
Recent developments for the photoinduced Ar–X bond dissociation reaction
Guanyinsheng
Qiu
*a,
Yuewen
Li
b and
Jie
Wu
*bc
aCollege of Biological, Chemical Science and Engineering, Jiaxing University, 118 Jiahang Road, Jiaxing 314001, China
bDepartment of Chemistry, Fudan University, 220 Handan Road, Shanghai 200433, China. E-mail: jie_wu@fudan.edu.cn
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
First published on 13th June 2016
Abstract
Recent developments in the photochemical carbon–carbon and carbon–heteroatom bond formation via photoinduced Ar–X bond dissociation are summarized. The transformations through the Ar–X bond dissociation enabled by photoenergy include nucleophilic substitution, arylation, alkylation, aminocarbonylation, aminosulfonylation and decarboxylative coupling reactions. Usually, the reactions undergo one-electron-transfer couplings and aryl radicals or cations are involved as the key intermediates. Under ultraviolet irradiation or visible light, the reactions of aryl halides with reactive partners proceed smoothly under mild conditions. In some cases, the transformations can proceed without any metals or photo-redox catalysts. Moreover, the broad reaction scope is demonstrated with good functional group tolerance.
 Guanyinsheng Qiu | Guanyinsheng Qiu was born in Jiangxi (China) in 1984. He received his BS (2008) and MS (2011) degrees from the Department of Chemistry at Jiangxi Normal University. In the year of 2011, he moved to Fudan University to pursue his Ph.D. under the guidance of Professor Jie Wu. After obtaining his Ph.D. degree in 2014, he has been working in Jiaxing University as a lecturer. During 2014–2015, he worked as a postdoctoral fellow in École Polytechnique Fédérale de Lausanne (supervisor: Prof. Jieping Zhu). His current research interest mainly focuses on the methods’ development for the synthesis of natural product-like compounds under palladium catalysis. |
 Yuewen Li | Yuewen Li was born in Jiangxi (China) in 1989. He received his B.Sc. from the Department of Chemistry at East China University of Technology in 2011; MS from the Department of Chemistry at Jiangxi Normal University in 2014. In the year of 2016, he moved to Fudan University to pursue his Ph.D. under the guidance of Professor Jie Wu. His current research interest mainly focuses on the photoinduced reactions for the synthesis of natural product-like compounds. |
 Jie Wu | Jie Wu received his B.Sc. in chemistry from Jiangxi Normal University in 1995 and he pursued his Ph.D. studies at Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, under the supervision of Prof. Xue-Long Hou (1995–2000). After conducting research as a postdoctoral fellow at Harvard University (2000–2001), he worked as a visiting scientist at the Aaron Diamond AIDS Research Center (2001–2002) and a staff scientist in VivoQuest, Inc. (2002–2004). In 2004, he moved to the Department of Chemistry at Fudan University and held the Professor rank two years later. He got the Thieme Chemistry Journals Award in 2010 and Dow Innovation Challenge Award in 2013. He serves as a member of the Editorial Advisory Board of ACS Combinatorial Science. |
1. Introduction
Over the last decade, photochemical reactions have attracted continuous interest, since novel transformations proceeding under mild reaction conditions could be expected.1,2 Usually, reactive radical or ionic species are involved in the reaction process. In most cases, photocatalysis is necessary and crucial for the successful conversion. For instance, Xiao and co-workers developed an efficient method for the generation of 4,5-dihydropyrazoles through the photocatalytic formation of an N-centered hydrazonyl radical.2d Transition-metal-free reactions enabled by photoenergy are also feasible. For example, the carboxylation of o-alkylphenyl ketones with CO2 under UV light or even solar light under catalyst-free conditions was developed by Murakami recently.2eThe transition-metal-catalyzed cross-coupling reactions of aryl halides are well known,3 and their applications in the total synthesis of complex natural products and pharmaceuticals have been demonstrated.4 The reaction usually proceeds initially through the oxidative addition of a metal to the Ar–X bond. Following transmetallation and reductive elimination, carbonylation or other transformations would result in the formation of new C–C and C–heteroatom bonds. Although the transition-metal-catalyzed cross-coupling reactions of aryl halides are efficient and powerful in organic synthesis, in some cases the transformations suffered from harsh reaction conditions and high temperature. Additionally, the disadvantages of this metal-catalyzed coupling approach in the drug discovery process are distinct, since it cannot avoid the contamination of metal salts in the final products.
So far, it is well recognized that the photoinduced Ar–X bond dissociation would generate the corresponding aryl radicals via electron transfer during the reaction process.5 For example, electron exchange was observed for the visible or long-wavelength ultraviolet excitation of alkali metal salts of cyclooctadiene with aryl halides, which afforded aryl radicals.5c Photoinduced reactions usually occur at room temperature. These mild conditions would broaden the applications of couplings of aryl halides in organic synthesis. Moreover, in some cases, the couplings could be performed under metal-free conditions since photocatalysis is not necessary. Usually, the broad reaction scope is demonstrated with good functional group tolerance. As mentioned above, this promising result would be attractive in the drug discovery process. Therefore, the photoinduced cross-coupling reactions of aryl halides as clean methods have been developed rapidly. This review will summarize the recent advances in the photochemical carbon–carbon and carbon–heteroatom bond formation via photoinduced Ar–X bond dissociation.
2. Mechanistic aspects
According to the reaction conditions in the presence or absence of a metal catalyst, two major mechanistic categories to deliver the aryl radicals from aryl halides via photoinduced Ar–X bond dissociation have been witnessed.5The cleavage of the Ar–X bond under UV-light irradiation would generate the aryl radical in the absence of a metal catalyst. The formation of an aryl radical through the mechanism of SRN1 is a photoinduced electron transfer from the anion of the nucleophile or the base to ArX, yielding an ArX radical anion. Subsequent fragmentation would result in the aryl radical, which would then couple with the nucleophile to provide the target molecules. As illustrated in Scheme 1, the aryl radical would be trapped by diverse nucleophiles to produce the desired products. The presence of a base such as potassium tert-butoxide would be necessary, which could trigger the reaction by photoinduced electron transfer to ArX.6
 | ||
| Scheme 1 Cleavage of Ar–X bond under UV irradiation. | ||
Another pathway to produce aryl radical could be achieved by a photoinduced redox process in the presence of a metal catalyst or organocatalyst. In the process, visible light would activate the catalyst to its excited state, which then would provide an electron to the aryl halide through single electron transfer (SET) to release an aryl radical. An example of haloarene–arene coupling is shown in Scheme 2. Compared with the UV irradiation as mentioned above, the excited state of the catalyst would act as the electron donor in the reaction.
 | ||
| Scheme 2 Formation of an aryl radical by a photoinduced redox process in the presence of a metal catalyst or organocatalyst. | ||
Based on these two major mechanistic categories, transformations through Ar–X bond dissociation including nucleophilic substitution, arylation, alkylation, aminocarbonylation, aminosulfonylation, and decarboxylative coupling reactions have been reported. Considering the convenience and clarity, this review is presented according to the types of reactions involving photoinduced Ar–X bond dissociation.
