
Transformable nanostructures of cholesteryl-containing rhomboidal metallacycles through hierarchical self-assembly†
Li-Jun
Chen
,
Bo
Jiang
and
Hai-Bo
Yang
*
Shanghai Key Laboratory of Green Chemistry and Chemical Processes, Department of Chemistry, East China Normal University, Shanghai 200062, P. R. China. E-mail: hbyang@chem.ecnu.edu.cn; Fax: +86 021 62235137
First published on 1st March 2016
Abstract
The precise morphological control of supramolecular structures is of pivotal importance in the fields of nanoscience and nanotechnology. We describe herein a successful example of transformable nanostructures from cholesteryl-containing rhomboidal metallacycles. Various interesting nanostructures such as flowers and tadpole shaped structures were successfully obtained through the hierarchical self-assembly of two well-defined rhomboidal metallacycles prepared by coordination-driven self-assembly. Variation of the linker structure and solvent composition was found to trigger such drastic morphological changes of aggregates as revealed by microscopy studies.
 Hai-Bo Yang | Hai-Bo Yang received his M.S. degree at Beijing Normal University in 2001 under the supervision of Prof. Ying Cheng. He obtained his Ph.D. degree at ICCAS in 2004 under the supervision of Prof. Mei-Xiang Wang. Then he joined Professor Peter J. Stang's group at the University of Utah as a Postdoctoral Fellow. He started his independent research as a full Professor at East China Normal University (ECNU) in Shanghai by the end of 2008. Prof. Yang is interested in well-controlled self-assembly of supramolecular organometallic architectures including functionalized metallacycles, metallodendrimers, and metallogels. He has authored or coauthored 63 publications in Chem. Soc. Rev., Proc. Natl. Acad. Sci. U. S. A., J. Am. Chem. Soc., Chem. Sci., etc. since his independent research at ECNU. He is now a member of the Advisory Board for Chem (Cell Press). |
Introduction
Self-assembly of small molecular building blocks into complex supramolecular architectures with specific size, shape and function (“bottom-up” strategy) is ubiquitous in chemistry, materials science and biology.1 It should be noted that the precise morphological control of supramolecular structures is of pivotal importance in the fields of nanoscience and nanotechnology since materials morphology plays an essential role in determining the functionality of the final devices.2 Thus, the construction of the ordered nanostructures through supramolecular self-assembly via noncovalent interactions has attracted considerable attention from chemists in recent decades.3 Among the widely used weak interactions, metal–ligand coordination has proven to be one of the most efficient driving-forces on constructing well-ordered complexes, especially the well-defined, discrete metallosupramolecular structures from two-dimensional (2-D) polygons to three-dimensional (3-D) cages, prisms, and polyhedra.4–6Recently, we have demonstrated that the introduction of functional moieties that possess other noncovalent interactions on the discrete metallacycles could induce hierarchical self-assembly of metallacycles to produce a variety of functional materials such as metallohydrogels and stimuli-responsive supramolecular polymers because of the noninterfering orthogonal nature of metal coordination and other weak noncovalent interactions (e.g. hydrogen-bonding, electrostatic forces, hydrophobic and hydrophilic interactions, van der Waals forces, etc.).7 However, although some interesting nanostructures such as nanofibers, nanoparticles and vesicle-like structures have been obtained from hierarchical self-assembly of discrete metallacyles in our previous study,8 the method to precisely control the morphology of these assemblies has been very limited. In particular, the realization of transformable nanostructures of discrete metallacycles is still challenging.
