
Considering organic mechanisms and the optimization of current flow in an electrochemical oxidative condensation reaction†
Derek T.
Rensing
,
Bichlien H.
Nguyen
and
Kevin D.
Moeller
*
Department of Chemistry, Washington University in St. Louis, St. Louis, MO 63130, USA. E-mail: moeller@wustl.edu
First published on 5th July 2016
Abstract
Oxidative condensation reactions are potentially valuable tools for the valorization of lignin derived materials. They are used in the two step conversion of raw sawdust into benzothiazole and benzimidazole ring skeletons. The reactions are also excellent candidates for use in paired electrochemical reactions that utilize the cathodic half-reaction to make the hydrogen gas needed for the conversion of lignin derived materials into indanones. However, the optimized oxidative condensation reactions used to date require the use of low currents that inhibit their utility in paired electrochemical reactions. In this manuscript, the optimization of current for the oxidative condensation is examined and this optimization is used to illustrate how the optimization of an electrochemical reaction relies on a traditional look at organic mechanisms.
Electrochemical reactions can provide a sustainable alternative for conducting a wide variety of oxidation and reduction reactions.1,2 In this context, we have recently employed a series of electrochemical oxidation and reduction reactions in the processing of lignin derived materials into more valuable synthetic substrates.3 In one of the more intriguing examples, the hydrogen gas needed in the synthesis of a lignin derived indanone was made at the cathode of an electrochemical reaction used to oxidize an alcohol (Scheme 1). In this paired electrolysis,4,5 both reactions afforded a high yield of product. The reaction demonstrated how the cathodic reaction required by an oxidation could be used for the on-site generation of a chemical reagent needed elsewhere in the larger synthetic effort.
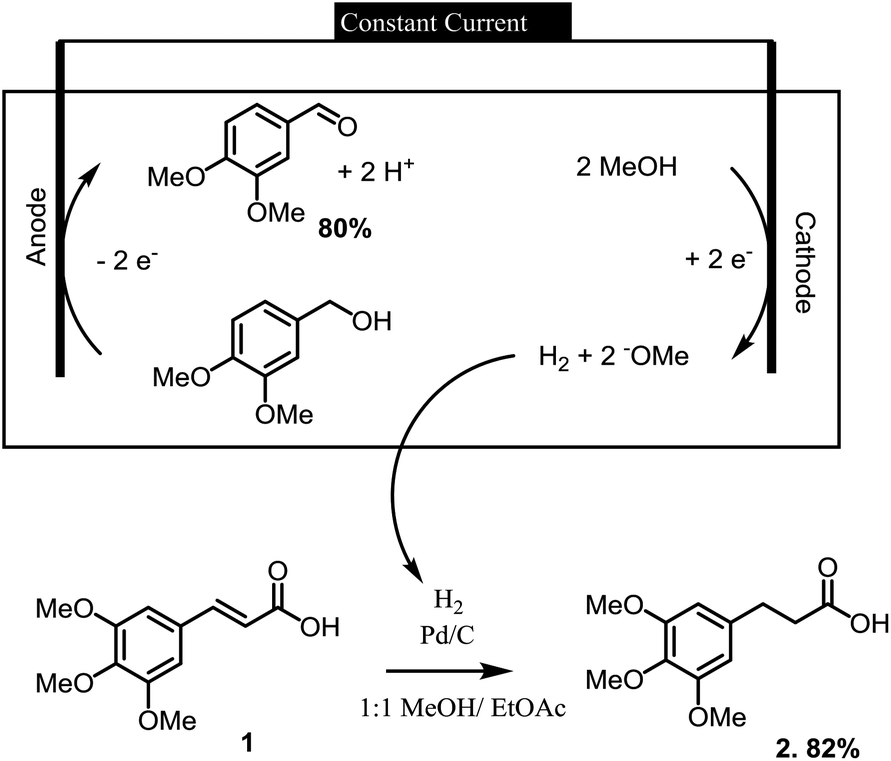 | ||
| Scheme 1 A paired electrolysis for the processing of lignin derived materials. | ||
Paired electrochemical reactions of this nature are intriguing because they offer a potentially general opportunity to optimize both the atom and energy economy of oxidation reactions by taking advantage of the necessary reduction reaction to make a variety of different chemical reagents needed for other synthetic steps in a sequence. Cathodic reactions can be used to generate not only hydrogen, but carbon monoxide, syngas, carbon anions, bases, etc.1 However, the optimization of a paired electrochemical reaction for the generation of a chemical reagent at the cathode does require one to think more broadly about the electrolysis reaction. Consider the oxidative condensation reaction shown in Scheme 2. This reaction was used to convert lignin derived syringealdehyde into a benzothiazole derivative in order to demonstrate how a biologically relevant molecular scaffold6 can be synthesized from raw sawdust in just two steps.3 It is tempting to suggest that this reaction would provide a better partner for demonstrating the paired valorization of lignin derived materials shown in Scheme 1 because the oxidative condensation converts the aldehyde into a new molecule not typically associated with lignin.