3. Photoinduced Ar–X bond dissociation reaction
3.1 Nucleophilic substitution
Nucleophilic substitution of aryl halides with various nucleophiles is an efficient route for the construction of a C–C bond and C–heteroatom bond.7 The nucleophiles including phenols, aryl thiols, N-heterocycles, and others have been employed successfully.The Ullmann reaction of amines with aryl halides promoted by a stoichiometric copper reagent at elevated temperature is a known method for the C–N bond formation. In the past century, many improved processes related to the formation of a C–N bond have emerged in organic synthesis. In 2012, Peter and Fu reported an exciting result, which discovered that the photoinduced Ullmann C–N coupling could proceed under unusually mild conditions by using a stoichiometric or a catalytic amount of copper salt (Scheme 3).8a A single-electron transfer mechanism was proposed, which was demonstrated by experimental support. They found that a photoinitiated step would promote the C–X cleavage to generate a radical intermediate. Although only C–N bond formation with carbazole was described, it could be expected that this promising and practical photoinduced process might be applied to the construction of other carbon–heteroatom bonds. In 2015, Kobayashi and co-workers also reported the Ullmann-type C–N cross-coupling reactions between carbazole derivatives and aryl iodides in the presence of an iridium-based photocatalyst and a copper salt under blue light emitting diode irradiation.8b
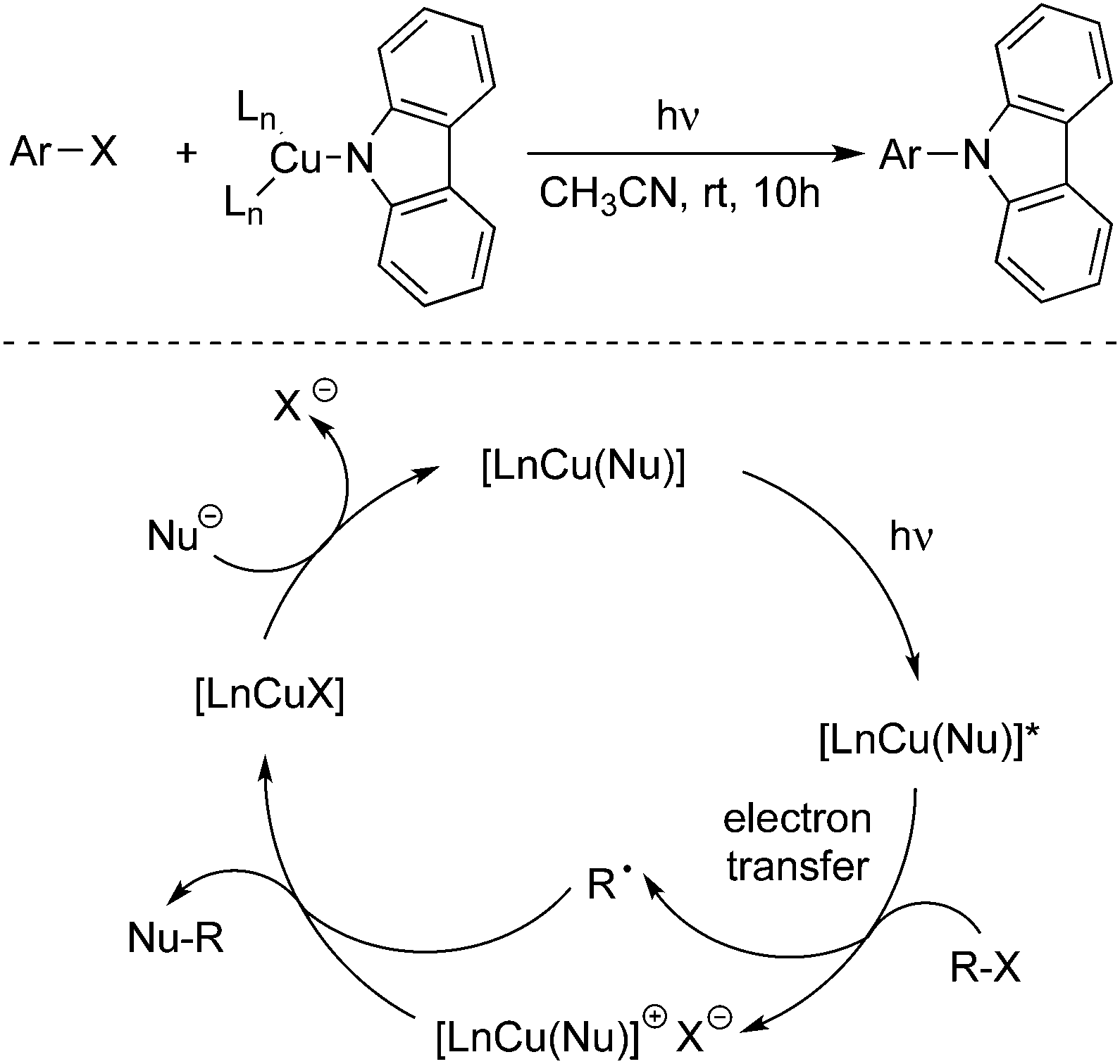 | ||
| Scheme 3 Photoinduced Ullmann C–N coupling. | ||
Peter and Fu further extended the above photoinduced copper-catalyzed cross-coupling reaction of nitrogen nucleophile to other N-heterocycles (Scheme 4).8c As expected, the C–N bond formation process occurred under mild conditions. It was found that the reactions of carbazoles with iodobenzene and alkyl halides worked well. Additionally, other nitrogen nucleophiles such as indoles, benzimidazoles, and imidazoles were all good coupling partners during the reactions. Moreover, not only aryl iodides but also aryl bromide and activated aryl chloride were suitable for the reactions. Ligands and additives were not necessary for this photoinduced C–N bond formation, and the transformation was performed in the presence of an inexpensive catalyst (CuI) at room temperature. The moisture could be tolerated and a variety of functional groups could be retained during the reaction process.
 | ||
| Scheme 4 Photoinduced copper-catalyzed cross-coupling reaction of nitrogen nucleophiles with aryl halides. | ||
In 2013, Peter and Fu described a photo-induced copper(I)-catalyzed method for the coupling of aryl thiols with aryl halides (Scheme 5).9a The same catalytic system – inexpensive copper(I) iodide – was used as a precatalyst without the addition of a ligand. This catalytic system was effective for a range of aryl thiols and aryl halides. Different functional groups including nitro, amino, cyano, pyridinyl and thiophenyl groups were all tolerated under the copper-catalyzed conditions. Usually, the non-photo-induced, copper-catalyzed processes proceeded through a concerted mechanism, and the reaction temperature was high. This photo-induced copper-catalyzed C–S cross-coupling reaction of an unactivated aryl iodide could even be performed at −40 °C. The mechanism was explored in detail, and the studies revealed that these photo-induced C–S cross-couplings occurred through a SET/radical pathway for C–X bond cleavage (via a Cu(I)–thiolate).9b
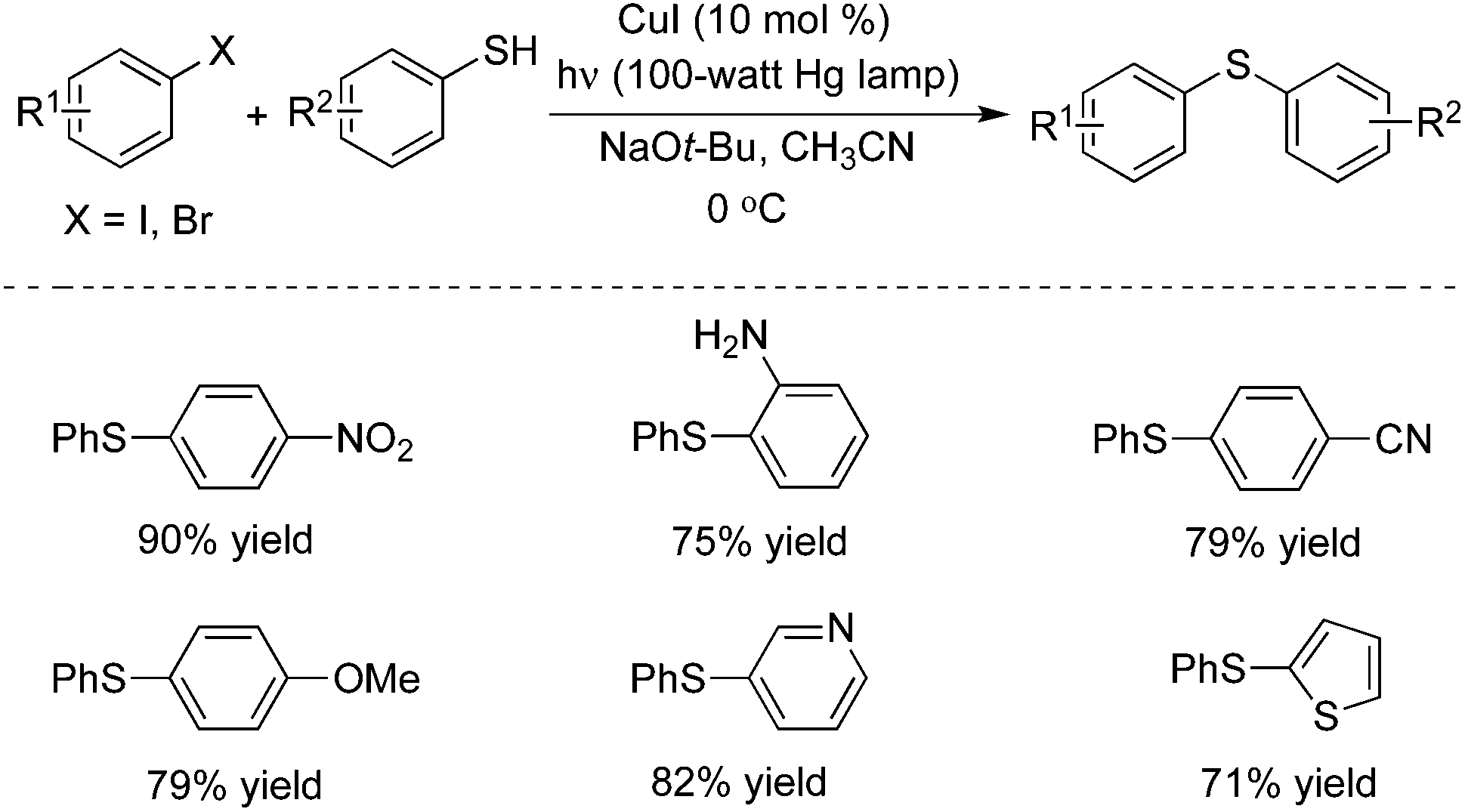 | ||
| Scheme 5 Photo-induced copper(I)-catalyzed coupling of aryl thiols with aryl halides. | ||
This powerful method could also be applied to the carbon–oxygen bond formation.10a Aryl iodides reacted with various phenols giving rise to the corresponding diaryl ethers in good yields under the photo-induced conditions (Scheme 6). In the presence of a copper pre-catalyst (CuI) and DBU (1,8-diazabicyclo[5.4.0]undec-7-ene), a range of phenols and aryl iodides could be coupled at room temperature, and different functional groups were tolerated. For example, the ester-substituted product was afforded in 61% yield, and 2-phenoxypyridine was obtained in 76% yield. It is know that most copper-catalyzed cross-couplings require an elevated reaction temperature. This method provides an efficient route to the diaryl ethers under mild conditions. A possible mechanism was proposed, which indicated that a Cu(I)–phenoxide complex would be involved as a viable intermediate during the photo-induced C–O bond formation process. MacMillan and co-workers described an efficient carbon–oxygen coupling reaction of alcohols and aryl bromides in the presence of photoredox and nickel catalysis.10b The mechanism was explored, which revealed that the preferred oxidation states of nickel alkoxides could be modulated by visible-light-excited photoredox catalysts in an operative catalytic cycle.
