
Bismuth(III) triflate-catalysed tandem cyclisations towards complex polycyclic ethers†
Pierrick
Ondet
,
Luisa
Lempenauer
,
Elisabet
Duñach
* and
Gilles
Lemière
*
Institut de Chimie de Nice, UMR 7272, Université Nice Sophia Antipolis, CNRS, Parc Valrose, 06108 Nice Cedex 2, France. E-mail: gilles.lemiere@upmc.fr; dunach@unice.fr
First published on 22nd June 2016
Abstract
Herein is described a double cyclisation of α-hydroxy enol ethers with a tethered olefinic double bond, readily obtained from simple ketone precursors. Interesting bridged polycyclic compounds with an additional oxaspirocycle are formed under mild conditions using a low loading of recyclable bismuth(III) triflate as the catalyst. Some experimental evidence accounts for concerted (or pseudo-concerted) polycyclisation.
Bridged polycyclic systems constitute common structural motifs present in a great number of naturally occurring organic molecules. These attractive scaffolds have always fascinated organic chemists promoting the emergence of various elegant synthetic strategies.1 Although many methodologies have been developed, most of them require several steps to interconnect the strained polycyclic systems and the different rings are often formed independently. The rapid and controlled generation of molecular complexity starting from simple precursors is one of the main challenges in modern organic synthesis. In this context, catalytic cycloisomerisation reactions represent undeniably versatile, economic and sustainable tools.2
Among the bridged systems, those containing a heteroatom such as oxygen are important scaffolds in many biologically active compounds.3 In the course of our investigations on the cycloisomerisation of polyunsaturated substrates using metal triflates,4 we discovered an intriguing activity for the latter, allowing for a very straightforward increase in molecular complexity. Indeed, in the presence of a metal triflate, α-hydroxy enol-ether 1a was successfully transformed into 2a, featuring a saturated bridged bicyclic structure with an additional oxaspirocycle. Compound 2a was obtained as a single isomer with respect to the spatial orientation of the two oxygen atoms present in the tricyclic system (Scheme 1).5
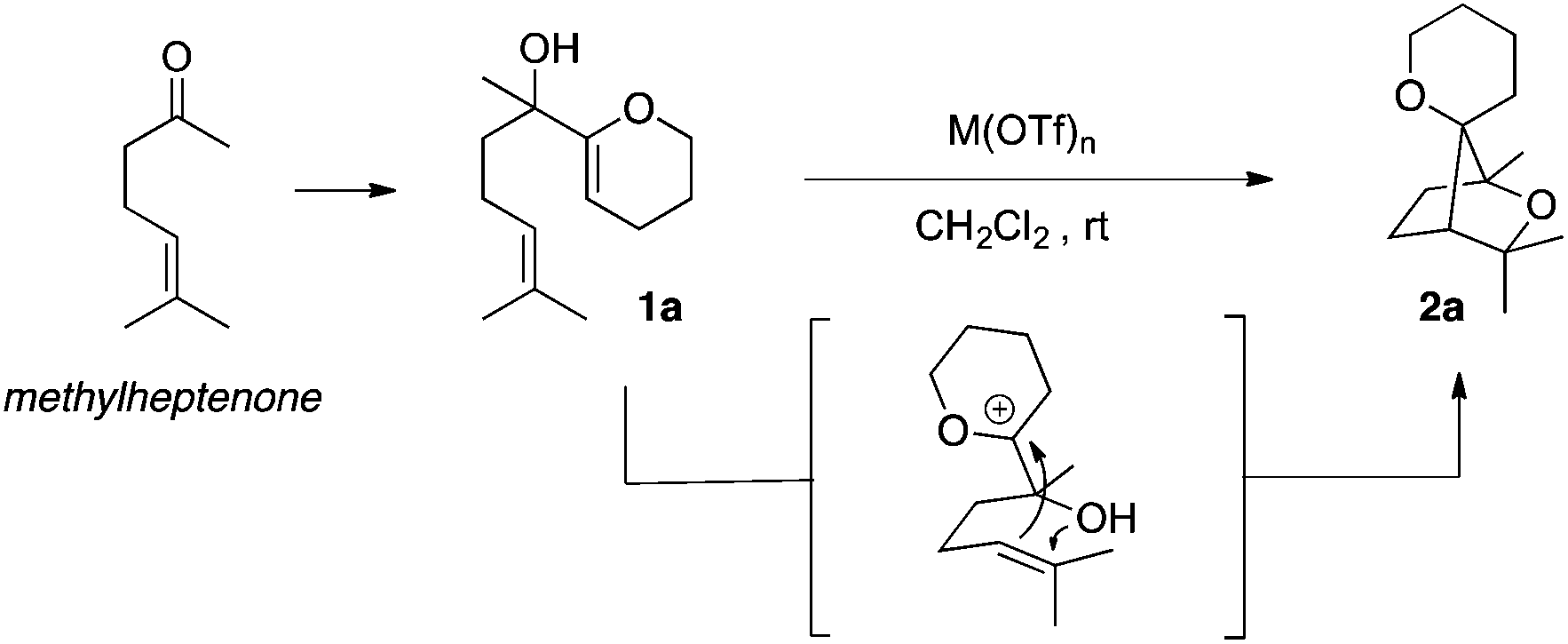 | ||
| Scheme 1 Formation of oxabicyclo[2.2.1]heptane 2a. | ||
Optimisation of the catalytic system was performed for the cycloisomerisation of dienol 1a assessing the activity of various metallic salts (Table 1). Several metal triflates used at a catalytic loading of 1 mol% in dichloromethane allowed for a complete conversion of the starting material in less than an hour (Table 1, entries 1–5). Among these catalysts, bismuth(III) triflate6,7 was found to be the most effective and 2a was obtained in 73% yield after only thirty minutes at room temperature. When the less reactive bismuth(III) tosylate or chloride was applied, full conversion of 1a was not reached even after six hours of reaction (Table 1, entries 6 and 7). Interestingly, triflic acid did also catalyse the formation of 2a, however with a decreased yield of 60%.