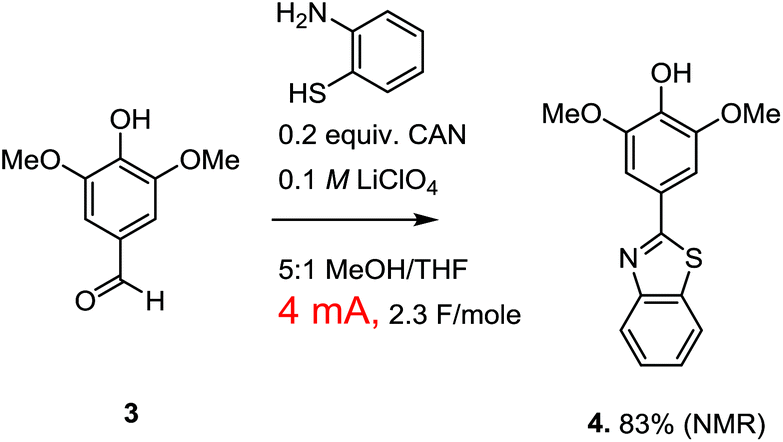 | ||
| Scheme 2 An oxidative condensation. | ||
While a paired electrochemical reaction involving an oxidative condensation is easy to propose (and easy to demonstrate in the lab), there is a rather serious practical problem with the suggestion. The optimized oxidative condensation is conducted with the passage of only 4 mA of current with the use of a reticulated vitreous carbon (RVC/100 PPI) anode having dimensions of 1 × 0.5 × 0.5 cm. The low current density was required in order to avoid over-oxidation of the product. This over-oxidized product appeared from the start of the reaction, and led to recovery of the starting material when the total charge passed through the cell was limited to that required for the oxidative condensation (2 F mol−1). Therefore, the overoxidized material was not a result of the reaction proceeding too long, but rather a result from a loss of selectivity during the reaction. Attempts to circumvent this problem with changes in reaction conditions, the use of different mediators with lower oxidation potentials, or the use of direct oxidation methods that adjust to the potential of the most easily oxidized material in solution all met with failure.7 So, the current density was kept low and the over-oxidation problem avoided. The use of a mediator (CAN) for the reaction gave a slightly better yield, so the indirect method was used for the final optimized version of the reaction. However, while the low current density works great for the oxidation reaction, it also means that the current flow through the cell is too low to generate meaningful amounts of a reagent at the cathode in a timely fashion. For a paired electrochemical reaction that aims to use the cathode for the on-site generation of chemical reagents, the optimized reaction was not really optimal.
So how can the current flow through a paired electrochemical reaction be increased and a cathodic process optimized without over-oxidizing the product generated at the anode?