 | ||
| Scheme 6 Photo-induced copper(I)-catalyzed coupling of aryl iodides with phenols. | ||
Li and co-workers described the conversion of aryl, heteroaryl, and vinyl bromides (or chlorides) into the corresponding iodides under photo-promoted metal-catalyst-free conditions (Scheme 7).11 During the reaction process, reactive aryl radicals were generated under ultraviolet irradiation. The direct activation of carbon–halogen bonds was observed, and the transformation proceeded through electron transfer between halide species. The mild and metal-catalyst-free conditions were attractive, since the aryl halide exchange process usually proceeded in the presence of a ligand catalyzed by copper or nickel salt, which is called the aromatic Finkelstein reaction.12 The broad reaction scope was demonstrated, and the corresponding aryl iodides were produced in good to excellent yields at room temperature. Different sensitive groups such as hydroxyl, amino, and cyano groups were retained. It was found that the presence of a catalytic amount of elemental iodine was essential, which could promote the reaction significantly. This result was interesting, since the synthesis of functionalized aryl iodides by light was a great challenge. The transformation was previously considered impossible due to their photo-lability. Thus, this work provides a practical and green protocol to diverse aryl iodides.
 | ||
| Scheme 7 Photo-induced metal-catalyst-free aromatic Finkelstein reaction. | ||
4-Hydroxycoumarins were utilized as nucleophiles as well in the photoinduced reactions with aryl halides. In 2013, Baumgartner and co-workers described a photoinduced direct arylation of 4-hydroxycoumarins with aryl halides (including iodobenzene, iodonaphthalene, 4-iodoanisole and 2-iodoanisole) in the presence of potassium tert-butoxide and dimethyl sulfoxide (Scheme 8).13 This reaction worked well to afford the corresponding 3-aryl-4-hydroxycoumarins in good yields. The procedure could be extended to the reaction of 1,2-diiodobenzene with 4-hydroxycoumarin, leading to the formation of polycyclic 6H-benzofuro[3,2-c]chromen-6-one.
 | ||
| Scheme 8 A photoinduced direct arylation of 4-hydroxycoumarins with aryl halides. | ||
Rossi reported a photo-stimulated reaction of unactivated aromatic halides with nucleophiles derived from the naphthyl system (Scheme 9).14a 1- and 2-naphthylamide, 2-naphthoxide, 2-naphthalenethiolate, and 2-naphthaleneselenate ions were employed as the nucleophiles. The reaction underwent the SRN1 mechanism of nucleophilic substitution, similar to the above transformation. The results are shown below. A theoretical study was performed to illustrate the regiochemistry of the reaction. The scope of the above photo-stimulated reaction was extended to o-dihalobenzenes. 2-Naphthoxide ions, 2-naphthalenethiolate ions, and 2-naphthaleneselenolate ions were used as the nucleophiles. The substitution product on carbon 1 of the naphthyl moiety was obtained, which, in some cases, further acted as an intermediate to furnish the ring closure product.14b
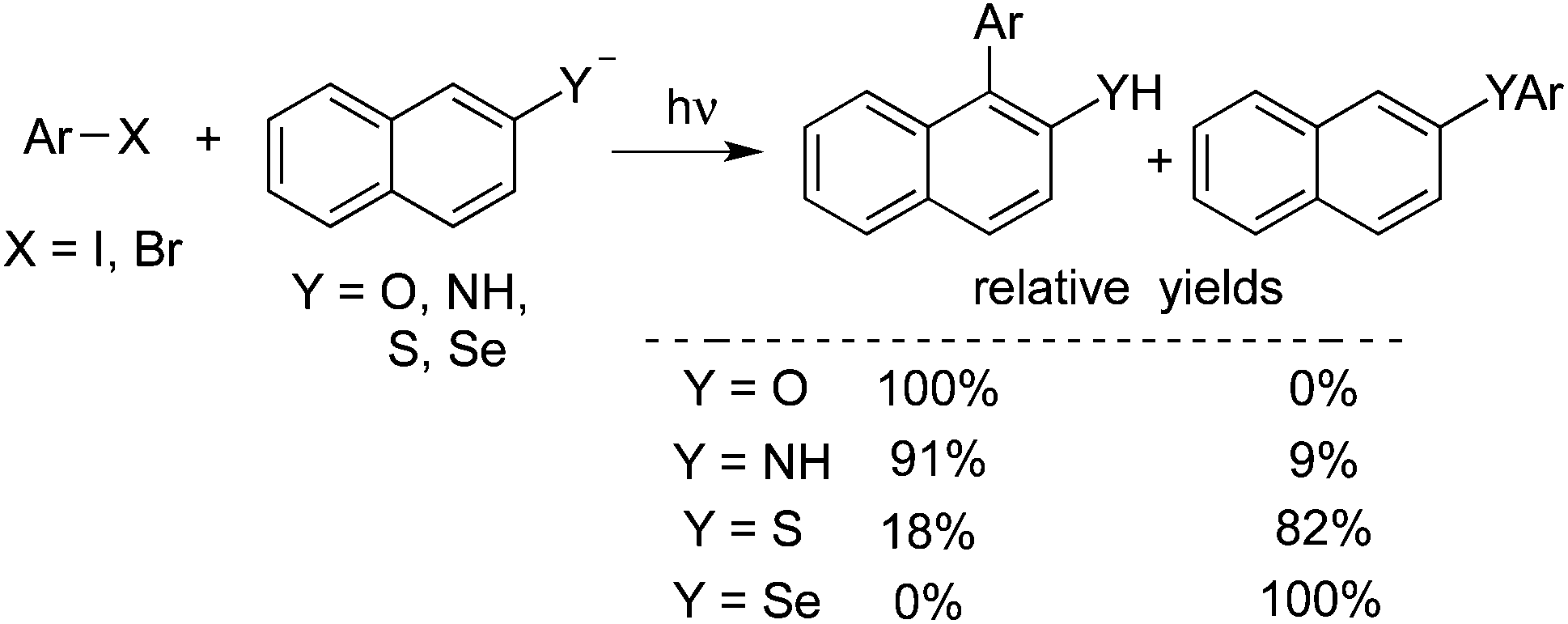 | ||
| Scheme 9 A photo-stimulated reaction of unactivated aromatic halides with nucleophiles derived from the naphthyl system. | ||
Peñéñory and co-workers found that arene thiolate ions could be generated through a photoinduced reaction of aryl halides with thiourea anions in DMSO.15 Following aliphatic nucleophilic substitution, SRN1 reaction, oxidation or protonation would produce aryl methyl sulfides, diaryl sulfides, diaryl disulfides, and aryl thiols. The result was interesting since the commercially available and inexpensive thiourea was utilized in a one-pot two-step process for the synthesis of aromatic sulfur compounds. Subsequently, potassium thioacetate was used as a replacement of thiourea. The photoinduced reactions of potassium thioacetate with aryl halides afforded aryl methyl sulfides and diaryl sulfides as well.16 The aromatic substitution would provide arene thiolates, which were then quenched with methyl iodide to produce aryl methyl sulfides in 26–59% yields. During the process, diaryl sulfides were obtained as well, although in low yields. The same group further extended the above method for the generation of arylselenide ethers under the photoinduced conditions (Scheme 10).17 During the transformation, selenobenzamide, selenourea, and selenocyanate anions were used as the nucleophiles, which reacted with aryl halides affording areneselenolate ions under ultraviolet irradiation in the presence of a base in DMSO. The in situ generated areneselenolate anions were then trapped by MeI, which would undergo an aliphatic nucleophilic substitution to produce aryl methyl selenides in good to excellent yields. Various aryl halides with electron-donating or electron-withdrawing substituents (including nitro and carbonyl groups) on the aromatic ring were all tolerated under the copper-catalyzed conditions. Competitive pathways were inevitable, which provided arenes and diarylselenide ethers. According to an electron-transfer mechanism, they also discovered that aryl radicals could be generated by a concerted or consecutive C–X bond fragmentation when the fluorescence excited state of a 2-naphthoxide ion was quenched by aromatic halides.18
 | ||
| Scheme 10 Photo-induced synthesis of arylselenide ethers. | ||
In the presence of a base, N-(2-halophenyl)indolo carboxamides would undergo an intramolecular O-arylation under irradiation. This transformation might take place through the indolyl radical and the aryl halide radical anion via photoinduced intramolecular electron transfer (Scheme 11). The following pathways were all possible: path a would proceed through a fast dehalogenation to form a biradical, which would then undergo a radical–radical collapse to produce the O-arylation product.19a In path b, an intramolecular SN(ET)Ar* reaction would occur, followed by dehalogenation to afford the final product. Additionally, the photostimulated C–O cyclization of anions was applied for the synthesis of 6-substituted 2-pyrrolyl and 2-indolyl benzoxazoles starting from 2-pyrrole carboxamides, 2-indole carboxamides, and 3-indole carboxamides.19b The corresponding products were afforded in moderate to good yields (41–100%) in DMSO and liquid ammonia. A theoretical analysis using DFT methods and the B3LYP functional was performed, which could explain the regiochemical outcome of these reactions. Photoreaction of N-(o-halophenyl)acetamide in basic acetonitrile could also proceed through a similar process. The intramolecular substituted product 2-methylbenzoxazole would be obtained as expected.20
 | ||
| Scheme 11 Intramolecular O-arylation of N-(2-halophenyl)indolo carboxamides under irradiation. | ||
3.2 Arylation
The photolysis of 4-chloro-N,N-dimethylaniline with ary halide, furan, pyrrole, or thiophene in acetonitrile would afford 2-(4-N,N-dimethylaminophenyl) heterocycles.21a,b The in situ formed triplet aryl cation was responsible for the regio- and chemo-selectivity of the process. Albini and co-workers further described the synthesis of the sterically crowded biphenyl compounds including tetra-ortho-substituted biphenyls via a chemoselective activation of Ar–H bonds in methylbenzenes enabled by photoenergy (Scheme 12).21c 2,2,2-Trifluoroethanol (TFE) was employed as the solvent in the reactions. This metal-free process is appealing, compared with other methods using transition-metal catalysis. | ||
| Scheme 12 Synthesis of the sterically crowded biphenyl compounds via a chemoselective activation of Ar–H bonds in methylbenzenes enabled by photoenergy. | ||
The synthesis of biaryls could be achieved as well by the photo-induced direct C–H arylation of benzene and thiophene in the presence of t-BuOK (Scheme 13).22 The broad reaction scope was demonstrated for this photo- and base-promoted homolytic aromatic substitution. Aryl iodides with different substituents including electron-donating and electron-withdrawing groups coupled with arenes efficiently. Heteroaryl halides, aryl bromides and chlorides were all good partners during the transformation.