| Entry | Catalysta (mol%) | Timeb | Yieldc (%) |
|---|---|---|---|
| a General screening procedure: the catalyst was added to a solution of 1a in anhydrous CH2Cl2 (0.1 M) at room temperature. b Time until full conversion of 1a (TLC). c Yields determined by GC using dodecane as an internal standard. d Conversion was not complete. | |||
| 1 | Al(OTf)3 (1) | 40 min | 64 |
| 2 | Cu(OTf)2 (1) | 40 min | 62 |
| 3 | Fe(OTf)2 (1) | 2 h | 61 |
| 4 | Fe(OTf)3 (1) | 40 min | 60 |
| 5 | Bi(OTf) 3 (1) | 30 min | 73 |
| 6 | Bi(OTs)3 (1) | 6 hd | 1 |
| 7 | BiCl3 (1) | 6 hd | 49 |
| 8 | TfOH (1) | 15 min | 60 |
Thus, we further explored the scope of the polycyclisation using the triflate salt of bismuth, which is a relatively inexpensive and non-toxic metal. In addition, and as opposed to triflic acid, it has been established that this metallic species could be conveniently recovered by a simple aqueous extraction/evaporation process and reused in several catalytic cycles.8
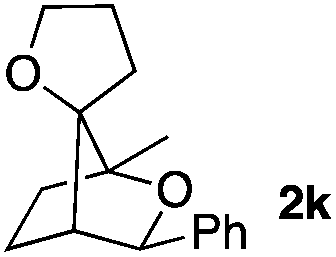
Consequently, some variations of the enol ether, carbinol and olefin part moiety were also made. The starting tertiary alcohols were easily synthesised in one step from the corresponding ketones via addition of lithiated enol ether. To our delight, the tandem cyclisation occurred with cyclic enol ethers such as dihydropyranyl and dihydrofuranyl derivatives 1a and 1b to lead to bridged-polycyclic compounds 2a and 2b in isolated yields of 62% and 86% respectively, using only 0.1 mol% of Bi(OTf)3 in the latter case (Table 2, entries 1 and 2). While the cyclisation of 1a occurred with a total diastereoselectivity in favour of the trans compound, two diastereoisomers were obtained for the analogous five-membered ring 1b, predominantly yielding the cis-isomer. It is worth mentioning that the polarity of the two isomers was noticeably different and they could be easily separated by flash chromatography for full characterisation. Moreover, the cyclisations could be performed on the gram scale. Acyclic enol ether derivative 1c was also readily cyclised to afford 2-oxabicyclo[2.2.1]heptane derivative 2c in 82% yield (Table 2, entry 3). Ethyl derivatives 1d and 1e were successfully cyclised into 2d and 2e, respectively (Table 2, entries 4 and 5). Interestingly, alcohols 1f and 1g bearing a phenyl substituent could be used as well. In both cases, elimination of the benzylic alcohol did not occur under the Lewis acidic conditions and polycyclic compounds 2f and 2g were obtained in 67% and 99% yields, respectively (Table 2, entries 6 and 7). Here again, the cyclisation of dihydropyranyl substrate 1f occurred with an excellent