Cholesterol is a fascinating molecule that has been widely exploited in supramolecular chemistry and materials science in many ways.9 Remarkably, the unique self-aggregation properties of cholesterol derivatives through weak van der Waals interactions make them a good candidate for the construction of liquid crystals, gels, and the ordered nanostructures, most of which have displayed the tunable morphology upon the change of molecular structures or the external environment conditions.10 Herein we present the successful example of transformable nanostructures from functionalized rhomboidal metallacycles though hierarchical self-assembly by introducing cholesteryl moieties into the discrete rhomboidal metallacycles. The chemical structures of the ligands employed in this study are shown in Scheme 1. Two well-defined rhomboidal metallacycles decorated with cholesteryl units were obtained from these ligands through metal–ligand interaction. The structures of the obtained metallacycles were well characterized by one-dimensional multinuclear (1H and 31P) and two-dimensional (1H–1H COSY, 1H–1H NOESY, and DOSY) NMR spectroscopy, mass spectrometry and elemental analysis. Subsequently, the presence of cholesteryl units enabled the orientation and aggregation of the metallacycles to form the ordered nanostructures via van der Waals forces. Notably, it was found that the morphology of the metallacycle aggregates was well tuned by changing the solvent polarity. In addition, the self-assembly behaviour could also be effectively controlled through the simple modification of the linker units. Such precise morphological control of the supramolecular nanostructures from discrete metallacycles has been less explored. The present work would provide further insight into the design of functional nanoscale materials with fine-tuning morphologies.
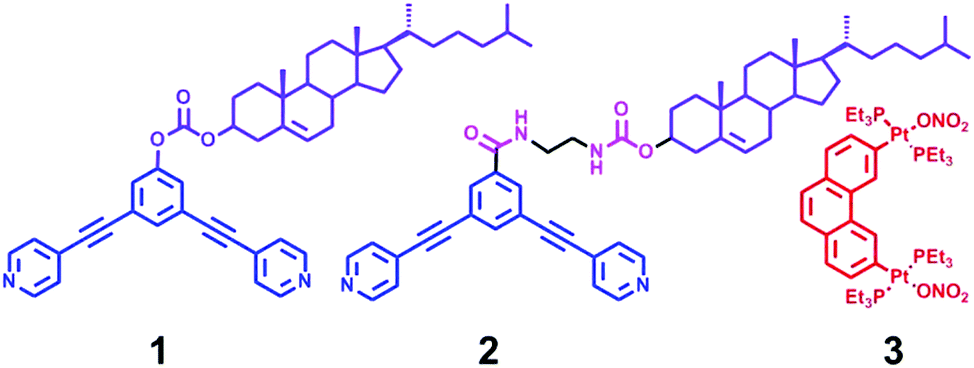 | ||
| Scheme 1 Chemical structures of ligands 1–3. | ||
Results and discussion
Synthesis and characterization
The 120° cholesteryl-containing dipyridyl donor ligand 1 was prepared through the acylation reaction of 3,5-bis(pyridin-4-ylethynyl)phenol (BisPy-OH), which was prepared in the same way as the procedure in the literature,11 with cholesteryl chloride in 86% yield (Scheme 2). In order to investigate the influence of the chemical structure on aggregation behaviour, an analogous ligand 2, which contained amide groups as a linker, was also prepared. Amidation of 3,5-bis(pyridin-4-ylethynyl)benzoic acid with cholesteryl (2-aminoethyl)carbamate produced ligand 2 in 67% yield (Scheme 3). With these newly designed 120° precursors in hand, the self-assembly of a new family of cholesteryl containing rhomboidal metallacycles was then investigated. Stirring a mixture of 1 or 2 with an equimolar amount of known 60° di-Pt(II) acceptor 3 resulted in the self-assembly of a [2 + 2] rhombus 4 or 5, respectively (Schemes 4, S1 and S2, ESI†). Self-assembly of the two-component metallacycles 4 and 5 was firstly monitored by multinuclear NMR spectroscopy (1H and 31P), which supported the formation of discrete, highly symmetric metallacycle species. For instance, each 31P{1H} NMR spectrum of metallacycles 4 and 5 displayed a sharp singlet (ca. 14.71 ppm for 4 and 14.69 ppm for 5), shifted upfield from the starting platinum acceptors 3 by approximately 4.43 and 4.41 ppm, respectively (Fig. 1). This change as well as the concomitant decrease in the coupling of flanking 195Pt satellites (ca. ΔJ = −208.9 Hz for 4 and ΔJ = −195.9 Hz for 5) is consistent with the back-donation from Pt transition metal atoms. Moreover, the 1H NMR spectrum exhibited significant downfield shifts of pyridyl proton signals (ca. α-HPy, 0.53–0.55 ppm; β-HPy, 0.58–0.64 ppm, Fig. S1 and S2, see the ESI†) associated with the loss of electron density upon coordination of the nitrogen atom with platinum metal centers.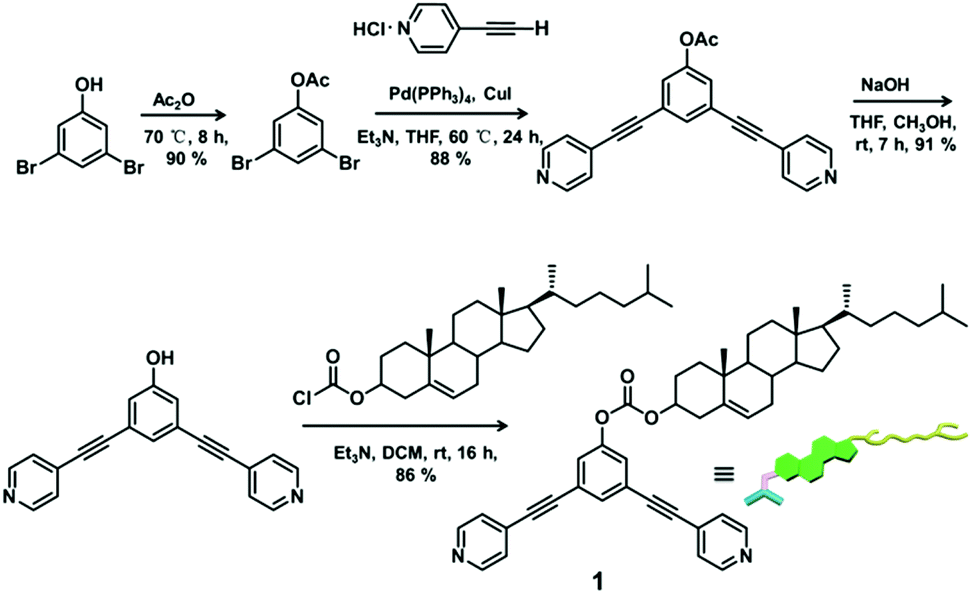 | ||
| Scheme 2 Synthetic procedure of cholesteryl-containing dipyridine ligand 1. | ||
 | ||
| Scheme 3 Synthetic procedure of cholesteryl-containing dipyridine ligand 2. | ||
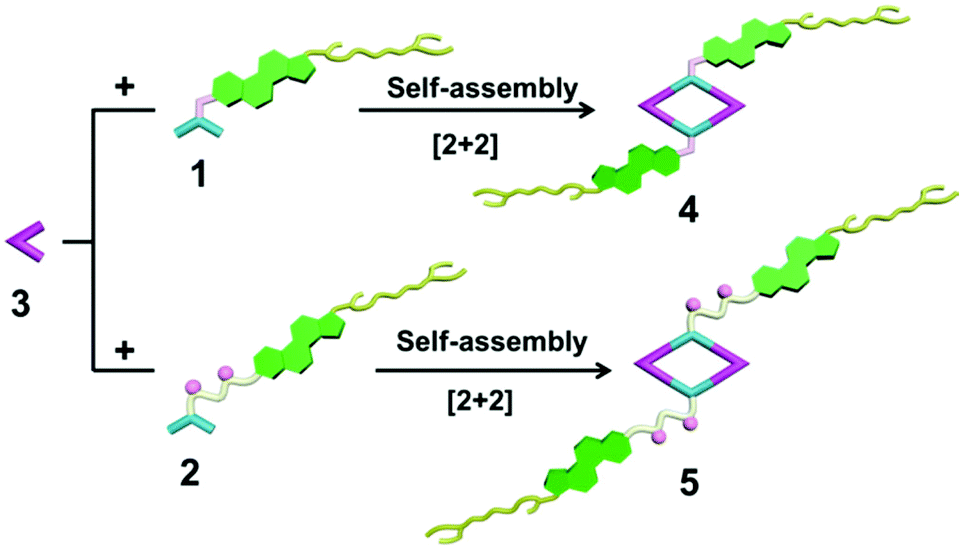 | ||
| Scheme 4 Graphical representation of the [2 + 2] rhomboidal metallacycles 4 and 5. | ||
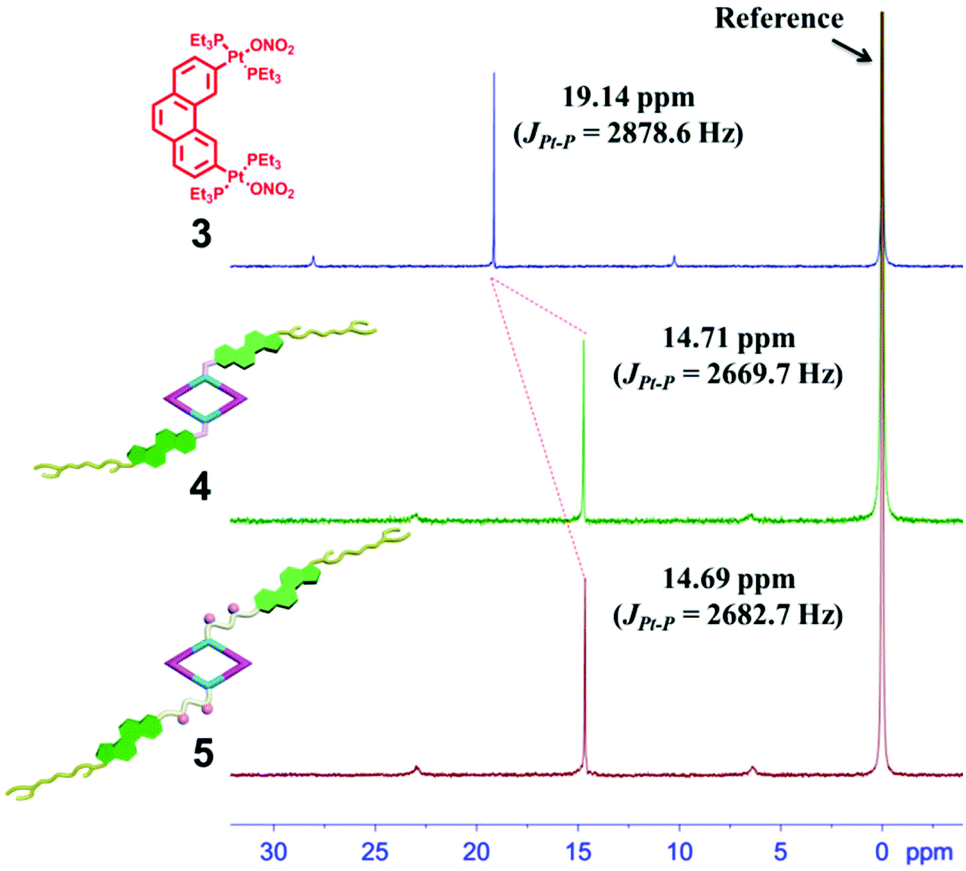 | ||
| Fig. 1 The 31P{1H} NMR spectra (400 MHz, acetone-d6, 298 K) of the 60° di-Pt(II) acceptor 3 and the [2 + 2] rhombus 4 and 5. | ||
Notably, two doublets for α- and β-hydrogen nuclei on the pyridine rings of the rhomboidal complexes 4 and 5 were observed in the 1H NMR spectrum (Fig. S1 and S2†), which might have been caused by the hindered rotation about the Pt–N (pyridyl) bond in a small-sized metallacycle. Further characterization with two-dimensional spectroscopy techniques (1H–1H COSY and NOESY) is in agreement with the formation of the discrete metallacycles 4 and 5 based on the formation of N–Pt bonds (Fig. S3–S6†). For example, the presence of cross peaks between the α-H proton on pyridine and PEt3 protons in the NOESY spectrum of each complex authenticated the formation of Pt–N coordination bonds (Fig. S5 and S6†).