 | ||
| Scheme 13 Synthesis of biaryls by photo-induced direct C–H arylation of benzene and thiophene in the presence of t-BuOK. | ||
Under visible-light irradiation, a direct arylation of unactivated arenes or heteroarenes with aryl halides catalysed by Ir(bpy)3 (tri(2-phenylpyridine)iridium(III)) was described in the presence of potassium tert-butoxide and dimethyl sulfoxide (Scheme 14).23 During the reaction process, Ir(ppy)3 was crucial as an effective photoredox catalyst in the transformation. Although the coupling reactions of aryl iodides with arenes could be performed at room temperature, elevated temperature was required for the couplings of aryl bromides. Aryl chlorides were not good reactive partners under the conditions. A possible reaction pathway was reported, which involved the homolytic aromatic substitution.
 | ||
| Scheme 14 Synthesis of biaryls through an Ir-catalyzed photoinduced reaction of aryl halides with arenes. | ||
Direct arylation of sesamol with aryl halides by a photoinduced reaction could be easily achieved. Several 6-arylsesamol derivatives were synthesized, with an expectation to evaluate the possible changes in their antioxidant properties (Scheme 15).24 A ring-closure product bearing a tetracyclic aromatic condensed ring system was obtained when the reaction was extended to o-dihalobenzenes. The antioxidant activity of these compounds towards a 1,1-diphenyl-2-picrylhydrazyl radical was evaluated by using sesamol as the reference compound.
 | ||
| Scheme 15 Direct arylation of sesamol with aryl halides by a photoinduced reaction. | ||
Guo and co-workers also reported the preparation of biaryls through a direct aromatic C–H functionalization of unactivated arenes with aryl iodides under metal-free process conditions (Scheme 16).25 In the reaction, 20 mol% of TMEDA (tetramethylethylenediamine) was used as the catalyst, and the transformation proceeded at room temperature leading to the corresponding products in good yields. The sensitive groups could be tolerated under the copper-catalyzed conditions. For instance, [1,1′-biphenyl]-4-ol was obtained in 76%, and 4-nitro-1,1′-biphenyl was produced in 91% yield. A possible mechanism was proposed, which involved the amine-catalyzed single electron transfer initiated radical coupling under UV irradiation.
 | ||
| Scheme 16 Preparation of biaryls through a direct aromatic C–H functionalization of unactivated arenes with aryl iodides. | ||
In 2014 König and co-workers described the reduction of stable aryl chlorides by visible light in the presence of perylene diimides (PDI).26a The in situ generated aryl radicals were trapped by hydrogen atom donors to provide the corresponding arenes. Since the aryl radical was the key intermediate, therefore, N-heterocyclic pyrroles were selected as the reaction partner. The C–C coupling products were obtained by irradiating aryl halides with N-methylpyrrole in the presence of PDI. The reaction scope could be extended to other pyrrole derivatives, and the desired arylated products were generated in good to excellent yields. König also reported the photoinduced C–C bond-forming arylation reactions via aryl radical intermediates. They further developed a metal-free process for the perfluoroarylation by using visible light photoredox catalysis (Scheme 17).26b The direct arylation of simple arenes with fluorinated aryl bromides worked well under visible light catalyzed by eosin Y by a photoredox process. The broad reaction scope was demonstrated in fluorinated compounds and arenes. The synthetically valuable polyfluorinated biaryl structures could be constructed efficiently under mild reaction conditions. Mechanistic studies were performed, which revealed that the reaction proceeded through the polyfluorinated aryl radical and a bromide anion. The polyfluorinated aryl radical was generated from the photoreduction of eosin Y via its triplet state by triethylamine and subsequent electron transfer from the eosin Y radical anion to the polyfluorinated bromoarene.
 | ||
| Scheme 17 Perfluoroarylation under visible light photoredox catalysis. | ||
In 2015, Su and co-workers described two general protocols for the photoinduced metal-free direct arylation of unactivated arenes with aryl iodides (Scheme 18).27a For most substrates, both methods gave rise to the corresponding products in good to excellent yields. It was noteworthy that only aryl iodides were reactive during the transformation, and the C–F, C–Cl, and C–Br bonds could be retained under the arylation conditions. As illustrated in Scheme 18, the excited state of aryl iodide accepted an electron to produce an aryl radical and halo anion. However, according to the findings of Murphy and co-workers,27b–e there was another possible way to account for the formation of biaryl compounds through metal-free haloarene–arene coupling reactions. The presence of potassium tert-butoxide would afford a potent electron donor, which facilitated the coupling of halobenzenes with arenes.
 | ||
| Scheme 18 Photoinduced metal-free direct arylation of unactivated arenes with aryl halides. | ||
Recently, Goddard and co-workers described the generation of hetero-biaryl derivatives through pyrimidinyl and pyrazinyl radicals under moderate energetic irradiation conditions (Scheme 19).28 This C–C bond formation process is highly efficient, which afforded the corresponding products in good to excellent yields. The highly interesting functional group selectivities were observed. Only pyrimidinyl and pyrazinyl bromide was reactive during the reactions. The reactions were inert when pyrimidinyl chlorides or bromides were used as substrates. In the meantime, the experimental results supported the proposed radical process.
 | ||
| Scheme 19 Generation of hetero-biaryl derivatives through pyrimidinyl and pyrazinyl radicals. | ||
An intramolecular photochemical cross-coupling reaction of 3-acyl-2-halo-N-(ω-arylalkyl)indoles with 2-chloro-N-(ω-arylalkyl)pyrrole-3-carbaldehydes in acetone was developed, which afforded the indolo[2,1-a]isoquinolines, indolo[2,1-a][2]benzazepines, pyrrolo[2,1-a]isoquinolines, and pyrrolo[1,2-a]benzazepines in good yields (Scheme 20).29 When 2-chloro-N-(2-phenoxyethyl)pyrrole-3-carbaldehyde was used as the substrate in this photo-cyclization reaction, pyrrolo[1,2-d][1,4]benzoxazepine was constructed under the conditions. This photo-induced reaction proceeded through the C–X bond dissociation to produce the corresponding radical species, which then reacted with the arene leading to the final products.
 | ||
| Scheme 20 Photochemical reaction of 3-acyl-2-halo-N-(ω-arylalkyl)indoles with 2-chloro-N-(ω-arylalkyl)pyrrole-3-carbaldehydes. | ||
Very recently, photoinduced room-temperature azole C–H arylations were accomplished as well by Ackermann and co-workers (Scheme 21).30 The inexpensive copper(I) iodide was used as the catalyst, which showed high efficiency for the direct arylation of various aromatic and non-aromatic heterocycles. The ample reaction scope was explored, and the C–H arylation proceeded effectively on substrates being devoid of directing groups. Moreover, this approach was successfully applied for the step-economical access to the alkaloid natural products, balsoxin and texamine.