diastereoselectivity. More substituted polycyclic compounds 2h and 2i could as well be accessed from more hindered substrates 1h and 1i bearing a quaternary carbon center with a gem-dimethyl group adjacent to the carbinol (Table 2, entries 8 and 9). Finally, the substitution pattern of the olefinic double bond was further modified and the isopropylidene group on the methine was replaced by a benzylidene one. Polycyclic compounds 2j and 2k were obtained with a total control of the stereochemistry at the benzylic position (Table 2, entries 10 and 11). Here again compound 2j featuring a tetrahydropyranyl group was obtained as a trans-isomer, exclusively.
|
|
||||
|---|---|---|---|---|
| Entrya | Substrate | Product | trans/cis | Yieldb |
| a Conditions: Bi(OTf)3 was added to a solution of alcohol 1 in CH2Cl2 (0.1 M) and the mixture was stirred at room temperature until full conversion was reached. b Isolated yields. c The reaction was performed in CH3NO2. d The reaction was conducted with 0.1 mol% of Bi(OTf)3. | ||||
| 1 |
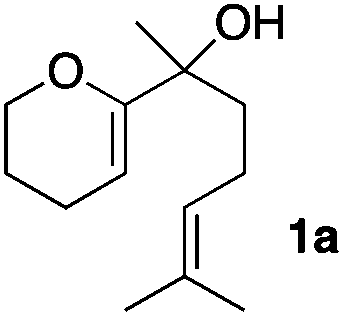
|
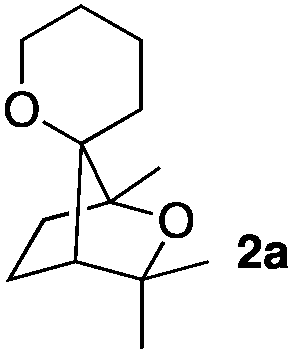
|
100/0 | 62% |
| 2 |
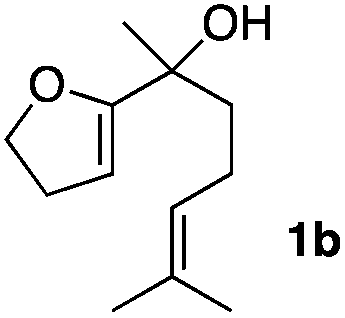
|
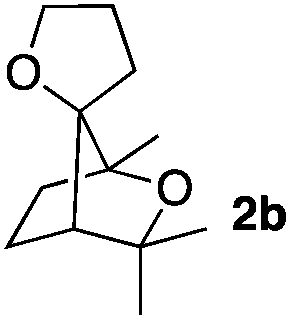
|
33/67 | 86%d |
| 3 |
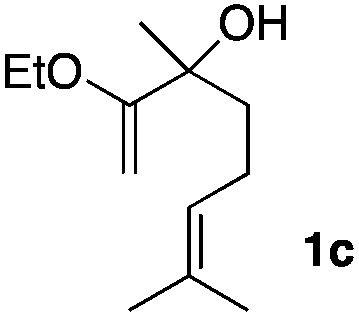
|
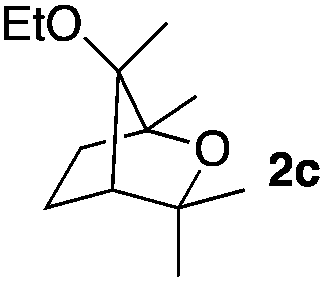
|
65/35 | 82%c |
| 4 |
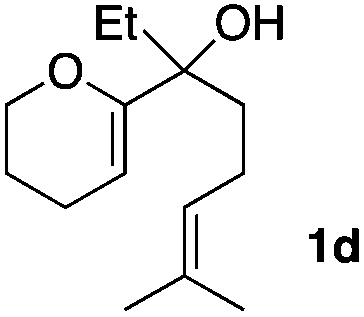
|
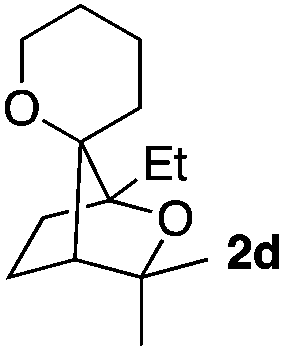
|
85/15 | 81%d |
| 5 |
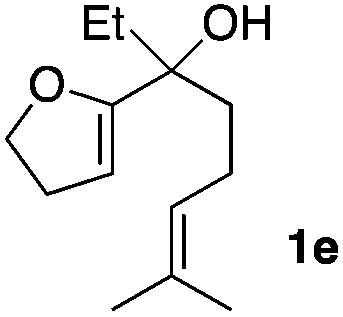
|
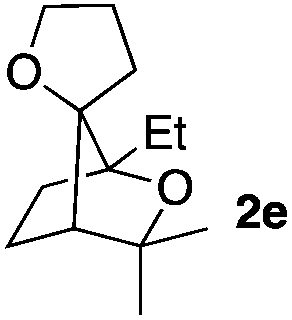
|
27/73 | 83%d |
| 6 |
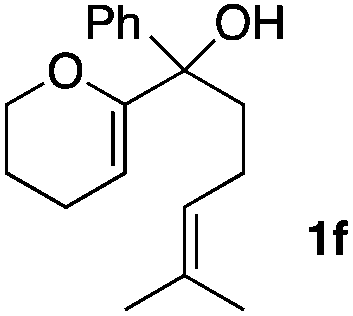
|
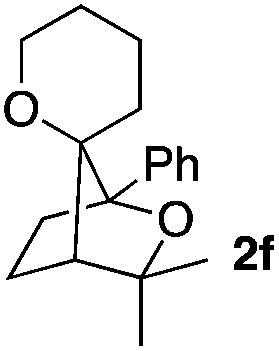
|
100/0 | 67%c,d |
| 7 |
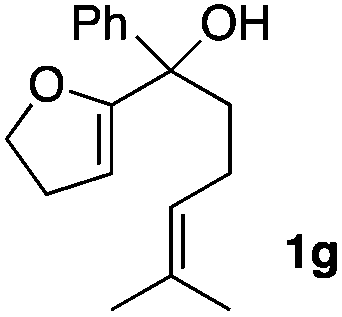
|
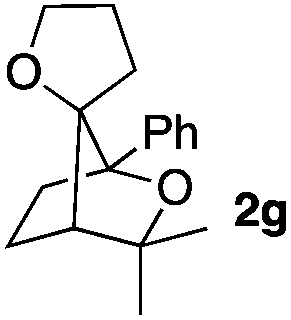
|
24/76 | 99%c,d |
| 8 |
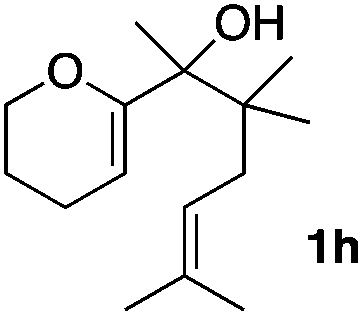
|
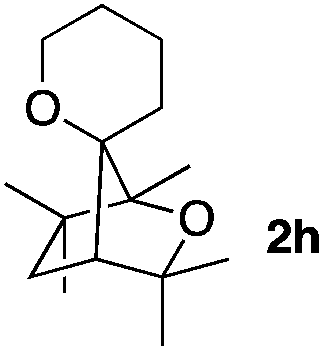
|
100/0 | 50% |
| 9 |
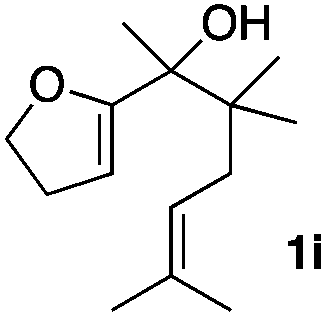
|
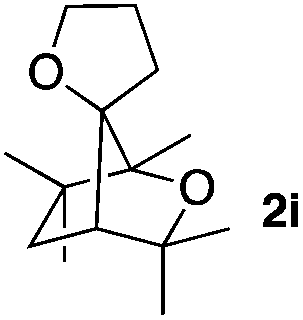
|
26/74 | 82%c |
| 10 |
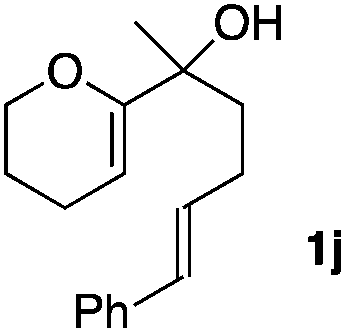
|
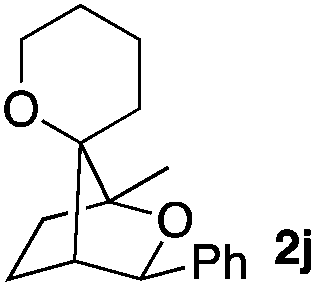
|
100/0 | 58% |
| 11 |
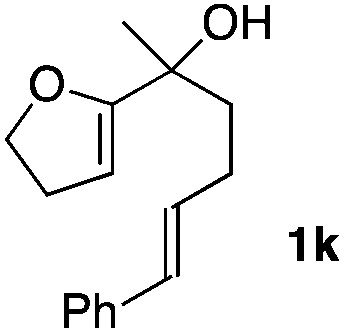
|
|
74/26 | 88% |
To extend the structural diversity of the products obtained with this protocol, we investigated the reactivity of substrates bearing a different substitution pattern. Replacing the prenyl group by a methallyl group led to a different cyclisation outcome, furnishing 7-oxabicyclo[2.2.1]heptane derivatives. In the presence of 1 mol% of Bi(OTf)3 in dichloromethane, dienol substrate 3a led to a nearly equal mixture of the saturated bridged polycycle 4a and monocyclised compound 5a.