The structures of metallacycles 4 and 5 were further confirmed by cold-spray ionization time-of-flight mass spectrometry (CSI-TOF-MS),12 which allows the assembly to remain intact during the ionization process while obtaining the high resolution required for isotopic distribution. For instance, the CSI-TOF-MS spectrum of the hexafluorophosphate salt rhomboid 4 revealed two peaks at m/z = 1892.34 and m/z = 1388.28, corresponding to [M − 2PF6]2+ and [M − 3PF6]3+ (Fig. S7†), respectively. Moreover, in the case of rhomboid 5, the peaks at m/z = 1962.81, m/z = 1259.97 and m/z = 908.73, corresponding to [M − 2PF6]2+, [M − 3PF6]3+ and [M − 4PF6]4+, respectively (Fig. 2), were observed. These peaks are isotopically resolved, and they are in agreement with the theoretical isotopic patterns. It should be noted that no obvious signals attributed to fragments or other assemblies were found in the full spectrum of both rhomboids 4 and 5 (Fig. S62 and S64†), supporting the formation of [2 + 2] rhomboids as the sole products in this study.
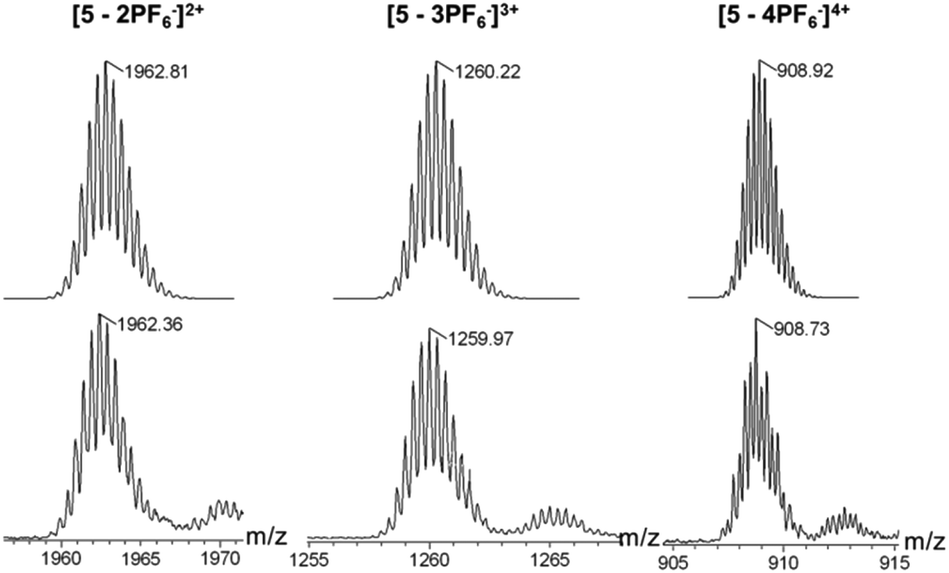 | ||
| Fig. 2 Theoretical (top) and experimental (bottom) CSI-TOF-MS results of [2 + 2] rhombus 5. | ||
With the aim to gain more structural information, a semiempirical molecular orbital method (PM6) was employed to optimize the geometry of the obtained metallacycles (Fig. 3). The optimized structure featured a roughly planar ring surrounded by flexible cholesteryl subunits at alternate corners. Metallacycles 4 and 5 resembled a well-defined rhombus featuring an approximately 0.8 × 1.2 nm cavity, with the outer radii of 2.7 nm and 3.1 nm, respectively. In order to obtain further insight into the structures of the resultant metallacycles, diffusion coefficient (D) of the metallacycles 4 and 5 was determined by two-dimensional diffusion-ordered 1H-NMR (2D-DOSY) experiments. A distinct band for both metallacycles 4 (Fig. S8†) and 5 (Fig. S9†) was observed, suggesting the formation of a single product. Moreover, 2D-DOSY NMR measurements for metallacycles 4 and 5 showed the expected D values trend with a slightly higher D value for 4 (5.62 × 10−9 m2 s−1), and a lower D value for 5 (5.01 × 10−9 m2 s−1).