 | ||
| Scheme 21 The copper(I)-catalyzed photoinduced room-temperature azole C–H arylations. | ||
Induced by strongly basic conditions, the phenanthridines could be generated from the photolysis of N-(o-iodo-benzyl)arylamines in liquid ammonia (or DMSO) and potassium tert-butoxide, as reported by Pierini and Rossi.31 A plausible mechanism involving a SRN1 single electron transfer process was described. Bräse developed an improved photochemical method for the synthesis of phenanthridines in two steps starting from anilines with 2-iodobenzyl bromides under neutral conditions.32a They further accomplished the synthesis of fluoro and catechol-protected substituted phenanthridine derivatives through a photochemical cyclization of N-benzylanilines.32b Interestingly, poor yields of cyclized products were obtained when the iodine atom was replaced by a bromine atom (Scheme 22). This photochemical approach was used effectively for the total synthesis of a natural product, trispheridine. They hypothesized that an aryl radical derived from the dissociation of the Ar–X bond would attack the phenyl group through 6-endo radical cyclization. Then deprotonation and oxidation would produce the desired phenanthridines. However, another possibility of the reaction pathway could not be excluded. The intermediate of the radical generated by SET would afford an aryl cation, which was then readily deprotonated with the formation of phenanthridines.
 | ||
| Scheme 22 A photochemical method for the synthesis of phenanthridines. | ||
The photochemical arylation and alkylation of 6,6′-dibromo-BINOL (BINOL = 2,2′-dihydroxy-1,1′-binaphthyl) under mild basic conditions was achieved when allyltrimethylsilane, ethyl vinyl ether, pyrrole, pyridine, thiophene, benzene, and indole were used as the coupling partners (Scheme 23).33 The BINOL chirality is preserved in the photoactivation process. This transformation was interesting, which proceeded under metal and protecting group free conditions leading to mono- and disubstituted 6-aryl/alkyl BINOLs.
 | ||
| Scheme 23 Photochemical arylation and alkylation of 6,6′-dibromo-BINOL. | ||
Biphenyl and phenanthrene could be obtained for the photolysis of iodoaromatic compounds.34a 3,4-Diarylthiophenes bearing a chlorine atom at the ortho-position of one of the aryl substituents proceeded via photocyclization in a mixed solvent of benzene and acetonitrile to afford the corresponding products in 60–82% yields.34b The position of a chlorine substituent on the aromatic ring determined the outcome of the photocyclization selectivity. The proposed mechanism indicated that the aryl radical would be the key intermediate during the photocyclization, and the conrotatory [4 + 2] process accompanied by elimination of HCl was excluded. Kim and co-workers described the photo-cyclization reaction of 2-halopyridinium salt tethered to an arene.34c A six-membered heterocyclic pyrido[2,1-a]-3H,4H-isoquinolinium salt could be obtained when 2-halophenethylpyridinium salts were used as the substrates under the photo-cyclization conditions. A photohomolytic radical mechanism was proposed for the cyclization, since the transient radical intermediates were detected with the laser flash photolysis facility. The studies revealed that the 2-halopyridinium salts tethered to a phenyl group with an ethylene linkage were less reactive than the 2-halopyridinium salts with a methylene group. The authors postulated that the excited singlet state might be involved in the reaction process of 2-halopyridinium salts tethered to the phenyl group with an ethylene linkage. For the photo-cyclization of the 2-halopyridinium salts with a methylene linkage, both the singlet and triplet states were involved. The regioselective arylation of azulene at the electron rich 1-position via photoirradiation of aryl iodide with azulene provided a facile route to diverse azulenes.34d Additionally, biaryl compounds were also produced by cyanoboronhydride in air and under photoirradiation conditions using a Xe lamp.34e
3.3 Borylation
Very recently, a metal- and additive-free, photo-induced borylation of haloarenes, including electron-rich fluoroarenes, was reported for the direct synthesis of arylboronic acids (Scheme 24).35 This borylation approach demonstrated a broad scope and functional group tolerance. Additionally, this method could be further extended to boronic esters and carried out on the gram scale as well as under flow conditions. Moreover, quaternary arylammonium salts could be used as the substrates as well under the standard conditions, as a replacement for haloarenes. This approach provides a simple and efficient route for the generation of arylboronic acids since the borylation produces arylboronic acids and esters in good to excellent yields under mild conditions. And also, the innocuous byproducts could be easily removed. A possible mechanism was proposed, which might proceed through homolytic substitution in the diboron reagents and sp2–sp3 Lewis base–diboron adducts by a photo-generated aryl radical and aryl triplet cation. | ||
| Scheme 24 Photo-induced borylation of haloarenes. | ||
3.4 Reactions with alkenes
Albini and co-workers reported that triplet phenyl cations could be generated via irradiation of 4-chloroaniline or its N,N-dimethyl derivative in polar solvents.36 When the cations were trapped by alkenes, the corresponding arylated products could be obtained in moderate to good yields (Scheme 25).36a B3LYP calculations were provided for illustration of the reaction process. Different coupling partners could be employed to afford interesting products. For example, 4-chloro-N,N-dimethylaniline reacted with 4-pentenoic acid via intramolecular nucleophilic trapping producing the expected lactam in 70% yield. | ||
| Scheme 25 Irradiation of 4-chloroaniline or its N,N-dimethyl derivative. | ||
The same group further reported the irradiation of N,N-dimethyl-4-chloroaniline in the presence of open-chain dienes in acetonitrile, giving rise to the addition of aminophenyl and chloro groups across one of the double bonds. When cyclic dienes were employed instead, a transannular cyclization occurred to form arylnortricyclene from norbornadiene and 1-arylbicyclo[3.3.0]octanes from 1,5-cyclooctadiene (Scheme 26).37a The mechanistic studies indicated that photoheterolysis of the chloroaniline was involved, yielding a 4-aminophenyl cation and its subsequent addition to a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond. It was found that the structure and medium determined the chemistry of the adduct cation (ion pairs in MeCN and solvated ions in CF3CH2OH). Photogeneration and reactivity of a 4-hydroxy(methoxy)phenyl cation with 2,3-dimethyl-2-butene, cyclohexene, and benzene were explored.37b It was found that 4-chlorophenol and 4-chloroanisole proceeded through a homolytic path in cyclohexane and a heterolytic path in alcohols. Triplet 4-hydroxy- and 4-methoxyphenyl cations were afforded via heterolysis of 4-chlorophenol and 4-chloroanisole in methanol and 2,2,2-trifluoroethanol. Then treatment of alkenes would give the arylated products in moderate to good yields.
C double bond. It was found that the structure and medium determined the chemistry of the adduct cation (ion pairs in MeCN and solvated ions in CF3CH2OH). Photogeneration and reactivity of a 4-hydroxy(methoxy)phenyl cation with 2,3-dimethyl-2-butene, cyclohexene, and benzene were explored.37b It was found that 4-chlorophenol and 4-chloroanisole proceeded through a homolytic path in cyclohexane and a heterolytic path in alcohols. Triplet 4-hydroxy- and 4-methoxyphenyl cations were afforded via heterolysis of 4-chlorophenol and 4-chloroanisole in methanol and 2,2,2-trifluoroethanol. Then treatment of alkenes would give the arylated products in moderate to good yields.
 | ||
| Scheme 26 Irradiation of N,N-dimethyl-4-chloroaniline with dienes. | ||
As mentioned above, photoheterolysis of electron-rich aromatic chlorides and fluorides would afford the phenyl cations, which would add to alkenes to form phenonium ions.38a,b Therefore, 3-butenoic, 4-pentenoic, and 5-hexenoic acids were employed in the photoinduced reactions of electron-donating substituted phenyl halides (Scheme 27).38c As expected, the electrophilic addition of a phenyl cation onto alkenoic acid occurred efficiently, followed by an intramolecular attack by the carboxylic group. This tandem process provided a one-step synthesis of various lactams. As shown in Scheme 25, the sensitive groups such as hydroxyl and amino groups were all tolerated under the copper-catalyzed conditions. Based on this approach, they further developed a three-component reaction of aromatic halides, alcohols, and vinyl ethers under photoinduced conditions, which provided an efficient route for the synthesis of 3-arylacetals and ketals.39
 | ||
| Scheme 27 Synthesis of lactams via photoinduced reactions. | ||
The photochemistry of 2,6-dimethyl-4-chlorophenol was further explored in methanol and trifluoroethanol by Albini and co-workers.40 Through product studies and transient absorption spectroscopy, it was found that a triplet hydroxyphenyl cation could be formed, which was subsequently trapped by allyltrimethylsilane to produce a phenonium ion and the allylated phenol. The mechanism was investigated in detail, and the photolysis of electron-donating substituted halobenzenes seemed to be promising for the mild generation of some classes of phenyl cations.