The ratio of these two isomers did not evolve even at higher temperature. Several solvents were screened and switching to more polar nitromethane allowed for the highly selective formation of the desired compound 4a (Scheme 2).
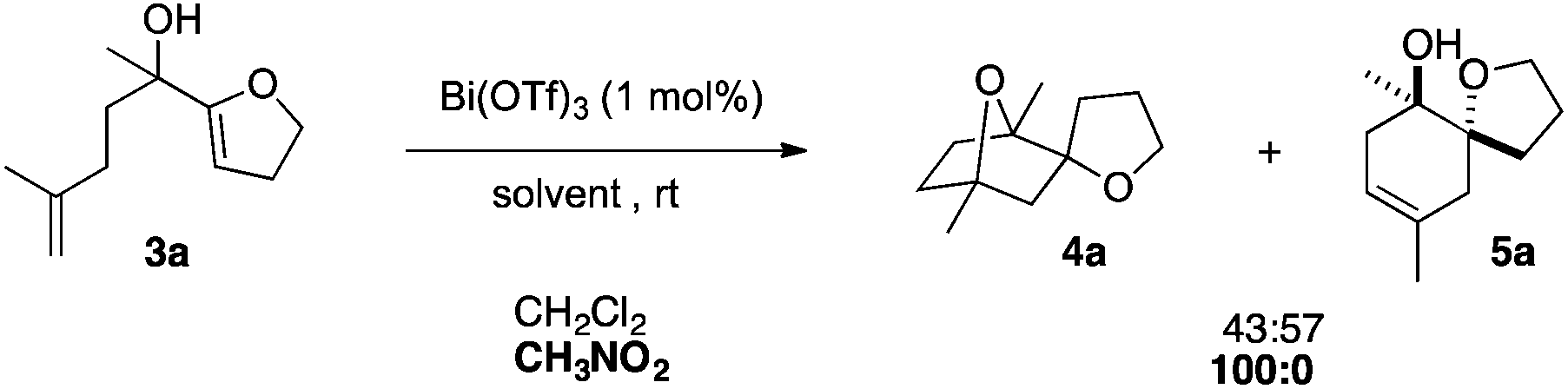 | ||
| Scheme 2 Solvent effect for the polycyclisation of methallyl derivative 3a. | ||
Under these conditions, a new set of products 4a–4d could be efficiently obtained with yields ranging from 78 to 95% (Scheme 3). The structure of these polycyclic compounds differs from the previous examples (Table 2). The polarity of the reacting double bond being inverted, the regioselectivity of the cyclisation changed. Furthermore, an excellent diastereoselectivity was observed in all cases and only diastereomers with the two oxygens oriented in an anti-fashion were formed.