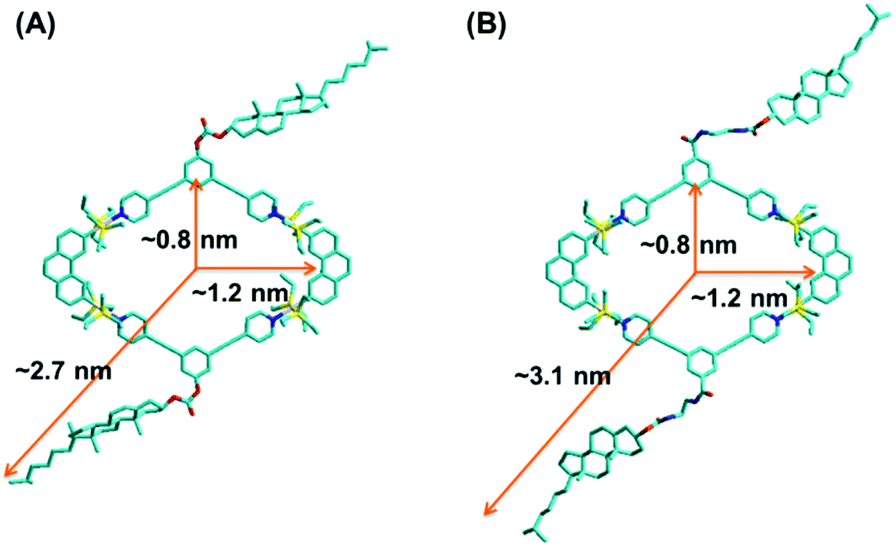 | ||
| Fig. 3 Simulated molecular models of [2 + 2] rhombus 4 (A) and 5 (B). | ||
Self-assembly on surface
The existence of cholesteryl groups outside the metallacycle skeleton stimulated us to investigate their hierarchical self-assembly behavior in various solvents by employing scanning electron microscopy (SEM). SEM samples were prepared by depositing the solution of metallacycles after 84 h incubation onto the freshly cleaned Si(111) surfaces followed by slow evaporation in air at room temperature. It was found that both rhomboidal metallacycles 4 and 5 in dichloromethane (DCM) displayed an irregular morphology after solvent evaporation possibly caused by their good solubility in DCM (Fig. 4a, S15† for 4 and Fig. 5a, S34† for 5). However, regarding the more polar solvents such as methanol, the SEM study revealed that rhomboid metallacycle 4 self-aggregated into rod-shaped structures extending over 100 μm in length (Fig. 4o and S30†). In addition, a spherical morphology with an average diameter of 160 nm was observed for metallacycle 5 (Fig. 5o and S49†) under the same conditions. | ||
| Fig. 4 SEM images of the aggregates of rhomboid metallacycle 4 formed in the CH2Cl2/CH3OH mixtures containing 0% (a), 10% (b), 20% (c), 25% (d), 30% (e), 35% (f), 40% (g), 45% (h), 50% (i), 55% (j), 65% (k), 70% (l), 80% (m), 90% (n), and 100% (o) methanol. Samples were prepared by spinning the solutions on a silicon substrate, dried under air and coating with Au. | ||
 | ||
| Fig. 5 SEM images of the aggregates of hexagonal metallacycle 5 formed in the CH2Cl2/CH3OH mixtures containing 0% (a), 20% (b), 25% (c), 30% (d), 35% (e), 40% (f), 45% (g), 50% (h), 55% (i), 60% (i), 65% (k), 70% (l), 80% (m), 90% (n), and 100% (o) methanol. Samples were prepared by spinning the solutions on a silicon substrate, dried under air and coating with Au. | ||
To verify the identity of the aggregate species for rhomboid metallacycles 4 and 5, electron microscopy and dynamic light scattering (DLS) have been employed. It was found that the metallacycles in the DCM solution featured average hydro-dynamic diameters (Dh) of ∼1.0 nm for 4 and 5 (Fig. S10 and S11†) without any evidence for further evolution of self-organization. However, in the solution of methanol, the Dh values were found to increase to 349 and 168 nm for 4 and 5, respectively (Fig. S12 and S13†), which indicated the formation of aggregation with large size. Moreover, the investigation of energy dispersive X-ray (EDX) spectroscopy of metallacycle 5 was carried out to obtain the further insight into the elemental composition of the obtained nanostructures. Elemental composition of carbon, oxygen, fluorine, platinum, and phosphorus were found from the aggregates of 5 in methanol (Fig. S14†), which strongly supported that these nanostructures were entirely generated from rhomboid metallacycle 5 through hierarchical self-assembly. These primary findings indicated that the aggregation morphologies of these complexes were heavily dependent on the solvent polarity.
In order to investigate the influence of solvent polarity, the morphologies of the rhomboid metallacycle 4 were firstly evaluated in the mixed DCM–methanol solvent systems with different solvent compositions but the same concentration at 0.2 mM. The aggregate morphology remained irregular in the mixed solvents with the methanol content less than 5%. When the methanol fraction was increased to 10%, some irregular spherical structures were observed (Fig. 4b and S16†). These spherical structures turned into regular fluff sphere-like structures (Fig. 4c and S17†) by increasing the methanol content to 20%, and further intertwined with each other (Fig. 4d and S18†) as a methanol fraction up to 25%. The entangled fluffy spheres were then transformed into fluff stick-like structures (Fig. 4e and S19†) in a 30% methanol solution. It is interesting to note that the formation of helical nanostructures was observed on the surface of fluff stick-like structures (Fig. 4f and S20†), which became smooth through a gradual process of annealing (Fig. 2g and S21†). Folding of the annealed sticks led to the formation of helical bundles (Fig. 4h and S22†), which further evolved into flowers through the sideway stacking of the sticks (Fig. 4i and S23†). These flower-like structures were maintained when the methanol fraction was between 55% and 90%, while the size and helical pitches varied along with the ratio change in DCM–methanol solution. Time-dependent morphology change of metallacycle 4 (CH2Cl2/CH3OH = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80 (v/v)) revealed that such flower-like structures were generated from small tablet like objects after 84 h growth (Fig. S31†) and remained stable for months. It was found that upon increasing the content of methanol, the size of the packing flowers decreased gradually, and the surface of the stick was becoming less wrinkly (Fig. 4j–n and S24–S28†). Finally, in pure methanol, all the helical morphologies disappeared and evolved into random sticks (Fig. 4o and S30†). Interestingly, evaporation of the solvents resulted in a similar morphology transformation process due to the polarity change of the mixed solvents during the evaporation (Fig. S32†). The presence of such a transformation process was confirmed from the analysis of transmission electron microscopy (TEM) (Fig. S33†) as well.
:80 (v/v)) revealed that such flower-like structures were generated from small tablet like objects after 84 h growth (Fig. S31†) and remained stable for months. It was found that upon increasing the content of methanol, the size of the packing flowers decreased gradually, and the surface of the stick was becoming less wrinkly (Fig. 4j–n and S24–S28†). Finally, in pure methanol, all the helical morphologies disappeared and evolved into random sticks (Fig. 4o and S30†). Interestingly, evaporation of the solvents resulted in a similar morphology transformation process due to the polarity change of the mixed solvents during the evaporation (Fig. S32†). The presence of such a transformation process was confirmed from the analysis of transmission electron microscopy (TEM) (Fig. S33†) as well.
The morphologies of analogous metallacycle 5 in DCM solutions containing different amounts of methanol were also investigated. As expected, morphology transformations accompanied by the change of the solvent polarity were observed as well. Starting from the irregular pores (Fig. 5a, b, S34 and S35†, 0–10%), the aggregates from 5 grew into the particles (Fig. 5c–g and S36–S40,† 20–40%) with the increase of methanol content. TEM analysis indicated that these spherical structures were hollow and showed a clear contrast between the interior and periphery (Fig. S50a†), which is consistent with the typical characteristic of vesicular structures. Such vesicular structures were then burst with a small hole (Fig. 5h and S41†) by continuously increasing the methanol fraction to 45%. Then nanotubes grew from the small hole, leading to the formation of tadpole-like nanostructures (Fig. 5i–k and S42–S44†). The length of the nanotube extended with the increase of solvent polarity, and the head part disappeared when the methanol fraction was up to 65% (Fig. 5l and S45†). Then, on the other side of the nanotube, small spheres were gradually generated (Fig. 5m and S46†). The diameter became larger in the more polar solution (Fig. 5n and o and S47–S49†). These small spherical structures were also hollow as evidenced by the TEM measurement (Fig. S50c†). Solutions of metallacycle 5 with different methanol fractions (e.g., 40% or 90%) displayed a similar time-dependent growth process (Fig. S51 and S52†), suggesting that all these nanostructures were generated under thermodynamic control as those for 4. Moreover, a reverse vesicle–tadpole–vesicle transformation was observed by diluting the solution of metallacyle 5 with dichloromethane (Fig. S53†), indicating the reversibility of this transformation process. Such morphology transformations between vesicles–nanotube–vesicles are of particular interest because vesicle-like structures usually featured the potential for controlled encapsulation and release of active substances (e.g., drugs, fragrances, and flavor additives) as we recently reported.8a
Interestingly, despite their structural similarity, the nanostructures generated from metallacycle 5 displayed totally different features compared with those of metallacycle 4 under the same conditions. For instance, when the volume ratio between CH2Cl2/CH3OH was 40/60, complex 5 aggregated into a tadpole-shaped nanostructure, while the flower-like features were observed for metallacycle 4. This phenomenon indicated that the amide linker in complex 5 represented a key structural factor toward the aggregation.