One of the things that we have learned through the years is that the key to optimizing electroorganic reactions is getting the organic chemistry correct. The planned initial electron-transfer reaction at the electrode happens as planned. What is needed is a way to control the chemistry prior to and subsequent to that electron-transfer. For anodic cyclization reactions, sometimes this has required alterations to the nature of the radical cation and the tuning of a cyclization reaction rate relative to competitive processes,8 sometimes it has meant taking care of a reactive intermediate after the cyclization and making sure it could follow a viable reaction pathway,9 and sometimes it has meant optimization of the rate at which a second electron is removed from the cyclic product.10 In all such cases, the optimization process begins with a careful look at a proposed reaction mechanism. For the oxidative condensation, the most likely mechanism is outlined in Scheme 3. In this mechanism, the first step is a condensation reaction between the electron-rich aldehyde and the thiol amine to form a thioaminal and water. The thioaminal is then oxidized to a radical cation that in turn undergoes deprotonation, the loss of a second electron, and the loss of a second equivalent of acid. The acid generated is neutralized by base generated at the cathode (the reduction of two equivalents of methanol to form hydrogen gas and two equivalents of methoxide). For this reaction, we do know that thioaminal 5 has a lower oxidation product than product 4. We know this because the direct electrolysis reaction using a constant current of 4 mA led to the desired product with no evidence of over-oxidation. The selectivity of a direct electrolysis reaction is based solely on potential. In such reactions, the working potential of the anode climbs until it matches the oxidation potential of the substrate in solution with the lowest oxidation potential. The potential at the anode then holds there until the substrate is consumed. So, if product 4 had a lower potential than the thioaminal, then the product would have oxidized faster than the thioaminal and the reaction would have afforded only over-oxidized material and recovered starting material after 2 F mol−1 no matter what current was used. So, how can a constant current reaction lose selectivity for the substrate with the lower oxidation potential from the start of the reaction?
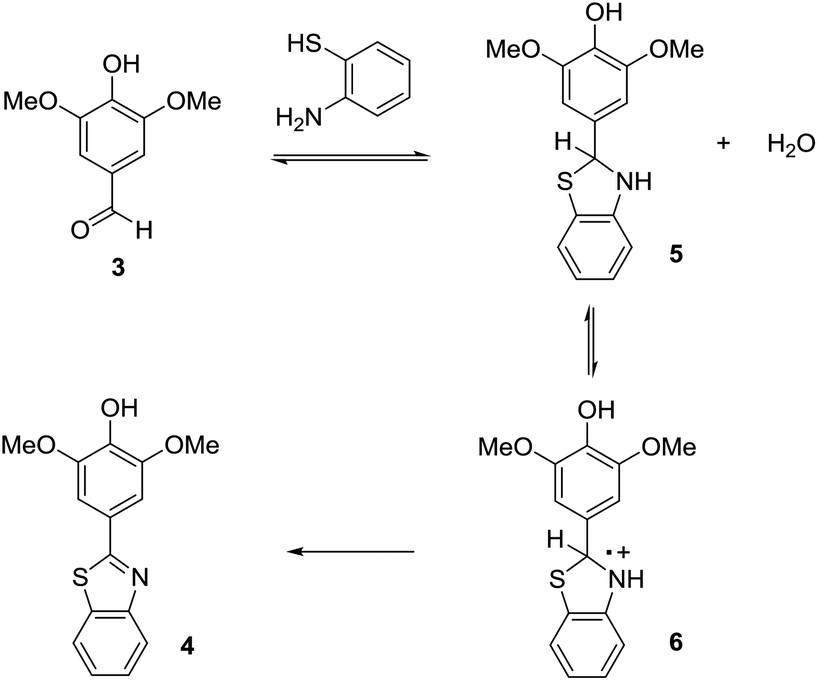 | ||
| Scheme 3 Proposed mechanism. | ||
The answer lies in the nature of a constant current electrolysis itself. As mentioned in the preceding paragraph, the working potential at the anode in a constant current electrolysis climbs until it matches that of the substrate with the lowest oxidation potential in solution and then remains there until the substrate is consumed. At that point, there is not enough material at the anode to satisfy the current that must be passed through the cell. As a result the working potential at the anode climbs again until it matches that of the substrate with the next highest oxidation potential in solution. For a constant current reaction to lose selectivity for the substrate with the lowest oxidation potential from the start of the reaction, there must never have been enough of that substrate to satisfy the current.