In 2014, Rossi and co-workers reported the synthesis of E-stilbenes through a photoinduced Heck-type reaction of aryl halides with alkenes (Scheme 28).41 This transformation proceeded at room temperature without the addition of transition metals. A range of aryl halides including ArI, ArBr, and ArCl is suitable as substrates. Additionally, the reaction is highly efficient in the presence of 18-crown-6 ether and t-BuOK without extra solvent, and attains completion in 15 min. A possible mechanism was proposed as well, which involved the formation of an aryl radical and its subsequent addition to an alkene.
 | ||
| Scheme 28 A photoinduced reaction of aryl halides with alkenes. | ||
The core of naphtho[b]furan can be found in many natural products and synthetic compounds. The conventional methods for access to this scaffold usually proceeded through cyclization catalysed by palladium or copper salts. In 2014, Yoshimi and co-workers described a simple approach for the synthesis of naphtho[b]furans from the photochemical reactions of the corresponding 1-bromo-2-naphthyl and 2-bromo-1-naphthyl allyl ethers (Scheme 29).42a A plausible mechanism was proposed for this photoreaction. As shown in Scheme 27, the homolytic C–Br bond cleavage in the triplet states of the substrates would occur to produce radical pairs. The aryl radical would go through 5-exo-cyclization and halogen atom capture to afford 2-halomethyl substituted naphthodihydrofuran. Subsequently, the photoinduced dehydrohalogenation of 2-halomethyl substituted naphthodihydrofuran provided an alkylidene dihydrofuran intermediate, which would then undergo tautomerization to generate naphtho[b]furan. 3-Methyl-2,3-dihydrobenzofurans could be obtained as well through a photoinduced electron transfer promoted reductive radical cyclization reaction of allyl 2-bromoaryl ethers.42b Another intramolecular example involved the radical cyclization of iodoaryl allyl azides with CO for the synthesis of spirocyclic indol-γ-lactams. The reaction required the use of Bu3SnH, AIBN (2,2′-azobisisobutyronitrile), and high pressure of carbon monoxide (80 atm).42c
 | ||
| Scheme 29 Synthesis of naphtho[b]furans via photochemical reactions. | ||
A similar result was observed by Rossi and co-workers when 1-allyloxy-2-bromobenzene or 1-allyloxy-2-chlorobenzene was used as the substrate under the photostimulated conditions. The desired 3-methyl-2,3-dihydro-benzofuran could be generated in high yield. The reactions of diallyl-(2-bromophenyl)amine and 2-allyloxy-1-halonaphthalenes worked efficiently as well, giving rise to the expected cyclized reduced products.43a They further discovered that several hydrogen donors could be utilized for reductive 6-exo cyclization in liquid ammonia, and also could be used for hydrodehalogenation reactions of aryl halides in DMSO.43b
Azole is a privileged scaffold in many natural products and pharmaceuticals. Weaver and co-workers reported a facile and efficient approach for access to a range of azoles through a photocatalytic reductive coupling of 2-bromoazoles with unactivated alkenes (Scheme 30). These Csp2–Csp3-coupled products are obtained in moderate to good yields.44 The mechanistic studies showed that the reaction occurred via photoinduced electron transfer from a tertiary amine to an aryl bromide. The in situ formed azole radical would subsequently react with an alkene with the formation of a C–C bond. Interestingly, the amine presented in the reaction system also acted as the final reductant. The broad reaction scope was demonstrated with good functional group tolerance. This method was successfully applied for the generation of complex molecules.
 | ||
| Scheme 30 Synthesis of azoles through a photocatalytic reductive coupling of 2-bromoazoles with unactivated alkenes. | ||
The visible light-induced reductive cyclization and hydrodehalogenation of aryl halides could be achieved under iridium catalysis. A broad range of aryl halides participated in the photocatalytic free-radical processes efficiently and furnished the products in excellent yield. Significant rate acceleration was observed when the light source was changed.45 In 2015, Jamison and co-workers applied the nickel/photoredox catalysis for the synthesis of indolines through a reaction of iodoacetanilides with alkenes. Couplings of both aliphatic and styrenyl olefins afforded 3-substituted indolines with high regioselectivity. In the reaction process, the photoredox catalyst was used as a controlled single electron transfer agent in multi-oxidation state nickel catalysis.46
3.5 Reactions with alkynes
Hwang and co-workers demonstrated that copper(I) chloride (CuCl) could catalyze the Sonogashira couplings of aryl halides with alkynes under blue LED light irradiation at room temperature (Scheme 31).47 This visible light-mediated Sonogashira cross-coupling reaction afforded the corresponding products in high yields. A range of aryl bromides/iodides and terminal alkynes are good coupling partners during the reaction process. However, the transformation worked poorly for aryl chlorides. The possibility of Pd contamination was ruled out with a control experiment. The mechanistic studies revealed that copper acetylide was the key light absorbing species, and played an important role in the photoinduced Sonogashira coupling reactions. The palladium-catalyzed Sonogashira coupling using a Ru/bipyridine complex as the energy transfer agent under irradiation of visible light was developed as well.48 In this catalytic system, the Ru/bipyridine complex was used as a replacement for the copper catalyst. | ||
| Scheme 31 Sonogashira couplings of aryl halides with alkynes under blue LED light irradiation. | ||
Albini and co-workers also developed a metal-free protocol for the mild alkynylation of aromatic compounds substituted with electron-donating groups (Scheme 32).49 The readily available aryl fluorides, aryl chlorides, aryl esters, aryl mesylates, and aryl phosphates were used as the substrates under photoheterolysis. The reaction proceeded through a phenyl cation, and the coupling products were obtained in good yields at room temperature. It was noteworthy that reactions of aryl fluorides worked well under the conditions, although the consumption of the starting material was incomplete. It is known that aryl fluorides are unreactive in the palladium-catalyzed Sonogashira reactions. This approach showed interesting results for the application of aryl fluorides.
 | ||
| Scheme 32 Alkynylation of aromatic compounds substituted with electron-donating groups under photoheterolysis. | ||
3.6 Insertion of carbon monoxide or sulfur dioxide
In 2015, Ryu and co-workers reported a catalyst-free aminocarbonylation of aryl iodides, in the presence of CO, with amines under photo-irradiation conditions by using a Xe-lamp (Scheme 33).50 This transition metal-free aminocarbonylation of aryl halides provided a good alternative to aromatic amides. During the reaction process, broad functional group tolerance was observed. Heteroaromatic amides could be generated as well under the photoinduced conditions. A hybrid radical/ionic chain process was proposed, which involved the electron transfer from a zwitterionic radical intermediate generated by nucleophilic attack of amines to an aroyl radical. However, this carbonylation required the use of relatively high pressure of carbon monoxide (70 atm). | ||
| Scheme 33 A catalyst-free aminocarbonylation of aryl iodides, in the presence of CO, with amines. | ||
In 2016, Wu and co-workers developed a photoinduced catalyst-free three-component reaction of aryl/alkyl halides, sulfur dioxide, and hydrazines (Scheme 34).51 This aminosulfonylation process through the insertion of sulfur dioxide under ultraviolet irradiation, proceeded at room temperature without any metals or photo-redox catalysts, leading to the corresponding N-aminosulfonamides in good to excellent yields. Good functional group tolerance was observed with the broad reaction scope. During the transformation, aryl iodides, bromide, and chlorides were all good reactive partners. The theoretical calculations for the possible mechanism were performed, which supported the radical process (DABCO: 1,4-diazabicyclo[2.2.2]octane;triethylenediamine; TBAI: tetra-n-butylammonium iodide).
 | ||
| Scheme 34 A photoinduced catalyst-free three-component reaction of aryl halides, sulfur dioxide, and hydrazines. | ||
3.7 Decarboxylative coupling
In 2013, MacMillan and co-workers developed a direct decarboxylative sp3–sp2 cross-coupling reaction of amino acids, as well as α-O- or phenyl-substituted carboxylic acids, with aryl halides in the presence of a combination of photoredox catalysis and nickel catalysis. Additionally, the cross-coupling reaction of Csp3–H in dimethylaniline with aryl halides via C–H functionalization was achieved in the meantime by using this photoredox-metal catalysis approach.52 In 2015, they further extended the visible-light-mediated photoredox and nickel catalysis to the direct decarboxylative arylation of α-oxo acids with aryl halides.53 The aryl and alkyl ketones could be easily obtained from α-oxo acid precursors via an acyl radical intermediate. This mild decarboxylative arylation was applied efficiently for the rapid synthesis of fenofibrate. The chiral benzylic amines in high enantiomeric excess could be obtained through an asymmetric decarboxylative Csp3–Csp2 cross-coupling reaction of α-amino acids with aryl halides in the presence of photoredox and nickel catalysts as well (Scheme 35).54 | ||
| Scheme 35 Photoinduced decarboxylation coupling reaction. | ||
3.8 Reduction of aryl halides
Unactivated aryl iodides catalyzed by fac-Ir(ppy)3 under visible light would generate the corresponding aryl radicals, which then undergo hydrogen atom abstraction or reductive cyclization (Scheme 36).55 The reaction occurred under mild reaction conditions, and exceptional functional group tolerance was displayed with high efficiency. It was noteworthy that the reaction time could be significantly shortened when the reaction took place in a flow reactor. | ||
| Scheme 36 Hydrogen atom abstraction of aryl iodides. | ||
In 2014 König and co-workers described the reduction of stable aryl chlorides by visible light in the presence of perylene diimides (PDI) (Scheme 37).26a The in situ generated aryl radicals were trapped by hydrogen atom donors to provide the corresponding arenes. This consecutive photoinduced electron transfer overcomes the energetic limitation of visible light photoredox catalysis. The experiments indicated that the excited PDI* would be reductively quenched by triethylamine, leading to PDI˙– and the radical cation of triethylamine (Et3N˙+). Upon the second excitation, the substrate would be reduced by PDI˙−*, giving rise to the aryl radical precursor (ArX˙−) with the formation of the neutral PDI. Subsequently, the aryl radical would abstract a hydrogen atom from either Et3N˙+ or the solvent to produce the final product.
