
Concurrent modifications of the C-terminus and side ring of thiostrepton and their synergistic effects with respect to improving antibacterial activities†
Shoufeng
Wang‡
*a,
Qingfei
Zheng‡
a,
Jianfeng
Wang
b,
Dandan
Chen
c,
Yunsong
Yu
b and
Wen
Liu
*ac
aState Key Laboratory of Bioorganic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: wangshoufeng@sioc.ac.cn; Tel: +86-21-54925539
bDepartment of Infectious Diseases, Sir Run Run Shaw Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang 310016, China
cHuzhou Center of Bio-Synthetic Innovation, 1366 Hongfeng Road, Huzhou 313000, China. E-mail: wliu@mail.sioc.ac.cn; Tel: +86-21-54925111
First published on 9th February 2016
Abstract
The double-mutant strain Streptomyces laurentii ΔtsrB/T was designed and constructed based on a recent understanding regarding the structure–activity relationship of thiostrepton (TSR) against prokaryotic pathogens. Five new C-terminally methylated TSR (CmTSR) derivatives that varied in the side-ring structure were obtained via the chemical feeding of quinaldic acid (QA) analogs. These derivatives provide new insights into the tolerance of QA incorporation in TSR biosynthesis. Certain members of the tested TSR derivatives, meanwhile, exhibited much better antibacterial activities than all currently known thiopeptide antibiotics.
Thiostrepton (TSR), one of the most structurally complex ribosomally synthesized and post-translationally modified peptides (RiPPs),1 possesses a variety of remarkable biological properties, including antibacterial,2 antitumor,3 antiplasmodial4 and immunosuppressive activities.5 Due to its intriguing structure and unique activities (e.g., its status as the only type of natural product known to inhibit tumor cell proliferation by binding the transcription factor FOXM1),6 TSR has long been a target molecule for synthetic chemists. In addition to the total synthesis accomplished by Nicolaou et al.,7 a number of semi-syntheses have been attempted with the objectives of improving TSR's bioactivity, developing new chemical probes, investigating TSR's structure–activity relationship (SAR) and/or relieving TSR's biochemical drawbacks (e.g., poor pharmacokinetics) for use in clinical settings.8 However, the complex architecture of TSR and the presence of simple sites for further functionalization pose tremendous challenges to chemical syntheses and semi-syntheses. In contrast, the fact that TSR originates from a ribosomally synthesized peptide provides tremendous opportunities for synthetic biologists to engineer TSR variants via precursor peptide gene modification.9 Mutagenesis10 and unnatural amino acid insertion11 have already been widely used in the molecular engineering of RiPP antibiotics. However, mutagenesis-induced modifications to the TSR's macro ring appear to frequently decrease the antibacterial activity,12 whereas changes to the quinaldic acid (QA)-containing side-ring system typically improve its biological activities.13
Focusing on the biologically relevant but tunable QA moiety, we previously conducted computer-aided molecular design and biosynthetically produced TSR-derived thiopeptide antibiotics via mutational biosyntheses.14 The obtained TSR derivatives that varied with respect to the QA moiety of the side ring not only possessed improved pharmaceutical properties14 but also exhibited a dual mode of action against intracellular pathogens (e.g., Mycobacterium marinum) that involves effects on both the host and the microbe.15 Furthermore, in our early studies on the functions of TsrB (an α/β hydrolase) and TsrC (an amidotransferase), we discovered that C-terminally methylated TSR (CmTSR, Fig. 1), an important intermediate involved in TSR biosynthesis, exhibited greater antibacterial activity (approximately 8-fold) than the parent compound TSR; this phenomenon could likely be attributed to CmTSR's better membrane permeation ability.16 In this study, given the potential synergistic effects of improving the antibacterial activity of TSR, we designed derivatives with modifications to both the side ring and the tail of TSR. These putative CmTSR compounds with regioselectively modified QA moieties in the side ring may exhibit stronger biological activities than the TSR derivatives that we have previously described.