This leads to an immediate question about the oxidative condensation reaction. What if the initial equilibrium favors the aldehyde and amino thiol substrates and only a small amount of the thioaminal is present in solution at any given time? The result would be the exact electrochemical behavior observed. Low current experiments with low demand for thioaminal substrate would be selective. High current experiments with high demand for thioaminal substrate would not be. If this was the case, then there would be two possible ways to address the situation (Scheme 4). One would be to drive the initial equilibrium to completion prior to the electrolysis reaction by the removal of water. This would insure that all of the substrate was converted to the thioaminal and available for the electrolysis. The second method would be to change the mechanism of the oxidation. If the reaction were mediated with a Cu(II)-oxidant, then the oxidation step would involve a β-hydride elimination from intermediate 7. Since the product cannot undergo a β-hydride elimination, no overoxidation would be possible. In such reactions, the Cu(II)-species generated at the anode is a catalyst that is never consumed. Hence, the working potential of the anode never climbs, and there is no danger of a direct oxidation of the product at the anode.
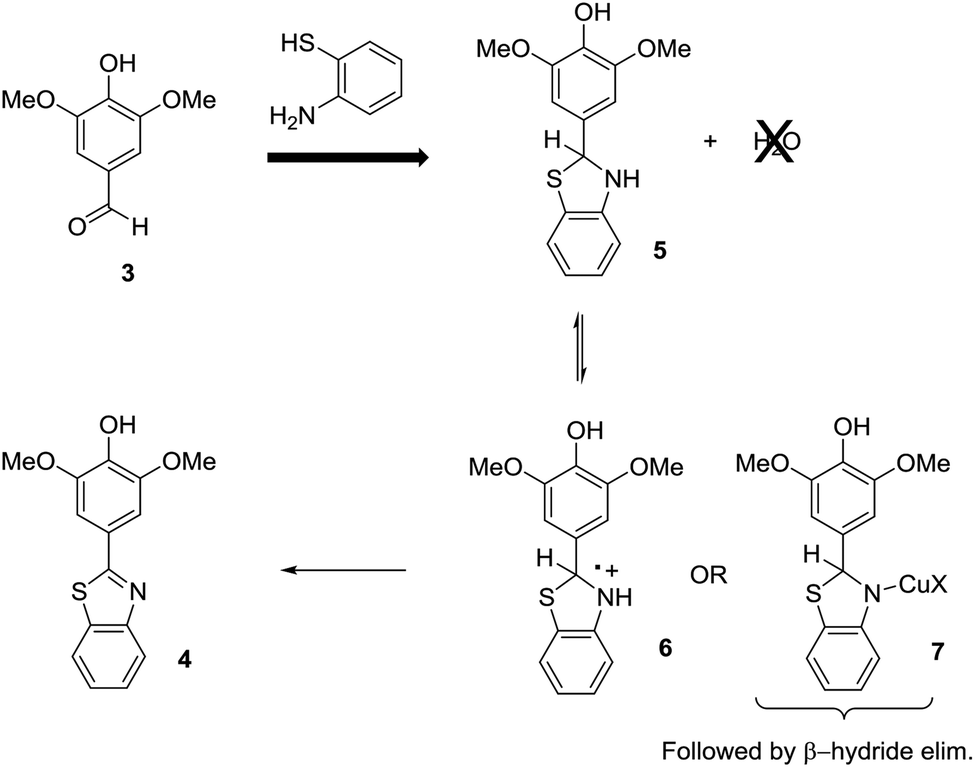 | ||
| Scheme 4 Proposed routes to improved current efficiency. | ||
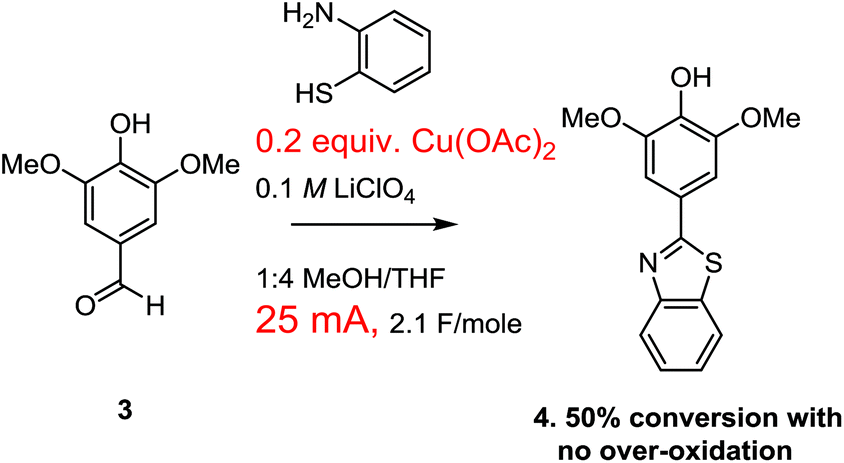
Our optimization attempts started with the use of a Cu(II)-mediator since the associated change in mechanism removed any possibility of over-oxidation (Scheme 5). To this end, the oxidative condensation was performed by passing 2.1 F mol−1 of a 25 mA current through a solution containing the aldehyde, the thiol amine, and 10 mol% of Cu(OAc)2. The current was selected to be the same as the successful paired electrolysis shown in Scheme 1. A 0.1 M LiClO4 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 methanol/THF was used as the electrolyte solution along with a RVC anode and cathode (for this study all RVC anodes were approximately 1.0 × 0.5 × 0.5 cm in size so that the relative currents used related directly to relative current density). The switch in solvent was made because the use of