and M. S. thank the University Grant Commission (UGC), New Delhi for their junior research fellowships (JRF). Also, we thank anonymous reviewers for their insightful comments and suggestions.References
- (a) T. Eicher and S. Hauptmann, The Chemistry of Heterocycles, Wiley-VCH, 2003 CrossRef; (b) K. N. Sundaramoorthy and S. K. Iyer, Synthesis of Phenanthridines, Azabicyclononanones and Cycloalkanones: Ammonium Acetate an Efficient Reagent for the One-pot Synthesis of Phenanthridines, Azabicyclononanones & Cycloalkanones, Lap Lambert Academic Publishing, GmbH KG, 2012 Search PubMed.
- (a) J.-W. Gu and X. Zhang, Org. Lett., 2015, 17, 5384 CrossRef CAS PubMed; (b) M. Ghosh, A. Ahmed, R. Singha and J. K. Ray, Tetrahedron Lett., 2015, 56, 353 CrossRef CAS; (c) A. Romo-Perez, L. D. Miranda and A. Garcia, Tetrahedron Lett., 2015, 56, 6669 CrossRef CAS.
- A. Pictet and A. Hubert, Ber. Dtsch. Chem. Ges., 1896, 29, 1182 CrossRef CAS.
- G. T. Morgan and L. P. Walls, J. Chem. Soc., 1931, 2447 RSC.
- L. Sripada, J. A. Teske and A. Deiters, Org. Biomol. Chem., 2008, 6, 263 CAS.
- (a) R. T. McBurney, A. M. Z. Slawin, L. A. Smart, Y. Yu and J. C. Walton, Chem. Commun., 2011, 47, 7974 RSC; (b) A. M. Linsenmeier, C. M. William and S. Brase, J. Org. Chem., 2011, 76, 9127 CrossRef CAS PubMed; (c) M. Tobisu, K. Koh, T. Furukawa and N. Chatani, Angew. Chem., Int. Ed., 2012, 51, 11363 CrossRef CAS PubMed.
- (a) R. Yanada, K. Hashimoto, R. Tokizane, Y. Miwa, H. Minami, K. Yanada, M. Ishikura and Y. Takemoto, J. Org. Chem., 2008, 73, 5135 CrossRef CAS PubMed; (b) L. R. Donaldson, D. Haigh and A. N. Hulme, Tetrahedron, 2008, 64, 4468 CrossRef CAS; (c) D. A. Candito and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 6713 CrossRef CAS PubMed; (d) T. Gerfaud, L. Neuville and J. Zhu, Angew. Chem., Int. Ed., 2009, 48, 572 CrossRef CAS PubMed; (e) L. Zhang, G. Y. Ang and S. Chiba, Org. Lett., 2010, 12, 3682 CrossRef CAS PubMed.
- N. Kaur, Synth. Commun., 2015, 45, 273 CrossRef CAS.
- W. Guo, S. Li, L. Tang, M. Li, L. Wen and C. Chen, Org. Lett., 2015, 17, 1232 CrossRef CAS PubMed.
- (a) W.-G. Shou, Y.-Y. Yang and Y.-G. Wang, J. Org. Chem., 2006, 71, 9241 CrossRef CAS PubMed; (b) M. E. Buden, V. B. Dorn, M. Gamba, A. B. Pierini and R. A. Rossi, J. Org. Chem., 2010, 75, 2206 CrossRef CAS PubMed; (c) Y. Wu, S. M. Wong, F. Mao, T. L. Chan and F. Y. Kwong, Org. Lett., 2012, 14, 5306 CrossRef CAS PubMed; (d) D. Nanni, P. Pareschi, C. Rizzoli, P. Sgarabotto and A. Tundof, Tetrahedron, 1995, 51, 9045 CrossRef CAS; (e) M. Tobisu, K. Koh, T. Furukawa and N. Chatani, Angew. Chem., 2012, 124, 11525 CrossRef; (f) J. Peng, T. Chen, C. Chen and B. Li, J. Org. Chem., 2011, 76, 9507 CrossRef CAS PubMed.