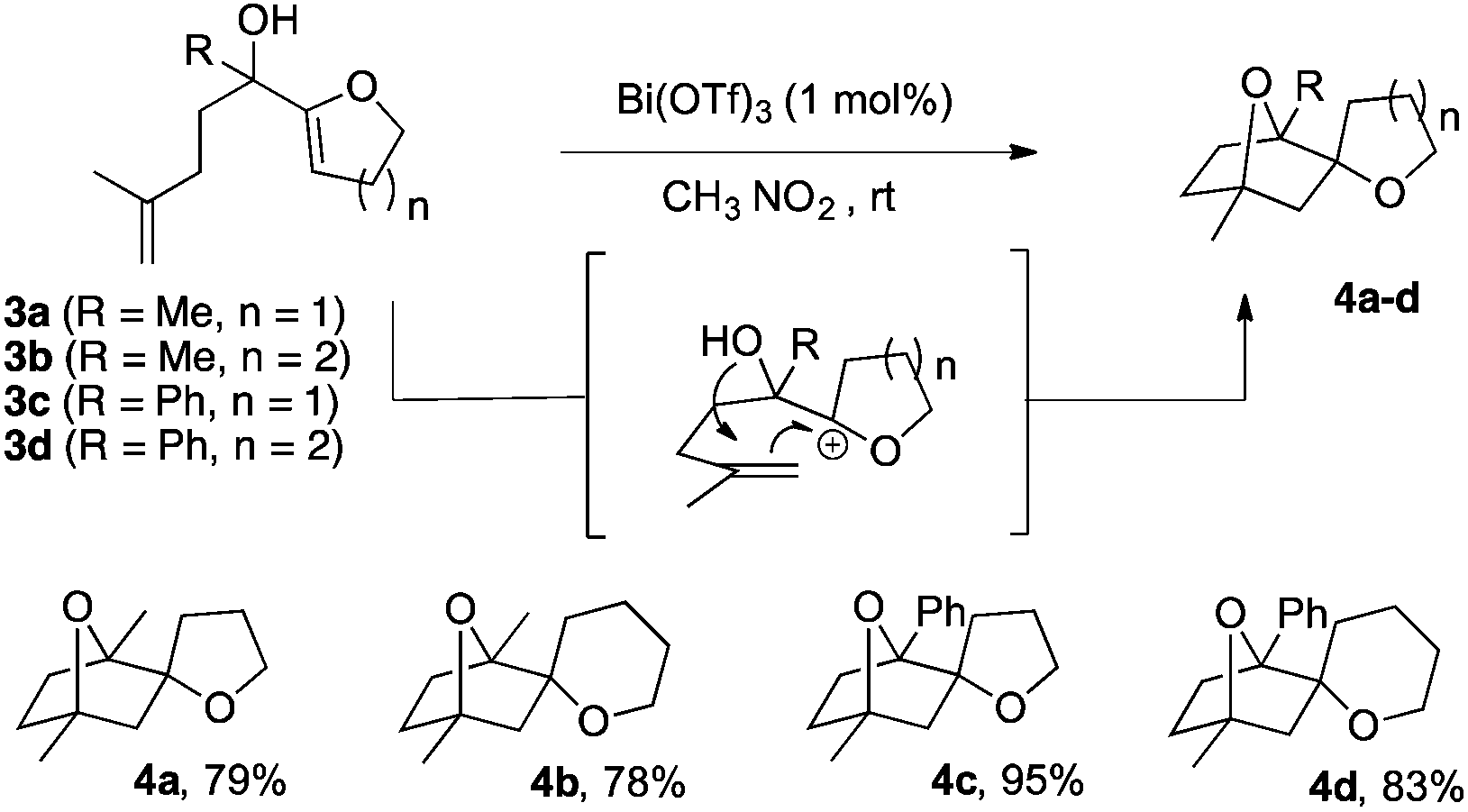 | ||
| Scheme 3 Bi(OTf)3-catalysed synthesis of oxabicyclo[2.2.1]heptanes 4. | ||
The bridged structure and the stereochemistry of cis-2g and compound 4d could be confirmed by X-ray crystallography (Fig. 1).
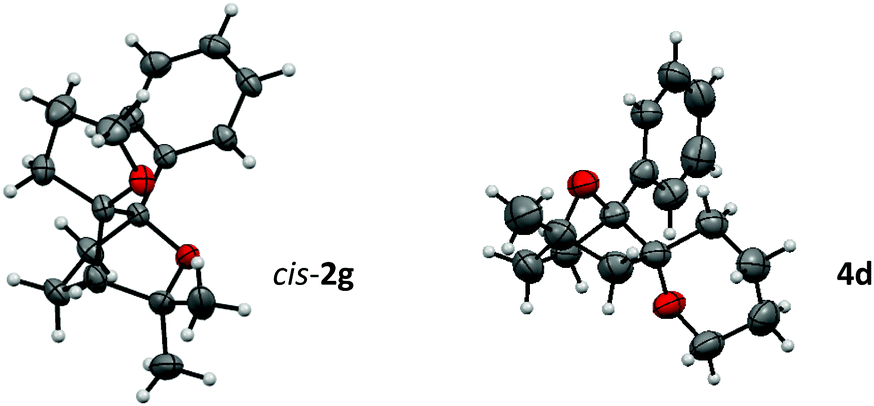 | ||
| Fig. 1 X-ray crystallographic structure of cis-2g and 4d. | ||
From a mechanistic point of view, two aspects were taken into consideration: (i) the role of the electrophilic bismuth(III) triflate in general and more specifically its behaviour towards the substrate and (ii) the cyclisation pathway to form the saturated bridged systems. With respect to the products obtained and the previous work in this field,7 it can be proposed that the electron-rich enol ether is activated first. Although bismuth(III) has been sporadically described as a π-Lewis acid,9 it is now commonly accepted that Bi(OTf)3 would act as a source of Brønsted acid,10 possibly through a Lewis acid-assisted Brønsted acid (LBA) system11 or as a triflic acid reservoir,12 the first possibility being energetically more favoured.13 In both cases, incipient protonation of the enol ether moiety would generate an oxocarbenium intermediate A (Scheme 4). From this reactive species, a stepwise mechanism, where the cyclisations occur as two single events would furnish oxabicyclo[2.2.1]heptane 2a. The first 5-exo-trig cyclisation has to proceed with a remarkably high diastereoselectivity in order to correctly place the alcohol in close proximity to the tertiary carbocation in B, to allow the second ring closure to occur. Intermediate B could eventually lose a proton to form the olefin C. Another pathway involving a (pseudo-) concerted polycyclisation may also be considered and accounts for the diastereoselectivity of these tandem cyclisations. In this case, the formation of the bridged system should be stereospecific.