less methanol seemed to coincide with less over-oxidation when the current was raised for the experiment shown in Scheme 2. This initial reaction using the Cu(II)-mediator formed the product with no evidence of over-oxidation. However, the reaction proceeded to only 50% conversion as measured by NMR integration of the crude product mixture. The reaction was not completely clean with several unidentified products formed. When the reaction was pushed to completion with the use of 7.3 F mol−1 of charge, noticeable amounts of side products were generated, but there was again no evidence for an over-oxidized product. However, the side-products generated made the product difficult to isolate.
:4 methanol/THF was used as the electrolyte solution along with a RVC anode and cathode (for this study all RVC anodes were approximately 1.0 × 0.5 × 0.5 cm in size so that the relative currents used related directly to relative current density). The switch in solvent was made because the use of less methanol seemed to coincide with less over-oxidation when the current was raised for the experiment shown in Scheme 2. This initial reaction using the Cu(II)-mediator formed the product with no evidence of over-oxidation. However, the reaction proceeded to only 50% conversion as measured by NMR integration of the crude product mixture. The reaction was not completely clean with several unidentified products formed. When the reaction was pushed to completion with the use of 7.3 F mol−1 of charge, noticeable amounts of side products were generated, but there was again no evidence for an over-oxidized product. However, the side-products generated made the product difficult to isolate.
 | ||
| Scheme 5 The reaction with a Cu(II)-mediator. | ||
Attempts to optimize the Cu(II)-mediated oxidation focused on providing more substrate for the reaction and avoiding the use of additional current. This was done by preforming the thioaminal by stirring the aldehyde and thioamine with magnesium sulfate for 2–18 h prior to the electrolysis. The reaction led to 55% conversion after 2.1 F mol−1 when 2 h was used for the dehydration step and 60% when 18 h was used for the dehydration. Hence, the length of time used for the dehydration had little effect on the final outcome of the reaction.