In order to explain the different behaviour of these two metallacycles, the in situ1H NMR spectra of both 4 and 5 in the mixed solvent of CD2Cl2–CD3OD were recorded to probe the aggregation process. As shown in Fig. S54 and S55,† upon increasing the methanol fraction, the proton signals around the aromatic region and PEt3 protons (–CH2 and –CH3) displayed a downfield shift in both 4 and 5. In addition, the protons on the linker moiety and a part of the cholesteryl moiety displayed an upfield shift in 5 while they remained unchanged in 4, indicating different aggregation modes for 4 and 5. Moreover, the involvement of hydrogen bonds during the aggregation process of 5 in DCM–methanol mixture solvents was confirmed by IR spectroscopy. It was found that the stretching vibrations of free NH groups that are usually found in the region between 3500 and 3600 cm−1 gradually disappeared. In addition, a slight shift of the signals corresponding to the stretching vibrations of amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (from 1700 cm−1 to 1719 cm−1) and the bending vibration of amide NH (from 1612 cm−1 to 1608 cm−1) was observed upon the increase of methanol fraction (Fig. S57†). All these findings were consistent with the existence of hydrogen bonding during the aggregation process.14
O (from 1700 cm−1 to 1719 cm−1) and the bending vibration of amide NH (from 1612 cm−1 to 1608 cm−1) was observed upon the increase of methanol fraction (Fig. S57†). All these findings were consistent with the existence of hydrogen bonding during the aggregation process.14
To further verify the aggregation mode of metallacycles 4 and 5, the optical studies of metallacycles in the mixed solvents of CH2Cl2–methanol were carried out. With the increase of methanol content, the UV-vis absorption spectra of metallacycle 4 displayed a slight hypsochromic shift of the absorption maximum accompanied by a new absorption shoulder at ca. 365 nm with a clear isosbestic point (Fig. S58a†), indicating the aggregation of metallacycle 4.13 However, in the case of metallacycle 5, no significant solvatochromism was observed with the increase of solvent polarity, which does not give a direct clue for its aggregation mode (Fig. S58b†).
As a matter of fact, the self-assembly of cholesterol-based metallacycles is based primarily on weak forces (van der Waals and hydrogen bonding), thus the solvent plays a critical role during the process of aggregation. This property might result from a subtle balance between solubility and insolubility of the metallacycle in solution. In this respect, the chemical nature, such as the length and flexibility of the linking group of the cholesterol units, has a large impact. In the case of rhomboid metallacycle 4 that has two cholesteryl moieties, one-dimensional stacking of the fully extended molecules could be involved in the formation of a twisted central column of stacked macrocycle units stabilized by cholesterol–cholesterol interactions at each extremity, thus leading to the ultimate formation of helical bundles and flowers (Fig. 6a). As for the analogous metallacycle 5 containing amide units as a linker, the growth of aggregates relied mainly on the hydrogen bonding between amide moieties. Firstly, the bilayer structures (type I) were formed and then bent and enlaced to generate the hollow spheres. Subsequently, the bilayer structures were gradually destroyed upon the addition of a polar solvent and transformed into another kind of bilayer (type II), in which most amide units pointed out of the solvent molecule, resulting in the formation of another vesicle (Fig. 6b). Although the exact formation mechanism for these different hierarchical superstructures is still not very clear at present, the obtained phenomena have already shown the power of the solvent and chemical structures for well-controlled supramolecular nanostructures from discrete metallacycles through hierarchical self-assembly.
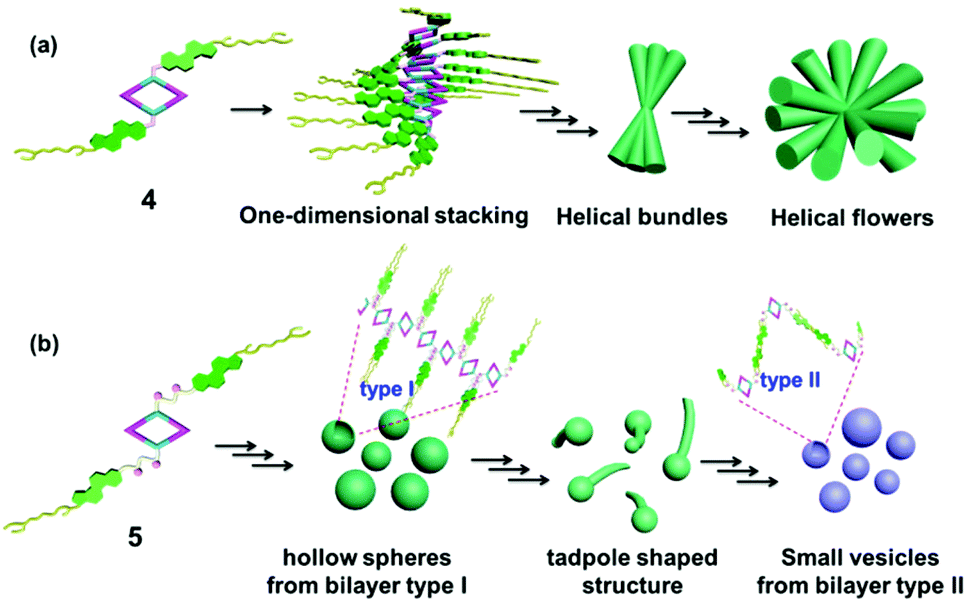 | ||
| Fig. 6 Schematic representation of the possible aggregation mode of rhomboid metallacycles 4 (a) and 5 (b). | ||
Conclusions
In conclusion, two cholesteryl-containing rhomboidal metallacycles 4 and 5 were prepared, which were further demonstrated to undergo hierarchical self-assembly in dichloromethane–methanol mixtures through van der Waals forces and hydrogen bonding. It was found that self-assembly can be effectively controlled by the variation of solvent polarity. For the rhomboid metallacycle 4, different morphologies such as helical bundles and flowers were produced. Interesting morphological transformations have been demonstrated by employing electronic micrographs (SEM and TEM). Simple modification of the precursor ligand with an amide group has resulted in an analogous metallacycle 5, which was found to form different macroscopic structures yet fine-tuned by solvent polarity. In short, this study provides a highly efficient approach to precisely control the shape and size of nanoscale structures containing discrete metallacycles as the main scaffolds through the simple modification of chemical structure and solvent polarity. It will be very helpful to gain a better understanding on how to control the self-organization of small building blocks on a large scale and prepare novel functional materials.Experimental section