 | ||
| Scheme 37 The reduction of aryl halides by visible light in the presence of perylene diimides (PDI). | ||
3.9 1,5-H Transfer reaction of o-anilide aryl iodides
Very recently, an intramolecular 1,5-H transfer reaction of o-anilide aryl iodides was developed via a visible-light photoredox catalysis approach, as reported by Xu and Liang (Scheme 38).56 This transformation proceeded under mild reaction conditions, and exceptional functional group tolerance was observed with good to excellent yields. The practicability of this method was demonstrated in the meantime for the concise synthesis of (±)-coerulescine and (±)-physovenine. | ||
| Scheme 38 Intramolecular 1,5-H transfer reaction of o-anilide aryl iodides. | ||
4. Conclusions
In summary, we have described the recent developments in photochemical carbon–carbon and carbon–heteroatom bond formation via photoinduced Ar–X bond dissociation. The transformations through the Ar–X bond dissociation enabled by photoenergy include nucleophilic substitution, arylation, alkylation, aminocarbonylation, aminosulfonylation and decarboxylative coupling reactions. Usually, the reactions undergo one-electron-transfer couplings and aryl radicals or cations are involved as the key intermediates. Under ultraviolet irradiation or visible light, the reactions of aryl halides with reactive partners proceed smoothly under mild conditions. In some cases, the transformations can proceed without any metals or photo-redox catalysts. Moreover, the broad reaction scope is demonstrated with good functional group tolerance. These findings establish the feasibility of using easily available aryl halides through a convenient and environmentally benign photochemical route. The extremely mild reaction conditions combined with experimental ease will make the photo-induced Ar–X bond dissociation attractive for further applications.Acknowledgements
Financial support from the National Natural Science Foundation of China (no. 21372046, 21532001 and 21502069) is gratefully acknowledged. We thank Prof. Tianning Diao (New York University) for English review.Notes and references
- For reviews, see: (a) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef CAS PubMed; (b) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed; (c) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (d) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef CAS PubMed; (e) L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687 RSC; (f) Y.-M. Xi, H. Yi and A.-W. Lei, Org. Biomol. Chem., 2013, 11, 2387 RSC; (g) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (h) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 985 CrossRef CAS PubMed; (i) M. N. Hopkinson, B. Sahoo, J.-L. Li and F. Glorius, Chem. – Eur. J., 2014, 20, 3874 CrossRef CAS PubMed; (j) J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632 CrossRef CAS PubMed; (k) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Chem. Soc. Rev., 2016, 45, 2044 RSC; (l) S. Protti, M. Fagnoni and D. Ravelli, ChemCatChem, 2015, 7, 1516 CrossRef CAS; (m) M. D. Karkas, B. S. Matsuura and C. R. J. Stephenson, Science, 2015, 349, 1285 CrossRef CAS PubMed; (n) R. Brimioulle, D. Lenhart, M. M. Maturi and T. Bach, Angew. Chem., Int. Ed., 2015, 54, 3872 CrossRef CAS PubMed; (o) D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97 RSC; (p) T. P. Yoon, ACS Catal., 2013, 3, 895 CrossRef CAS PubMed; (q) M. A. Ischay and T. P. Yoon, Eur. J. Org. Chem., 2012, 3359 CrossRef CAS; (r) N. Hoffmann, ChemSusChem, 2012, 5, 352 CrossRef CAS PubMed; (s) J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617 CrossRef CAS PubMed.
- For selected examples, see: (a) J. A. Terrett, J. D. Cuthbertson, V. W. Shurtleff and D. W. C. MacMillan, Nature, 2015, 524, 330 CrossRef CAS PubMed; (b) C. C. Nawrat, C. R. Jamison, Y. Slutskyy, D. W. C. MacMillan and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 11270 CrossRef CAS PubMed; (c) J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87 CrossRef CAS PubMed; (d) X.-Q. Hu, J.-R. Chen, Q. Wei, F.-L. Liu, Q.-H. Deng, A. M. Beauchemin and W.-J. Xiao, Angew. Chem., Int. Ed., 2014, 53, 12163 CrossRef CAS; (e) Y. Masuda, N. Ishida and M. Murakami, J. Am. Chem. Soc., 2015, 137, 14063 CrossRef CAS PubMed; (f) M. El Khatib, R. A. M. Serafim and G. A. Molander, Angew. Chem., Int. Ed., 2016, 55, 254 CrossRef CAS PubMed; (g) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433 CrossRef CAS PubMed; (h) H.-Q. Do, S. Bachman, A. C. Bissember, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 2162 CrossRef CAS PubMed; (i) A. C. Bissember, R. J. Lundgren, S. E. Creutz, J. C. Peters and G. C. Fu, Angew. Chem., Int. Ed., 2013, 52, 5129 CrossRef CAS PubMed.
- Transition Metal Reagents and Catalysts, ed. J. Tsuji, John Wiley & Sons, Ltd, 2000 Search PubMed.
- (a) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed; (b) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4490 CrossRef CAS PubMed.
- (a) A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef CAS PubMed; (b) A. Studer and D. P. Curran, Nat. Chem., 2014, 6, 765 CrossRef CAS PubMed; (c) M. A. Fox and N. J. Singletary, J. Org. Chem., 1982, 47, 3413 Search PubMed.
- For selected examples, see: (a) L. C. Schmidt, J. E. Argüello and A. B. Peñéñory, J. Org. Chem., 2007, 72, 2936 CrossRef CAS PubMed; (b) R. A. Rossi, A. B. Pierini and A. B. Peñéñory, Chem. Rev., 2003, 103, 71 CrossRef CAS PubMed; (c) J. I. Bardagí, V. A. Vaillard and R. A. Rossi, in Encyclopedia of Radicals in Chemistry, Biology & Materials, ed. C. Chatgilialoglu and A. Studer, John Wiley & Sons Ltd, Chichester, UK, 2012, pp. 333–364 Search PubMed.
- (a) J. F. Bunnett, Acc. Chem. Res., 1978, 11, 413 CrossRef CAS; (b) C. Karapire and I. Siddik, in CRC Handbook of Organic Photochemistry and Photobiology, ed. W. M. Horspool and F. Lenci, CRC Press, Boca Raton, 2nd edn, 2004 Search PubMed.
- (a) S. E. Creutz, K. J. Lotito, G. C. Fu and J. C. Peters, Science, 2012, 338, 647 CrossRef CAS PubMed; (b) W.-J. Yoo, T. Tsukamoto and S. Kobayashi, Org. Lett., 2015, 17, 3640 CrossRef CAS PubMed; (c) D. T. Ziegler, J. Choi, J. M. Muñoz-Molina, A. C. Bissember, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2013, 135, 13107 CrossRef CAS PubMed.
- (a) C. Uyeda, Y. C. Tan, G. C. Fu and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 9548 CrossRef CAS PubMed; (b) M. W. Johnson, K. I. Hannoun, Y. Tan, G. C. Fu and J. C. Peters, Chem. Sci., 2016 10.1039/C5SC04709A.
- (a) Y. C. Tan, J. M. Munoz-Molina, G. C. Fu and J. C. Peters, Chem. Sci., 2014, 5, 2831 RSC; (b) J. A. Terrett, J. D. Cuthbertson, V. W. Shurtleff and D. W. C. MacMillan, Nature, 2015, 524, 330 CrossRef CAS PubMed.
- L. Li, W. Liu, H. Zeng, X. Mu, G. Cosa, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2015, 137, 8328 CrossRef CAS PubMed.
- (a) A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 14844 CrossRef CAS PubMed; (b) A. Casitas, M. Canta, M. Sola, M. Costas and X. Ribas, J. Am. Chem. Soc., 2011, 133, 19386 CrossRef CAS PubMed; (c) A. A. Cant, R. Bhalla, S. L. Pimlott and A. Sutherland, Chem. Commun., 2012, 48, 3993 RSC; (d) M. Chen, S. Ichikawa and S. L. Buchwald, Angew. Chem., Int. Ed., 2015, 54, 263 CrossRef CAS PubMed.
- S. A. Rodríguez and M. T. Baumgartner, Tetrahedron Lett., 2010, 51, 5322 CrossRef.
- (a) A. B. Pierini, M. T. Baumgartner and R. A. Rossi, J. Org. Chem., 1991, 56, 580 CrossRef CAS; (b) M. T. Baumgartner, A. B. Pierini and R. A. Rossi, J. Org. Chem., 1993, 58, 2593 CrossRef CAS.
- J. E. Argüello, L. C. Schmidt and A. B. Peñéñory, Org. Lett., 2003, 5, 4133 CrossRef PubMed.
- L. C. Schmidt, V. Rey and A. B. Peñéñory, Eur. J. Org. Chem., 2006, 2210 CrossRef CAS.
- L. M. Bouchet, A. B. Peñéñory and J. E. Argüello, Tetrahedron Lett., 2011, 52, 969 CrossRef CAS.
- J. E. Argüello and A. B. Peñéñory, J. Org. Chem., 2003, 68, 2362 CrossRef PubMed.
- (a) V. A. Vaillard, R. A. Rossi and J. E. Argüello, Org. Biomol. Chem., 2012, 10, 9255 RSC; (b) V. A. Vaillard, J. F. Guastavino, M. E. Budeń, J. I. Bardagí, S. M. Barolo and R. A. Rossi, J. Org. Chem., 2012, 77, 1507 CrossRef CAS PubMed.