- (a) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef CAS PubMed; (b) P. V. Pham, D. A. Nagib and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2011, 50, 6119 CrossRef CAS PubMed; (c) C. Dai, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2011, 3, 140 CrossRef CAS PubMed; (d) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 6174 CrossRef PubMed; (e) Z. Zhang, X. Tang and W. R. Dolbier Jr., Org. Lett., 2015, 17, 4401 CrossRef CAS PubMed; (f) B. Zhang and A. Studer, Chem. Soc. Rev., 2015, 44, 3505 RSC.
- (a) H. Jiang, Y. Cheng, R. Wang, M. Zheng, Y. Zhang and S. Yu, Angew. Chem., Int. Ed., 2013, 52, 13289 CrossRef CAS PubMed; (b) B. Zhang, C. Muck-Lichtenfeld, C. G. Daniliuc and A. Studer, Angew. Chem., 2013, 125, 10992 CrossRef.
- L. Gu, C. Jin, J. Liu, H. Dinga and B. Fana, Chem. Commun., 2014, 50, 4643 RSC.
- T. Xiao, L. Li, G. Lin, Q. Wang, P. Zhang, Z.-W. Mao and L. Zhou, Green Chem., 2014, 16, 2418 RSC.
- (a) R. Alonso, P. J. Campos, B. Garcia and M. A. Rodriguez, Org. Lett., 2006, 8, 3521 CrossRef CAS PubMed; (b) R. Alonso, A. Caballero, P. J. Campos and M. A. Rodriguez, Tetrahedron, 2010, 66, 8828 CrossRef CAS; (c) H. Jiang, X. An, K. Tong, T. Zheng, Y. Zhang and S. Yu, Angew. Chem., Int. Ed., 2015, 54, 1 CrossRef.
- R. Wang, H. Jiang, Y. Cheng, A. A. Kadi, H.-K. Fun, Y. Zhang and S. Yu, Synthesis, 2014, 2711 CAS.
- X. Sun and S. Yu, Org. Lett., 2014, 16, 2938 CrossRef CAS PubMed.
- Isocyanide Chemistry: Applications in Synthesis and Material Science, ed. V. Nenajdenko, Wiley-VCH, 2012 Search PubMed.
- R. T. Morrison and R. N. Boyd, in Organic Chemistry, ed. A. Bacon, Boston, MA, 5th edn, 1987 Search PubMed.
- (a) D. P. Hari, P. Schroll and B. J. Konig, J. Am. Chem. Soc., 2012, 134, 2958 CrossRef CAS PubMed; (b) T. Xiao, X. Dong, Y. Tang and L. Zhou, Adv. Synth. Catal., 2012, 354, 3195 CrossRef CAS; (c) M. Majek and A. J. V. Wangelin, Chem. Commun., 2013, 49, 5507 RSC; (d) M. Majek, F. Filace and A. J. V. Wangelin, Beilstein J. Org. Chem., 2014, 10, 981 CrossRef CAS PubMed.
- (a) F. Mo, G. Dong, Y. Zhanga and J. Wang, Org. Biomol. Chem., 2013, 11, 1582 RSC; (b) S. Gowrisankar and J. Seayad, Chem. – Eur. J., 2014, 20, 12754 CrossRef CAS PubMed; (c) P. Maity, D. Kundu and B. C. Ranu, Eur. J. Org. Chem., 2015, 1727 CrossRef CAS.
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Butterworth-Heinemann, Oxford, 6th edn, 2009 Search PubMed.
- Under the optimized reaction conditions N-(2-aminoaryl)-phenylmethanimine, N-(2-aminoaryl)-methylphenylmethanimine and N-(2-aminoaryl)-ethylphenylmethanimine, respectively, derived from o-phenylenediamine and benzaldehyde, acetophenone and propiophenone afforded a very poor yield (15–20%) of the expected phenanthridine product. It may be due to the rapid cis–trans isomerism occurred in these unsymmetrical imines.
- (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (b) L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General aspects, experimental procedure, characterization data for products and references. See DOI: 10.1039/c6qo00275g |
| This journal is © the Partner Organisations 2016 |