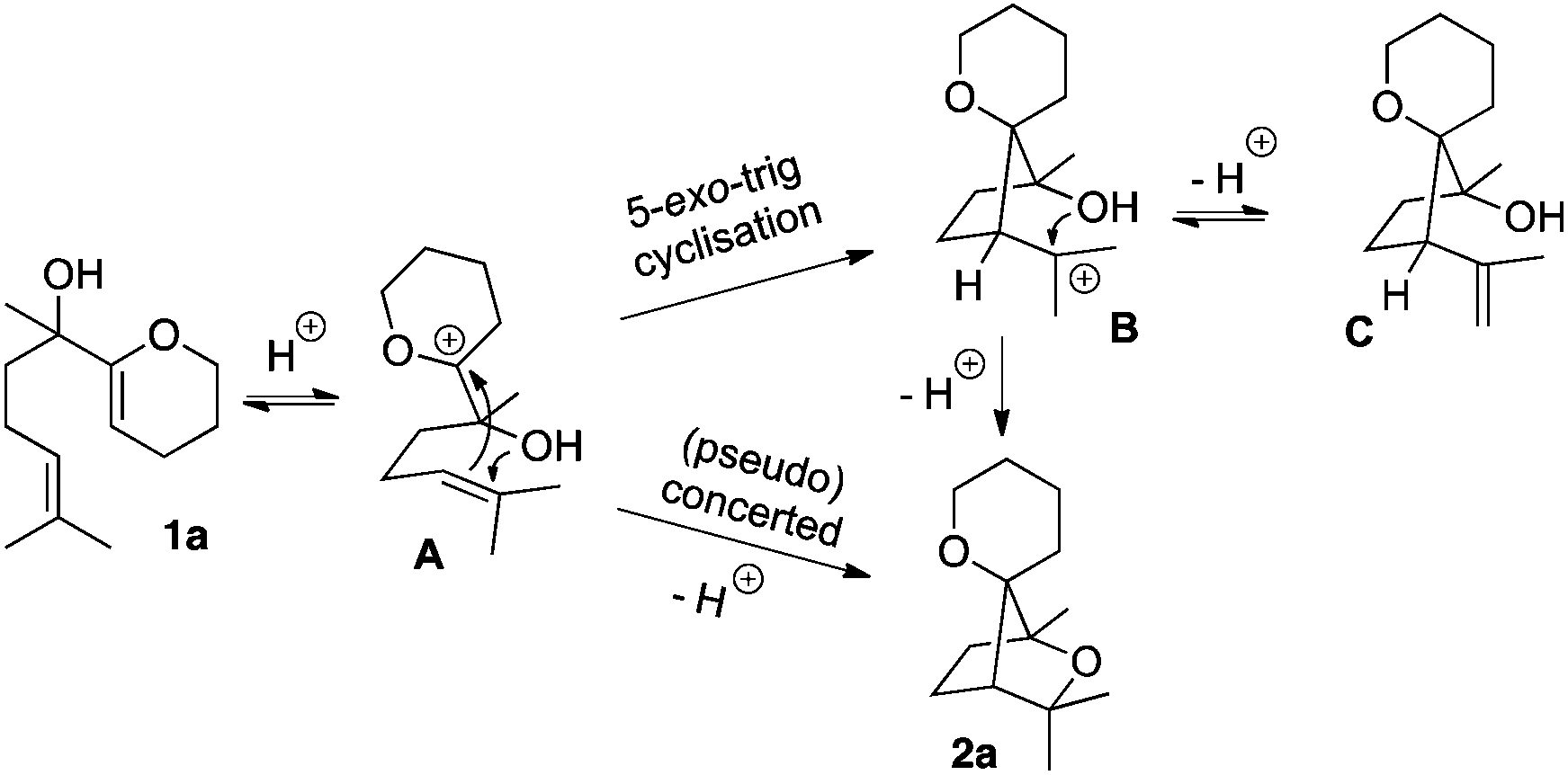 | ||
| Scheme 4 Mechanistic aspects in the polycyclisation of 1a. | ||
A set of experiments was further carried out in order to get some mechanistic insights. We first examined whether cyclohexenol 5a, obtained from the cyclisation of 3a (Scheme 2), was potentially involved in the formation of 4a. When 5a was subjected to 1 mol% of Bi(OTf)3 in dichloromethane or nitromethane, compound 4a was not formed and the only isolable product was the aromatic compound 6, resulting from the elimination of alcohol and tetrahydrofuran ring opening (Scheme 5). This fact might be a possible indication for a concerted cyclisation of 3 to 4.
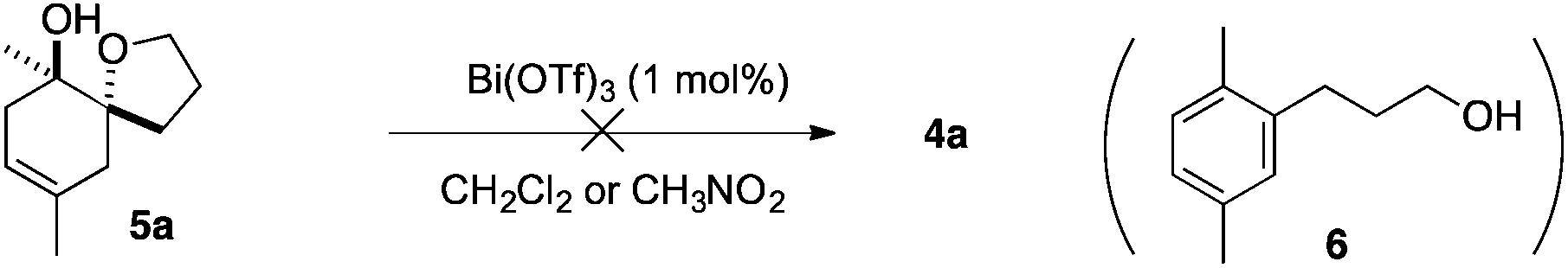 | ||
| Scheme 5 Attempts to synthesise 4a from 5a. | ||
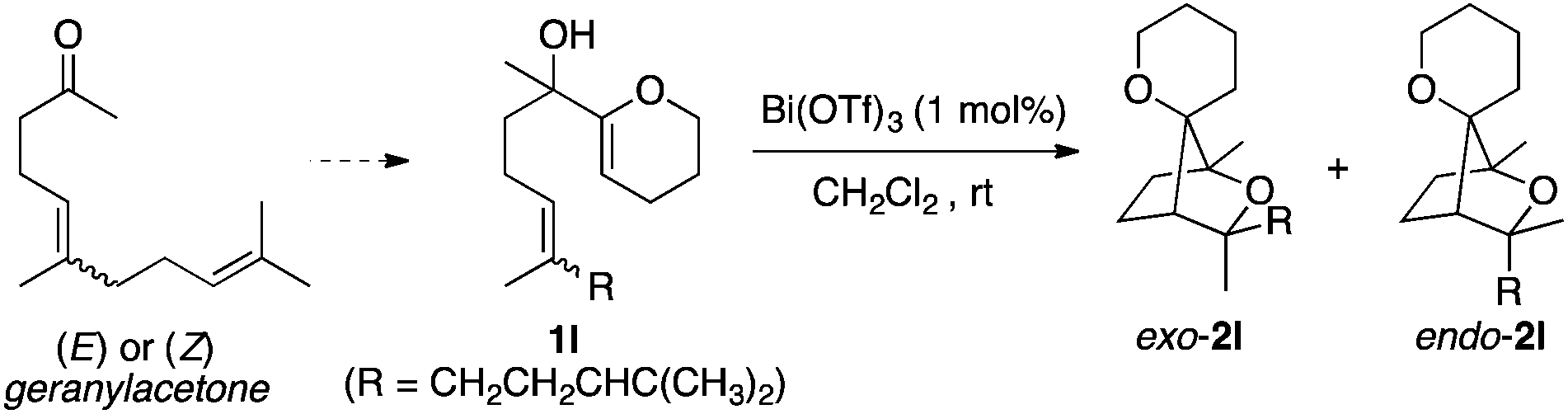
In order to get more information about the stereospecificity of the double cyclisation leading to 2-oxabicyclo(2.2.1)heptanes of type 2, we studied the cyclisation of (E) and (Z) isomers of 1l. Biomimetic cationic cyclisations of polyenes are known to proceed with a high level of stereospecificity according to the Stork–Eschenmoser paradigm.14 The concerted nature of the cyclisation is thought to be dependent on the reactivity of the terminating nucleophile.15 While a nearly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereoisomers exo-2l/endo-2l was obtained from a 45/55 E/Z mixture of substrate 1l (Table 3, entry 1), cyclisation of almost diastereomerically pure (E)-1l or (Z)-1l predominantly led to exo-2l or endo-2l, respectively (Table 3, entries 2 and 3). These results give evidence for a concerted (or pseudo-concerted) polycyclisation. It is worth noting that examples of stereospecific cation-based cyclisations of polyenols have been mainly reported for the synthesis of fused ether-containing polycycles through an anti-addition.15,16 In the present case, a concerted syn-addition onto the double bond is observed.
:1 mixture of diastereoisomers exo-2l/endo-2l was obtained from a 45/55 E/Z mixture of substrate 1l (Table 3, entry 1), cyclisation of almost diastereomerically pure (E)-1l or (Z)-1l predominantly led to exo-2l or endo-2l, respectively (Table 3, entries 2 and 3). These results give evidence for a concerted (or pseudo-concerted) polycyclisation. It is worth noting that examples of stereospecific cation-based cyclisations of polyenols have been mainly reported for the synthesis of fused ether-containing polycycles through an anti-addition.15,16 In the present case, a concerted syn-addition onto the double bond is observed.