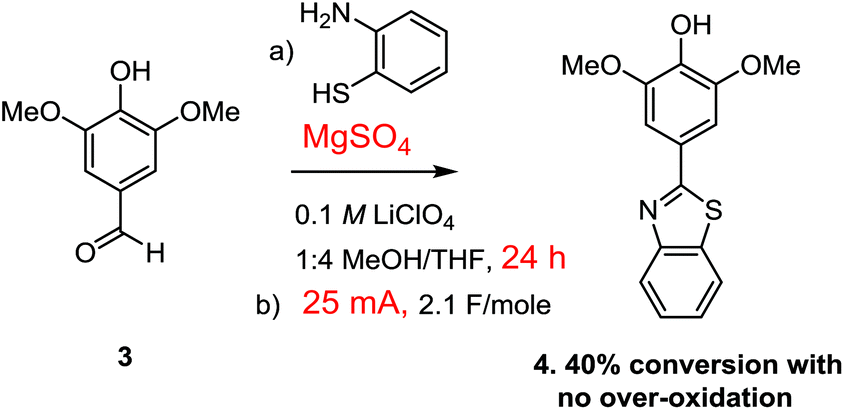
The use of the Cu(II)-mediator was abandoned when it was discovered that pre-formation of the thioaminal prior to the electrolysis also allowed for the use of direct oxidation conditions without any observable over-oxidation (Scheme 6). In this case, the reaction was extremely clean with only the starting thioaminal and product evident in the proton NMR of the crude reaction mixture (see the ESI† for details).
 | ||
| Scheme 6 A direct electrolysis following pre-formation of the thioaminal. | ||
However, once again the reaction did not proceed to completion, this time affording about 40% completion after 2.1 F mol−1 of charge had been passed through the cell. This was a very interesting observation. If 2.1 F mol−1 of charge was passed through the reaction and the reaction went to only 40% completion with no over-oxidation or generation of any side-products, then where did the rest of the current go?
This is a situation that we have encountered previously. For example, the alcohol trapping of a styrene radical cation by an alcohol is a very clean reaction that also initially afforded a very high mass balance and poor conversion.10a In that case, the low current efficiency was the result of a very stable radical cation intermediate and the use of an undivided cell. The radical cation survived long enough to migrate across the cell and get reduced at the cathode. The current in the reaction was being consumed by the unproductive oxidation and re-reduction of the substrate. In the current case, the same situation might arise if radical cation 6 was stable enough to reach the cathode in the undivided cell used.
Initial evidence that this was also the problem for the current reaction was obtained when the electrolyte concentration of the reaction was increased. Additional electrolyte provides additional counterions for the cations generated at the anode, a change that can shift an equilibrium at the electrode surface toward the oxidized product10a and slow migration of the charged species to the cathode. In the current case, an increase in electrolyte concentration from 0.1 M to 0.5 M improved the conversion of the reaction to 56%.
While the use of more electrolyte provided a better conversion, it was still not high and the use of a divided electrolysis cell seemed like it would provide a very straight-forward solution to the problem. The use of a divided cell removes any chance for contact between the radical cation and the cathode, and therefore any chance of the radical cation being reduced back to the starting material. For the oxidative condensation, the use of a simple H-cell (Fig. 1) improved the reaction greatly. In this reaction, both chambers of the H-cell were charged with an 0.5 M lithium perchlorate in 4:1 THF/methanol electrolyte solution, and then the starting syringealdehyde and amino thiol added to the anodic chamber along with some MgSO4 for the condensation reaction.pp As in the earlier cases, the reaction was initially allowed to stir for a couple of hours prior to the electrolysis in order to insure formation of the thioaminal. When 25 mA of current was passed through the cell for 2.1 F mol−1, the reaction went to 100% completion. It was clear that the divided cell solved the conversion issue encountered with the undivided cell and the presence of the cathode in the reaction.