- Y. Park, M. Song, M. Kim and J. Kwon, Bull. Korean Chem. Soc., 2002, 23, 9 CrossRef.
- (a) B. Guizzardi, M. Mella, M. Fagnoni and A. Albini, Tetrahedron, 2000, 56, 9383 CrossRef CAS; (b) M. Fagnoni, M. Mella and A. Albini, Org. Lett., 1999, 1, 1299 CrossRef CAS; (c) V. Dichiarante, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2007, 46, 6495 CrossRef CAS PubMed.
- M. E. Buden, J. F. Guastavino and R. A. Rossi, Org. Lett., 2013, 15, 1174 CrossRef CAS PubMed.
- Y. Cheng, X. Gu and P. Li, Org. Lett., 2013, 15, 2664 CrossRef CAS PubMed.
- S. A. Rodríguez, M. A. Nazareno and M. T. Baumgartner, Aust. J. Chem., 2013, 66, 1334 CrossRef.
- X. L. Zheng, L. Yang, W. Y. Du, A. S. Ding and H. Guo, Chem. – Asian J., 2014, 9, 439 CrossRef CAS PubMed.
- (a) I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725 CrossRef CAS PubMed; (b) A. U. Meyer, T. Slanina, C.-J. Yao and B. König, ACS Catal., 2016, 6, 369 CrossRef CAS.
- (a) J. Kan, S. Huang, H. Zhao, J. Lin and W. Su, Sci. China: Chem., 2015, 58, 8 Search PubMed; (b) S. O'Sullivan, E. Doni, T. Tuttle and J. A. Murphy, Angew. Chem., Int. Ed., 2014, 53, 474 CrossRef PubMed; (c) S. S. Hanson, E. Doni, K. T. Traboulsee, G. Coulthard, J. A. Murphy and C. A. Dyker, Angew. Chem., Int. Ed., 2015, 54, 11236 CrossRef CAS PubMed; (d) E. Cahard, F. Schoenebeck, J. Garnier, S. P. Y. Cutulic, S. Zhou and J. A. Murphy, Angew. Chem., Int. Ed., 2012, 51, 3673 CrossRef CAS PubMed; (e) J. P. Barham, G. Coulthard, R. G. Kane, N. Delgado, M. P. John and J. A. Murphy, Angew. Chem., Int. Ed., 2016, 55, 4492 CrossRef CAS PubMed.
- J. Ruch, A. Aubin, G. Erbland, A. Fortunato and J.-P. Goddard, Chem. Commun., 2016, 52, 2326 RSC.
- S. C. Lu, X. X. Zhang, Z. J. Shi, Y. W. Ren, B. Li and W. Zhang, Adv. Synth. Catal., 2009, 351, 2839 CrossRef CAS.
- F. Yang, J. Koeller and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 4759 CrossRef CAS PubMed.
- (a) M. E. Buden, V. B. Dorn, M. Gamba, A. B. Pierini and R. A. Rossi, J. Org. Chem., 2010, 75, 2206 CrossRef CAS PubMed; (b) J. K. Laha, S. M. Barolo, R. A. Rossi and G. D. Cuny, J. Org. Chem., 2011, 76, 6421 CrossRef CAS PubMed; (c) S. M. Barolo, X. Teng, G. D. Cuny and R. A. Rossi, J. Org. Chem., 2006, 71, 8493 CrossRef CAS PubMed; (d) V. A. Vaillard, M. E. Buden, S. E. Martín and R. A. Rossi, Tetrahedron Lett., 2009, 50, 3829 CrossRef CAS.
- (a) A. M. Linsenmeier, C. M. Williams and S. Bräse, J. Org. Chem., 2011, 76, 9127 CrossRef CAS PubMed; (b) A. M. Linsenmeier, C. M. Williams and S. Bräse, Eur. J. Org. Chem., 2013, 3847 CrossRef CAS.
- D. Verga, F. Doria, L. Pretali and M. Freccero, J. Org. Chem., 2010, 75, 3477 CrossRef CAS PubMed.
- (a) R. K. Sharma and N. Kharasch, Angew. Chem., Int. Ed., 1968, 7, 36 CrossRef CAS; (b) I. Schnapperelle and T. Bach, Chem. – Eur. J., 2014, 20, 9725 CrossRef CAS PubMed; (c) Y. T. Park, N. W. Song, C. G. Hwang, K. W. Kim and D. Kim, J. Am. Chem. Soc., 1997, 119, 10677 CrossRef CAS; (d) T.-I. Ho, C.-K. Ku and R. S. H. Liu, Tetrahedron Lett., 2001, 42, 715 CrossRef CAS; (e) T. Kawamoto, A. Sato and I. Ryu, Org. Lett., 2014, 16, 2111 CrossRef CAS PubMed.
- A. M. Mfuh, J. D. Doyle, B. Chhetri, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2016, 138, 2985 CrossRef CAS PubMed.
- (a) M. Mella, P. Coppo, B. Guizzardi, M. Fagnoni, M. Freccero and A. Albini, J. Org. Chem., 2001, 66, 6344 CrossRef CAS PubMed; (b) B. Guizzardi, M. Mella, M. Fagnoni, M. Freccero and A. Albini, J. Org. Chem., 2001, 66, 6353 CrossRef CAS PubMed; (c) B. Guizzardi, M. Mella, M. Fagnoni and A. Albini, J. Org. Chem., 2003, 68, 1067 CrossRef CAS PubMed; (d) P. Coppo, M. Fagnoni and A. Albini, Tetrahedron Lett., 2001, 42, 4271 CrossRef CAS.
- (a) B. Guizzardi, M. Mella, M. Fagnoni and A. Albini, Chem. – Eur. J., 2003, 9, 1549 CrossRef CAS PubMed; (b) S. Protti, M. Fagnoni, M. Mella and A. Albini, J. Org. Chem., 2004, 69, 3465 CrossRef CAS PubMed.
- (a) V. Dichiarante and M. Fagnoni, Synlett, 2008, 787 CAS; (b) M. Fagnoni, Lett. Org. Chem., 2006, 3, 253 CrossRef CAS; (c) S. Protti, M. Fagnoni and A. Albini, J. Am. Chem. Soc., 2006, 128, 10670 CrossRef CAS PubMed.
- S. Lazzaroni, S. Protti, M. Fagnoni and A. Albini, Org. Lett., 2009, 11, 349 CrossRef CAS PubMed.
- I. Manet, S. Monti, M. Fagnoni, S. Protti and A. Albini, Chem. – Eur. J., 2005, 11, 140 CrossRef PubMed.
- J. F. Guastavino, M. E. Buden and R. A. Rossi, J. Org. Chem., 2014, 79, 9104 CrossRef CAS PubMed.
- (a) Y. Suzuki, Y. Okita, T. Morita and Y. Yoshimi, Tetrahedron Lett., 2014, 55, 3355 CrossRef CAS; (b) Y. Yoshimi, H. Kanai, K. Nishikawa, Y. Ohta, Y. Okita, K. Maeda and T. Morita, Tetrahedron Lett., 2013, 54, 2419 CrossRef CAS; (c) M. Ueda, Y. Uenoyama, N. Terasoma, S. Doi, S. Kobayashi, I. Ryu and J. A. Murphy, Beilstein J. Org. Chem., 2013, 9, 1340 CrossRef PubMed.
- (a) S. E. Vaillard, A. Postigo and R. A. Rossi, J. Org. Chem., 2004, 69, 2037 CrossRef CAS PubMed; (b) J. I. Bardagí, S. E. Vaillard and R. A. Rossi, Tetrahedron Lett., 2006, 47, 3149 CrossRef.
- A. Arora, K. A. Teegardin and J. D. Weaver, Org. Lett., 2015, 17, 3722 CrossRef CAS PubMed.
- H. Kim and C. Lee, Angew. Chem., Int. Ed., 2012, 51, 12303 CrossRef CAS PubMed.
- S. Z. Tasker and T. F. Jamison, J. Am. Chem. Soc., 2015, 137, 9531 CrossRef CAS PubMed.
- A. Sagadevana and K. C. Hwang, Adv. Synth. Catal., 2012, 354, 3421 CrossRef.
- M. Osawa, H. Nagai and M. Akita, Dalton Trans., 2007, 827 RSC.
- S. Protti, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2005, 44, 5675 CrossRef CAS PubMed.
- T. Kawamoto, A. Sato and I. Ryu, Chem. – Eur. J., 2015, 21, 14764 CrossRef CAS PubMed.
- Y. Li, D. Zheng, Z. Li and J. Wu, Org. Chem. Front., 2016, 3, 574 RSC.
- (a) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2013, 339, 1593 CrossRef PubMed; (b) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS PubMed.
- L. Chu, J. M. Lipshultz and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 7929 CrossRef CAS PubMed.
- Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832 CrossRef CAS PubMed.
- J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854 CrossRef CAS PubMed.
- J.-Q. Chen, Y.-L. Wei, G.-Q. Xu, Y.-M. Liang and P.-F. Xu, Chem. Commun., 2016, 52, 6455 RSC.
| This journal is © the Partner Organisations 2016 |