Conclusions
In conclusion, we have reported an efficient methodology for the straightforward synthesis of complex oxaspiro ethers featuring an additional bridged bi- or tricyclic system. These cycloisomerisations were carried out under very mild conditions at ambient temperature using a very low catalytic loading of cheap and recyclable bismuth(III) triflate. In many cases, the diastereoselectivity is excellent, allowing for the control of the relative configuration of three stereogenic centers in a single transformation. Preliminary mechanistic investigations highlighted a concerted polycyclisation for the construction of the 2-bicyclo[2.2.1]heptane scaffold through a syn-addition. Finally, polycyclic ethers are widely employed as fragrance compounds in perfume formulations.17 Most of the complex cyclic ethers synthesized via this methodology feature promising olfactory properties and therefore could potentially find some applications in the field of fragrance chemistry.Notes and references
- M. Presset, Y. Coquerel and J. Rodriguez, Chem. Rev., 2013, 113, 525 CrossRef CAS PubMed; M. Ruiz, P. López-Alvarado, G. Giorgi and J. C. Menéndez, Chem. Soc. Rev., 2011, 40, 3445 RSC; W. Zhao, Chem. Rev., 2010, 110, 1706 CrossRef PubMed.
- Y. Yamamoto, Chem. Rev., 2012, 112, 4736 CrossRef CAS PubMed; C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954 CrossRef PubMed; A. Fürstner, Chem. Soc. Rev., 2009, 38, 3208 RSC; E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326 CrossRef PubMed; V. Michelet, P. Y. Toullec and J.-P. Genêt, Angew. Chem., Int. Ed., 2008, 47, 4268 CrossRef PubMed; G. C. Lloyd-Jones, Org. Biomol. Chem., 2003, 1, 215 Search PubMed.
- M. Flores and D. Díez, Synlett, 2014, 1643 Search PubMed; A. Deiters and S. F. Martin, Chem. Rev., 2004, 104, 2199 CrossRef CAS PubMed; P. Vogel, J. Cossy, J. Plumet and O. Arjona, Tetrahedron, 1999, 55, 13521 CrossRef.
- G. Lemière and E. Duñach, Chem. – Eur. J., 2013, 19, 3270 CrossRef CAS PubMed; S. Antoniotti, S. Poulain-Martini and E. Duñach, Synlett, 2010, 2973 Search PubMed; S. Antoniotti, V. Dalla and E. Duñach, Angew. Chem., Int. Ed., 2010, 49, 7860 CrossRef PubMed.
- For similar cyclisations based on double Prins reaction see: M. E. Jung, S. Angelica and D. C. D'Amico, J. Org. Chem., 1997, 62, 9182 CrossRef CAS; X. Wang, J. Zheng, Q. Chen, H. Zheng, Y. He, J. Yang and X. She, J. Org. Chem., 2010, 75, 5392 CrossRef PubMed.