 | ||
| Fig. 1 The use of a divided cell. | ||
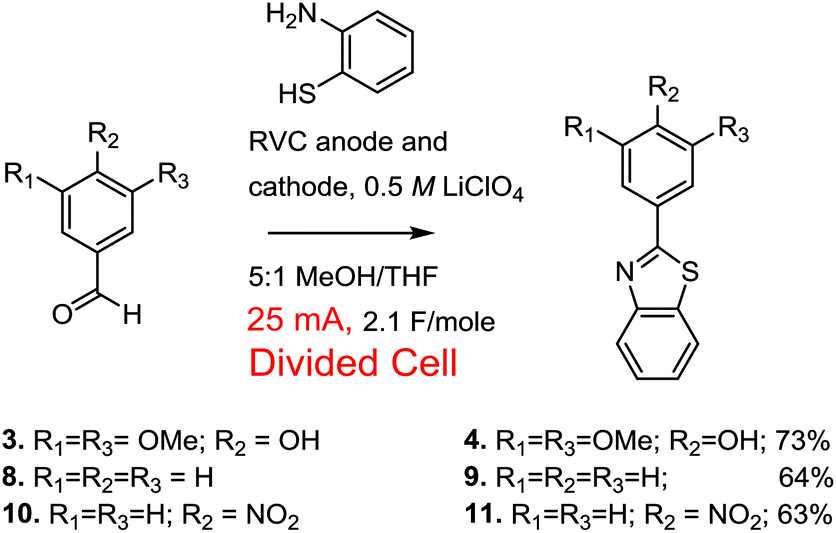
Interestingly, when the divided cell was used there was no need for the initial incubation period. Even without preforming the thioaminal (Scheme 7), the reaction led to 100% conversion and a 73% isolated yield of the product with no evidence of a monomeric over-oxidized product in the proton NMR of the crude reaction mixture. The lower than quantitative isolated yield appeared to mainly result from a small amount of polymer formation during the reaction and some product sensitivity toward the chromatographic purification method employed. The observation that preformation of the thioaminal was not needed when a divided cell was used was true even when the current passed through the cell was raised to 40 mA. While we have no direct evidence for why the reaction in the divided cell does not need to have the thioaminal preformed, we can speculate. The divided cell reaction will be more acidic than the reaction in an undivided cell (no base from the cathode). If this greater acidity leads to protonation of the amine lone-pair in the product, then it would protect the product from over-oxidation.
 | ||
| Scheme 7 Divided cell reactions. | ||
With the successful paired electrochemical reactions shown in Scheme 1 utilizing 25 mA of current, it was clear that the current flow through the oxidative condensation reaction was now sufficiently high for it to be compatible with use in paired electrolyses.
The optimized oxidative condensation conditions were also used to conduct the reaction with both benzaldehyde and a p-nitrobenzaldehyde. While the yields of the reactions were slightly lower than the reaction using the very electron-rich syringealdehyde, the reaction is compatible with both simple phenyl and electron-poor aryl aldehydes (Scheme 7).
In conclusion, the optimization of the electrochemical conditions used for an oxidative condensation reaction strongly benefited from a careful consideration of the reaction mechanism. The actual oxidation step occurred in the exact manner expected, but optimizing the effectiveness of that step required better control of the chemical intermediate presented to the electrode and then manipulation of the “downstream” intermediates following the oxidation. The result in this case is an optimization of the current that can be passed through the oxidative condensation reaction so that it can be utilized in future paired electrochemical reactions for the valorization of lignin derived materials.
Acknowledgements
This work is generously supported by the National Science Foundation (CHE-1463913 as well as CHE-1240194/CenSURF for lignin related efforts).References
- For a general discussion of electrochemical methods see: Organic Electrochemistry: Revised and Expanded, ed. O. Hammerich and B. Speiser, CRC Press, Boca Raton, FL, 5th edn, 2016 Search PubMed.
- For reviews of recent efforts see: (a) J. B. Sperry and D. L. Wright, Chem. Soc. Rev., 2006, 35(7), 605 RSC; (b) J. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108(7), 2265 CrossRef CAS PubMed; (c) B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12(12), 2099 RSC.
- B. H. Nguyen, R. J. Perkins, J. A. Smith and K. D. Moeller, J. Org. Chem., 2015, 80, 11953 CrossRef CAS PubMed.