Full experimental details are provided in the ESI.† The most important information is summarized briefly below.Synthesis of 1
To a solution of 3,5-bis(pyridin-4-ylethynyl)phenol (100.0 mg, 0.34 mmol) in 50 ml anhydrous dichloromethane was added an equivalent amount of NEt3 (50 μL). Cholesteryl chloride (151.6 mg, 0.34 mmol) in 20 ml anhydrous dichloromethane was then added dropwise to the reaction mixture under stirring at 0 °C for 1 h, followed by stirring for 15 h at room temperature. The solvent was reduced at room temperature under vacuum and the residue was purified by column chromatography on silica gel (acetone/petroleum ether = 2/3, Rf = 0.5) to give compound 1. Yield: 206.5 mg, 86% (pale yellow solid). Mp: 200–205 °C. 1H NMR (CDCl3, 400 MHz): δ 8.65 (d, J = 4.8 Hz, 4H, α-H), 7.64 (s, 1H, Ar–H), 7.45–7.49 (m, 6H, β-H + Ar–H), 5.44 (d, J = 4.0 Hz, 1H, CHC), 4.59–4.65 (m, 1H, OCH), 2.49–2.51 (m, 2H), 1.05–1.98 (m, 37H, Chol–H), 0.70 (s, 3H, Chol–Me); 13C NMR (CDCl3, 100 MHz): δ 11.85, 18.69, 19.26, 21.03, 22.81, 23.80, 24.26, 27.60, 28.00, 28.20, 31.81, 31.89, 35.76, 36.15, 36.54, 36.78, 37.88, 39.49, 42.29, 49.94, 56.10, 56.65, 79.57, 87.98, 92.22, 123.43, 123.79, 125.40, 125.76, 131.38, 132.61, 138.91, 149.19, 150.95, 152.39; MS (EI): 709.27 (M+), 710.28 [(M + 1)+]; HRMS (EI): Exact mass calcd for C48H56N2O3 [M]+: 709.4365, Found: 709.4386.
Synthesis of 2
Compounds 3,5-bis(pyridin-4-ylethynyl)benzoic acid (800.0 mg, 1.69 mmol) and 4-dimethylaminopyridine (DMAP) (20.7 mg, 0.17 mmol) were dispersed in 6 mL dried dichloromethane and 3 mL anhydrous DMF. A solution of N,N′-dicyclohexylcarbodiimide (DCC) (523.6 mg, 2.54 mmol) and compound cholesteryl (2-aminoethyl)carbamate (823.0 mg, 2.54 mmol) in dry dichloromethane (5.0 mL) was added dropwise to the dispersed solution at 0 °C under a nitrogen atmosphere. The mixture was stirred at room temperature for 24 h. Then, the mixture was filtered, and the filtrate was evaporated in a vacuum to dryness, and the residues were dissolved in dichloromethane. The dichloromethane solution was washed with water at least 10 times. The organic phase was separated and dried by using anhydrous magnesium sulfate. The dried and purified organic solution was concentrated in a vacuum to dryness. The residue was purified by column chromatography on silica gel (dichloromethane/methanol = 10/1, Rf = 0.3) to give compound 2. Yield: 902.1 mg, 67% (pale yellow solid). 1H NMR (CDCl3, 400 MHz): δ 8.63 (d, J = 3.6 Hz, 4H, α-H), 8.01 (s, 2H, Ar–H), 7.84 (s, 1H, Ar–H), 7.39 (d, J = 3.2 Hz, 4H, β-H), 7.27 (b, 1H, N–H), 5.31 (d, J = 4.8 Hz, 1H, CHC), 5.07 (b, 1H, N–H), 4.50–4.53 (m, 1H, OCH), 3.61 (b, 2H, CH2–NH), 3.49 (b, 2H, CH2–NH), 2.26–2.29 (m, 2H), 0.90–1.99 (m, 37H, Chol–H), 0.61 (s, 3H, Chol–CH3); 13C NMR (CDCl3, 100 MHz): δ 11.75, 19.17, 20.90, 22.49, 22.75, 23.80, 24.17, 27.93, 28.07, 28.12, 29.28, 29.62, 31.71, 31.76, 35.70, 36.09, 36.42, 36.87, 38.43, 39.59, 40.19, 41.83, 42.19, 41.83, 42.19, 49.86, 56.05, 56.53, 75.01, 76.68, 77.00, 77.32, 88.07, 91.78, 122.59, 123.13, 125.51, 130.66, 130.94, 135.19, 137.13, 139.44, 149.76, 157.80, 165.96; MS (ESI): 779.34 (M+), 780.36 [(M + 1)+]; HRMS (ESI): Exact mass calcd for C51H62N4O3 [M]+: 779.4895, Found: 779.4916.
Synthesis of [2 + 2] rhombus 4
The dipyridyl donor ligand 1 (3.24 mg, 4.57 μmol) and the organoplatinum 60° acceptor 3 (5.31 mg, 4.57 μmol) were weighed accurately in a glass vial. To the vial was added 2.0 mL DCM, and the reaction solution was stirred at room temperature for 18 h to yield a homogeneous yellow solution. Then acetone (3.0 mL) was added, followed by the addition of a saturated aqueous solution of KPF6 into the bottle with continuous stirring (10 min) to precipitate the product. The reaction mixture was centrifuged, washed several times with water, and dried. The pale-yellow product 4 (8.55 mg, >99%) was collected and redissolved in acetone-d6 for NMR analysis. 1H NMR (acetone-d6, 400 MHz): δ 9.18 (d, 2H, J = 5.6 Hz, α-H), 9.16 (d, 2H, J = 5.6 Hz, α′-H), 8.78 (s, 2H, Phen–H), 8.05 (s, 1H, Ar–H), 8.03 (d, 2H, J = 5.2 Hz, β-H), 7.98 (d, 2H, J = 5.6 Hz, β′-H), 7.67–7.79 (m, 8H, Phen–H + Ar–H), 5.48 (s, 1H, CHC), 4.57–4.61 (m, 1H, OCH), 2.52–2.54 (m, 2H), 1.88–1.89 (m, 3H, Chol–H), 1.52 (br, 31H, Chol–H + CH2-P), 0.87–1.28 (m, 66H, Chol–H + CH3), 0.75 (s, 3H, Chol–Me). 31P NMR (acetone-d6, 161.9 MHz): δ 14.71 (s, JPt–P = 2669.7 Hz); CSI-MS: m/z: 1213.26 [M − 3PF6]3+, 1892.34 [M − 2PF6]2+. Anal. Calcd (%) for C172H248F24N4O6P12Pt4 C: 50.69, H: 6.13, N: 1.37; Found: C: 50.45, H: 6.12, N: 1.45.