- For reviews on bismuth catalysis: T. Ollevier, Org. Biomol. Chem., 2013, 11, 2740 Search PubMed; J. M. Bothwell, S. W. Krabbe and R. S. Mohan, Chem. Soc. Rev., 2011, 40, 4649 RSC; H. Gaspard-Iloughmane and C. Le Roux, Eur. J. Org. Chem., 2004, 2517 CrossRef CAS.
- For recent publications on cyclisations using Bi(OTf)3: M.-Y. Chang, Y.-C. Cheng and Y.-J. Lu, Org. Lett., 2015, 17, 3142 CrossRef CAS PubMed; M.-Y. Chang, Y.-C. Cheng and Y.-J. Lu, Org. Lett., 2015, 17, 1264 CrossRef PubMed; P. Ondet, A. Joffrin, I. Diaf, G. Lemière and E. Duñach, Org. Lett., 2015, 17, 1002 CrossRef PubMed; D. Nitsch and T. Bach, J. Org. Chem., 2014, 79, 6372 CrossRef PubMed; I. Diaf, G. Lemière and E. Duñach, Angew. Chem., Int. Ed., 2014, 53, 4177 CrossRef PubMed; B. Cacciuttolo, P. Ondet, G. Lemière, S. Poulain-Martini and E. Dunach, Org. Chem. Front., 2014, 1, 765 RSC.
- G. Lemière, B. Cacciuttolo, E. Belhassen and E. Duñach, Org. Lett., 2012, 14, 2750 CrossRef CAS PubMed; B. Cacciuttolo, S. Poulain-Martini and E. Duñach, Eur. J. Org. Chem., 2011, 3710 CrossRef.
- M. Murai, N. Hosokawa, D. Roy and K. Takai, Org. Lett., 2014, 16, 4134 CrossRef CAS PubMed; Y. Nishimoto, M. Takeuchi, M. Yasuda and A. Baba, Angew. Chem., Int. Ed., 2012, 51, 1051 CrossRef PubMed; H. Qin, N. Yamagiwa, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2009, 128, 1611 CrossRef PubMed.
- R. F. Lambert, R. J. Hinkle, S. E. Ammann, Y. Lian, J. Liu, S. E. Lewis and R. D. Pike, J. Org. Chem., 2011, 76, 9269 CrossRef CAS PubMed; D. B. G. Williams and M. J. Lawton, Mol. Catal. A: Chem., 2010, 317, 68 CrossRef; A. Dzudza and T. J. Marks, Chem. – Eur. J., 2010, 16, 3403 CrossRef PubMed; D. C. Rosenfeld, S. Shekhar, A. Takemiya, M. Utsunomiya and J. F. Hartwig, Org. Lett., 2006, 8, 4179 CrossRef PubMed; T. C. Wabnitz, J.-Q. Yu and J. B. Spencer, Chem. – Eur. J., 2004, 10, 484 CrossRef PubMed; S. Kobayashi, S. Nagayama and T. Busujima, J. Am. Chem. Soc., 1998, 120, 8287 CrossRef.
- O. Kanno, W. Kuriyama, Z. J. Wang and F. D. Toste, Angew. Chem., Int. Ed., 2011, 50, 9919 CrossRef CAS PubMed; C. H. Cheon, O. Kanno and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 13248 CrossRef PubMed; L. Coulombel, M. Rajzmann, J.-M. Pons, S. Olivero and E. Duñach, Chem. – Eur. J., 2006, 12, 6356 CrossRef PubMed; H. Yamamoto and K. Futatsugi, Angew. Chem., Int. Ed., 2005, 44, 1924 CrossRef PubMed.
- M. Murai, K. Origuchi and K. Takaki, Org. Lett., 2014, 16, 3828 CrossRef CAS PubMed; F. Mathia and P. Szolcsanyi, Org. Biomol. Chem., 2012, 10, 2830 Search PubMed; T. Ollevier and E. Nadeau, Org. Biomol. Chem., 2007, 5, 3126 Search PubMed; R. Dumeunier and I. E. Markó, Tetrahedron Lett., 2004, 45, 825 CrossRef.
- P. Nava, Y. Carissan, J. Drujon, F. Grau, J. Godeau, S. Antoniotti, E. Duñach and S. Humbel, ChemCatChem, 2014, 6, 500 CrossRef CAS; J. Godeau, F. Fontaine-Vive, S. Antoniotti and E. Dunach, Chem. – Eur. J., 2012, 18, 16815 CrossRef PubMed.
- G. Stork and A. W. Burgstahler, J. Am. Chem. Soc., 1955, 77, 5068 CrossRef CAS; G. Gamboni, H. Schinz and A. Eschenmoser, Helv. Chim. Acta, 1954, 37, 964 CrossRef; A. Eschenmoser, L. Ruzicka, O. Jeger and D. Arigoni, Helv. Chim. Acta, 1955, 38, 1890 CrossRef.
- J. A. Feducia and M. R. Gagné, J. Am. Chem. Soc., 2008, 130, 592 CrossRef CAS PubMed; R. A. Yoder and J. N. Johnston, Chem. Rev., 2005, 105, 4730 CrossRef PubMed.
- A. Fürstner and L. Morency, Angew. Chem., Int. Ed., 2008, 47, 5030 CrossRef PubMed; R. L. Snowden, J. C. Eichenberger, S. M. Linder, P. Sonnay, C. Vial and K. H. Schulte-Elte, J. Org. Chem., 1992, 57, 9 CrossRef.
- P. Kraft, J. A. Bajgrowicz, C. Denis and G. Frater, Angew. Chem., Int. Ed., 2000, 39, 2980–3010 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental data, 1H- and 13C-spectra. CCDC 1452479 and 1452480. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00212a |
| This journal is © the Partner Organisations 2016 |