- For a review see: B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12(12), 2099 RSC.
- (a) C. A. Paddon, M. Atobe, T. Fuchigami, P. He, P. Watts, S. J. Haswell, G. J. Pritchard, S. D. Bull and F. Marken, J. Appl. Electrochem., 2006, 36(6), 617 CrossRef CAS; (b) X. Song, H. Du, Z. Liang, Z. Zhu, D. Duan and S. Liu, Int. J. Electrochem. Sci., 2013, 8(5), 6566 CAS; (c) D. Nematollahi and F. Varmaghani, Electrochim. Acta, 2008, 53(8), 3350 CrossRef CAS.
- (a) R. D. Carpenter, M. Andrei, O. H. Aina, E. Y. Lau, F. C. Lightstone, R. Liu, K. S. Lam and M. J. Kurth, J. Med. Chem., 2009, 5(1), 14 CrossRef PubMed; (b) A. Ammazzalorso, A. Giancristofaro, A. 'Angelo, B. De Filippis, M. Fantacuzzi, L. Giampietro, C. Maccallini and R. Amoroso, Bioorg. Med. Chem. Lett., 2011, 21, 4869 CrossRef CAS PubMed; (c) C. B. Vu, J. C. Milne, D. P. Carney, J. Song, W. Choy, P. D. Lambert, D. J. Gagne, M. Hirsch, A. Cote, M. Davis, E. Lainez, M. Meade, K. Normington, M. R. Jirousek and R. B. Perni, Bioorg. Med. Chem. Lett., 2009, 19, 1416 CrossRef CAS PubMed; (d) P. W. Bowyer, R. S. Gunaratne, M. Grainger, C. Withers-Martinez, S. R. Wickramsinghe, E. W. Tate, R. J. Leatherbarrow, K. A. Brown, A. A. Holder and D. F. Smith, Biochem. J., 2007, 408, 173 CrossRef CAS PubMed; (e) M. F. G. Stevens, C. J. McCall, P. Lelievald, P. Alexander, A. Richter and D. E. Davies, J. Med. Chem., 1994, 37(11), 1689 CrossRef CAS PubMed; (f) T. D. Bradshaw and A. D. Westwell, Curr. Med. Chem., 2004, 11, 1241 CrossRef; (g) E. Y. Song, N. Kaur, K. M. Park, Y. Jin, K. Lee, G. Kim, K. Y. Lee, J. S. Yang, J. H. Shin, K. Y. Nam, K. T. No and G. Han, Eur. J. Med. Chem., 2008, 43, 1519 CrossRef CAS PubMed.
- For examples of a constant current electrolysis automatically adjusting to the use of a variety of oxidants see: B. H. Nguyen, A. Redden and K. D. Moeller, Green Chem., 2014, 16, 69–72 RSC.
- (a) F. Tang and K. D. Moeller, J. Am. Chem. Soc., 2007, 129, 12414 CrossRef CAS PubMed; (b) F. Tang and K. D. Moeller, Tetrahedron, 2009, 65, 10863 CrossRef CAS; (c) A. Redden, R. J. Perkins and K. D. Moeller, Angew. Chem., Int. Ed., 2013, 52, 12865 CrossRef CAS PubMed.
- (a) J. D. Brandt and K. D. Moeller, Org. Lett., 2005, 7, 3553 CrossRef CAS PubMed; (b) C. M. Hudson, M. R. Marzabadi, K. D. Moeller and D. G. New, J. Am. Chem. Soc., 1991, 113, 7372 CrossRef CAS.
- (a) J. A. Smith and K. D. Moeller, Org. Lett., 2013, 15, 5818 CrossRef CAS PubMed; (b) R. J. Perkins, H.-C. Xu, J. M. Campbell and K. D. Moeller, Beilstein J. Org. Chem., 2013, 9, 1630 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data for all new compounds are provided along with a sample procedure for conducting the electrolysis. See DOI: 10.1039/c6qo00248j |
| This journal is © the Partner Organisations 2016 |