Synthesis of [2 + 2] rhombus 5
The dipyridyl donor ligand 2 (4.83 mg, 6.20 μmol) and the organoplatinum 60° acceptor 3 (7.21 mg, 6.20 μmol) were weighed accurately in a glass vial. To the vial were added 1.0 mL DCM and 1.0 mL acetone, and the reaction solution was stirred at room temperature for 18 h to yield a homogeneous yellow solution. Then acetone (3.0 mL) was added, followed by the addition of a saturated aqueous solution of KPF6 into the bottle with continuous stirring (10 min) to precipitate the product. The reaction mixture was centrifuged, washed several times with water, and dried. The pale-yellow product 5 (12.04 mg, >99%) was collected and redissolved in acetone-d6 for NMR analysis. 1H NMR (acetone-d6, 400 MHz): δ 9.18 (d, 2H, J = 5.6 Hz, α-H), 9.16 (d, 2H, J = 5.6 Hz, α′-H), 8.79 (s, 2H, Phen–H), 8.28 (s, 2H, Ar–H), 8.16 (s, 1H, Ar–H), 8.03 (d, 2H, J = 5.6 Hz, β-H), 8.00 (d, 2H, J = 5.6 Hz, β′-H), 7.67–7.79 (m, 6H, Phen–H), 6.45 (b, 1H, N–H), 5.36 (s, 1H, CHC), 4.40–4.45 (m, 1H, OCH), 3.57–3.60 (m, 2H, CH2–NH), 3.42 (b, 2H, CH2–NH), 2.20–2.31 (m, 2H), 1.84–1.87 (m, 3H, Chol–H), 1.52 (br, 31H, Chol–H + CH2), 0.87–1.28 (m, 66H, Chol–H + CH3), 0.74 (s, 3H, Chol–Me). 31P NMR (acetone-d6, 161.9 MHz): δ 14.69 (s, JPt–P = 2682.7 Hz); CSI-MS: m/z: 1962.36 [M − 3PF6]3+, 1259.97 [M − 2PF6]2+, 908.73 [M − PF6]2+. Anal. Calcd (%) for C178H260F24N8O6P12Pt4 C: 50.71, H: 6.22, N: 2.66, Found: C: 50.51, H: 6.21, N: 2.65.
SEM, TEM, EDX, and DLS experiments
SEM images were obtained using an S-4800 (Hitachi Ltd) with an accelerating voltage of 3.0–10.0 kV. Samples were prepared by dropping solutions onto a silicon wafer. To minimize sample charging, a thin layer of Au was deposited onto the samples before SEM examination. TEM images were recorded on a Tecnai G2 F30 (FEI Ltd). The sample for the TEM measurement was prepared by dropping the solution onto a carbon-coated copper grid. EDX measurements were performed using an S-4800 (Hitachi Ltd) with a voltage of 10.0 kV. DLS measurements were performed under Malvern Zetasizer Nano-ZS light scattering apparatus (Malvern Instruments, U.K.) with a He–Ne laser (633 nm, 4 mW).Acknowledgements
This work was financially supported by NSFC/China (no. 21322206 and 21132005) and the Key Basic Research Project of Shanghai Science and Technology Commission (no. 13JC1402200).Notes and references
- (a) J.-M. Lehn, Supramolecular Chemistry, Concepts and Perspecties, VCH, New York, 1995 Search PubMed; (b) J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, West Sussex, U.K., 2000 Search PubMed; (c) J. W. Steed, D. R. Turner and K. J. Wallace, Core Concepts in Supramolecular Chemistry and Nanochemistry, John Wiley & Sons, West Sussex, U.K., 2007 Search PubMed; (d) T. Nakanishi, Supramolecular Soft Matter: Applications in Materials and Organic Electronics, John Wiley & Sons, West Sussex, U.K., 2011 Search PubMed.
- (a) M. Shimomura and T. Sawadaishi, Curr. Opin. Colloid Interface Sci., 2001, 6, 11–16 CrossRef CAS; (b) M.-X. Wang, Acc. Chem. Res., 2012, 45, 182–195 CrossRef CAS PubMed; (c) D. Ajami and J. Rebek, Acc. Chem. Res., 2013, 46, 990–999 CrossRef CAS PubMed; (d) V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341–5370 RSC; (e) Y. Wang, H.-X. Lin, L. Chen, S.-Y. Ding, Z.-C. Lei, D.-Y. Li, X.-Y. Cao, H.-J. Liang, Y.-B. Jiang and Z.-Q. Tian, Chem. Soc. Rev., 2014, 43, 399–411 RSC; (f) N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed; (g) P. A. Gale and C. Caltagirone, Chem. Soc. Rev., 2015, 44, 4212–4227 RSC; (h) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- (a) J. M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763–4768 CrossRef CAS PubMed; (b) S. F. M. van Dongen, J. Clerx, K. Nørgaard, T. G. Bloemberg, J. J. L. M. Cornelissen, M. A. Trakselis, S. W. Nelson, S. J. Benkovic, A. E. Rowan and R. J. M. Nolte, Nat. Chem., 2013, 5, 945–951 CrossRef CAS PubMed; (c) D. A. Leigh, R. G. Pritchard and A. J. Stephens, Nat. Chem., 2014, 6, 978–982 CrossRef CAS PubMed; (d) S. Ko, S. K. Kim, A. Share, V. M. Lynch, J. Park, W. Namkung, W. Van Rossom, N. Busschaert, P. A. Gale, J. L. Sessler and I. Shin, Nat. Chem., 2014, 6, 885–892 CrossRef CAS PubMed; (e) J. Tian, T.-Y. Zhou, S.-C. Zhang, S. Aloni, M. V. Altoe, S.-H. Xie, H. Wang, D.-W. Zhang, X. Zhao, Y. Liu and Z.-T. Li, Nat. Commun., 2014, 5, 5574 CrossRef CAS PubMed; (f) S.-H. Li, H.-Y. Zhang, X. Xu and Y. Liu, Nat. Commun., 2015, 6, 7590 CrossRef PubMed; (g) B. Zheng, F. Klautzsch, M. Xue, F. Huang and C. A. Schalley, Org. Chem. Front., 2014, 1, 532–540 RSC; (h) S. Da Ros, A. Linden, K. K. Baldridge and J. S. Siegel, Org. Chem. Front., 2015, 2, 626–633 RSC; (i) T.-G. Zhan, B.-Y. Lu, F. Lin, T.-Y. Zhou, X. Zhao and Z.-T. Li, Org. Chem. Front., 2015, 2, 1578–1583 RSC.
- (a) P. J. Stang and B. Olenyuk, Acc. Chem. Res., 1997, 30, 502–518 CrossRef CAS; (b) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS PubMed; (c) D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349–358 CrossRef CAS PubMed; (d) B. H. Northrop, H.-B. Yang and P. J. Stang, Chem. Commun., 2008, 5896–5908 RSC; (e) A. M. Spokoyny, D. Kim, A. Sumrein and C. A. Mirkin, Chem. Soc. Rev., 2009, 38, 1218–1227 RSC; (f) G. R. Newkome and C. Shreiner, Chem. Rev., 2010, 110, 6338–6442 CrossRef CAS PubMed; (g) A. H. Clever and M. Shionoya, Coord. Chem. Rev., 2010, 254, 2391–2402 CrossRef; (h) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 110, 6810–6918 CrossRef PubMed; (i) M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC; (j) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed; (k) T. R. Cook, Y. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed; (l) M. Yoshizawa and J. K. Klosterman, Chem. Soc. Rev., 2013, 42, 1885–1898 Search PubMed; (m) S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419–432 RSC; (n) A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed; (o) L. Xu, Y.-X. Wang, L.-J. Chen and H.-B. Yang, Chem. Soc. Rev., 2015, 44, 2148–2167 RSC.
- (a) M. Han, R. Michel, B. He, Y.-S. Chen, D. Stalke, M. John and G. H. Clever, Angew. Chem., 2013, 125, 1358–1362 ( Angew. Chem., Int. Ed. , 2013 , 52 , 1319–1323 ) CrossRef; (b) W. Meng, T. K. Ronson and J. R. Nitschke, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10531–10535 CrossRef CAS PubMed; (c) M. Yamashina, M. M. Sartin, Y. Sei, M. Akita, S. Takeuchi, T. Tahara and M. Yoshizawa, J. Am. Chem. Soc., 2015, 137, 9266–9269 CrossRef CAS PubMed; (d) B. Roy, A. K. Ghosh, S. Srivastava, P. D'Silva and P. S. Mukherjee, J. Am. Chem. Soc., 2015, 137, 11916–11919 CrossRef CAS PubMed.
- (a) S. Chen, L.-J. Chen, H.-B. Yang, H. Tian and W. Zhu, J. Am. Chem. Soc., 2012, 134, 13596–13599 CrossRef CAS PubMed; (b) L. Xu, L.-J. Chen and H.-B. Yang, Chem. Commun., 2014, 50, 5156–5170 RSC; (c) L. Xu, Y.-X. Wang and H.-B. Yang, Dalton Trans., 2015, 44, 867–890 RSC; (d) W. Wang, Y.-X. Wang and H.-B. Yang, Org. Chem. Front., 2014, 1, 1005–1009 RSC; (e) S. Shanmugaraju and P. S. Mukherjee, Chem. – Eur. J., 2015, 21, 6656–6666 CrossRef CAS PubMed; (f) S. Shanmugaraju, S. A. Joshi and P. S. Mukherjee, Inorg. Chem., 2011, 50, 11736–11745 CrossRef CAS PubMed; (g) S. Shanmugaraju, A. K. Bar, K. Chi and P. S. Mukherjee, Organometallics, 2010, 29, 2971–2980 CrossRef CAS.
- (a) G.-Z. Zhao, L.-J. Chen, W. Wang, J. Zhang, G. Yang, D. Wang, Y. Yu and H.-B. Yang, Chem. – Eur. J., 2013, 19, 10094–10100 CrossRef CAS PubMed; (b) Z.-Y. Li, Y. Zhang, C.-W. Zhang, L.-J. Chen, C. Wang, H. Tan, Y. Yu, X. Li and H.-B. Yang, J. Am. Chem. Soc., 2014, 136, 8577–8589S CrossRef CAS PubMed; (c) N.-W. Wu, L.-J. Chen, C. Wang, Y.-Y. Ren, X. Li, L. Xu and H.-B. Yang, Chem. Commun., 2014, 50, 4231–4233 RSC; (d) B. Sun, M. Wang, Z. Lou, M. Huang, C. Xu, X. Li, L.-J. Chen, Y. Yu, G. L. Davis, B. Xu, H.-B. Yang and X. Li, J. Am. Chem. Soc., 2015, 137, 1556–1564 CrossRef CAS PubMed.
- (a) L.-J. Chen, G.-Z. Zhao, B. Jiang, B. Sun, M. Wang, L. Xu, J. He, Z. Abliz, H. Tan, X. Li and H.-B. Yang, J. Am. Chem. Soc., 2014, 136, 5993–6001 CrossRef CAS PubMed; (b) J. Zhang, R. Marega, L.-J. Chen, N.-W. Wu, X.-D. Xu, D. C. Muddiman, D. Bonifazi and H.-B. Yang, Chem. – Asian J., 2014, 9, 2928–2936 CrossRef CAS PubMed; (c) L.-J. Chen, Y.-Y. Ren, N.-W. Wu, B. Sun, J.-Q. Ma, L. Zhang, H. Tan, M. Liu, X. Li and H.-B. Yang, J. Am. Chem. Soc., 2015, 137, 11725–11735 CrossRef CAS PubMed.
- (a) V. Ajay Mallia and N. Tamaoki, Chem. Soc. Rev., 2004, 33, 76–84 RSC; (b) M. Žinic, F. Vögtle and F. Fages, Top. Curr. Chem., 2005, 256, 833–836 CrossRef; (c) C. Netto, H. E. Toma and L. H. Andrade, J. Mol. Catal. B:Enzym., 2013, 85–86, 71–92 CrossRef CAS; (d) J. W. Morzycki, Steroids, 2014, 83, 62–79 CrossRef CAS PubMed; (e) M. C. Rheinstadter and O. G. Mouritsen, Curr. Opin. Colloid Interface Sci., 2013, 18, 440–447 CrossRef CAS.
- (a) M. George and R. G. Weiss, Acc. Chem. Res., 2006, 39, 489–497 CrossRef CAS PubMed; (b) T. Coviello, P. Matricardi, C. Marianecci and F. Alhaique, J. Controlled Release, 2007, 119, 5–24 CrossRef CAS PubMed; (c) T. Vermonden, R. Censi and W. E. Hennink, Chem. Rev., 2012, 112, 2853–2888 CrossRef CAS PubMed; (d) A. Tam and V. Yam, Chem. Soc. Rev., 2013, 42, 1540–1567 RSC; (e) L. Gu, A. Faig, D. Abdelhamid and K. Uhrich, Acc. Chem. Res., 2014, 47, 2867–2877 CrossRef CAS PubMed; (f) A. Pessi, J. Pept. Sci., 2015, 21, 379–386 CrossRef CAS PubMed; (g) F. Ercole, M. R. Whittaker, J. F. Quinn and T. P. Davis, Biomacromolecules, 2015, 16, 1886–1914 CrossRef CAS PubMed.
- H.-B. Yang, A. M. Hawkridge, S. D. Huang, N. Das, S. D. Bunge, D. C. Muddiman and P. J. Stang, J. Am. Chem. Soc., 2007, 129, 2120–2129 CrossRef CAS PubMed.
- J. He, Z. Abliz, R. Zhang, Y. Liang and K. Ding, Anal. Chem., 2006, 78, 4737–4740 CrossRef CAS PubMed.
- (a) Z. Chen, U. Baumeister, C. Tschierske and F. Würthner, Chem. – Eur. J., 2007, 13, 450–465 CrossRef CAS PubMed; (b) S. Ghosh, X. Li, V. Stepanenko and F. Würthner, Chem. – Eur. J., 2008, 14, 11343–11357 CrossRef CAS PubMed.
- (a) A. Syamakumari, A. P. H. J. Schenning and E. W. Meijer, Chem. – Eur. J., 2002, 8, 3353–3361 CrossRef CAS PubMed; (b) J. Gershberg, F. Fennel, T. H. Rehm, S. Lochbrunner and F. Würthner, Chem. Sci., 2016, 7, 1729–1737 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qo00017g |
| This journal is © the Partner Organisations 2016 |