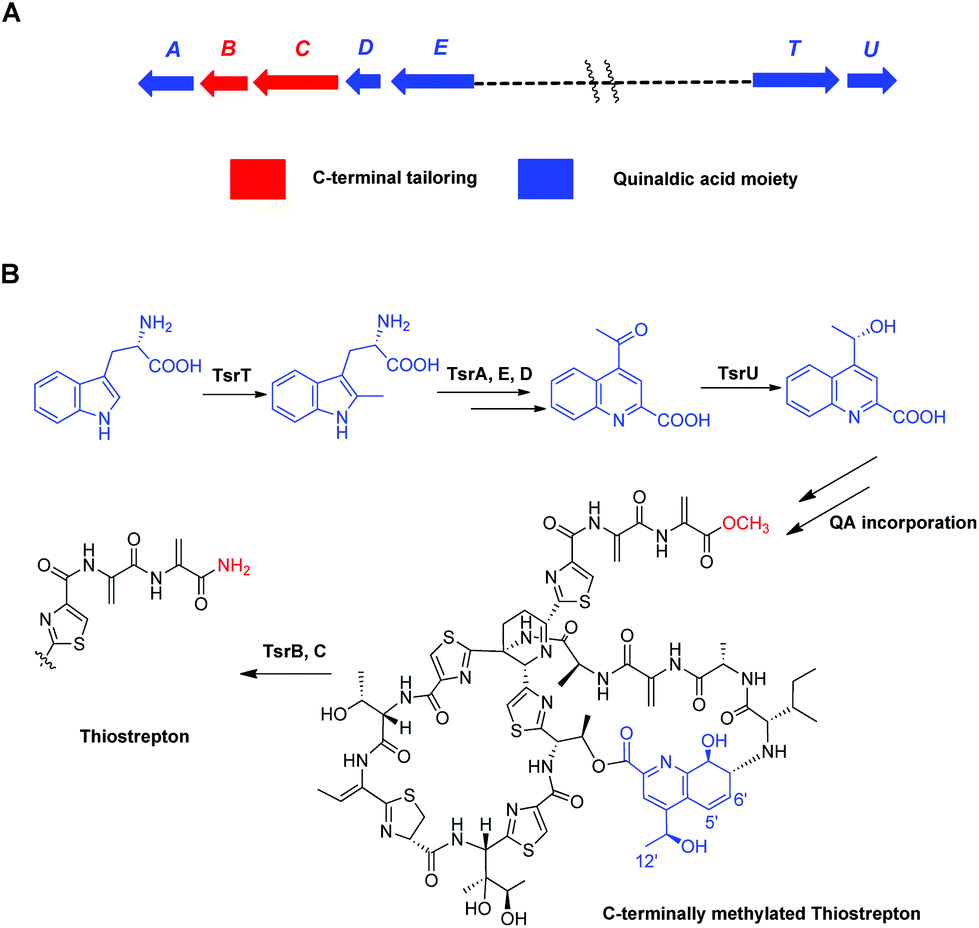 | ||
| Fig. 1 Biosynthesis of TSR and CmTSR. (A) The TSR biosynthetic gene cluster, in which the genes responsible for C-terminal tailoring and QA moiety formation are indicated in red and blue, respectively. (B) The biosynthetic pathway of the TSR QA-containing side ring and the biotransformation from CmTSR to TSR. | ||
To validate our hypothesis, we first conducted in-frame deletion of tsrT in the mutant strain Streptomyces laurentii ΔtsrB to construct the new double-mutant strain ΔtsrB/T (Fig. S3†). TsrB is a tailoring enzyme that contributes to the generation of the C-terminal amide structure during the final stage of TSR maturation, and a lack of the tsrB gene results in the production of CmTSR.16 TsrT has been described as a methyltransferase involved in the extremely early stages of the biosynthesis of QA, an important building block in the formation of TSR's side ring; the elimination of TsrT completely destroys the production of TSR.13a Thus, the newly constructed double-mutant strain ΔtsrB/T exhibited no CmTSR production, and the exogenous chemical feeding of QA restored CmTSR production (Fig. 2).
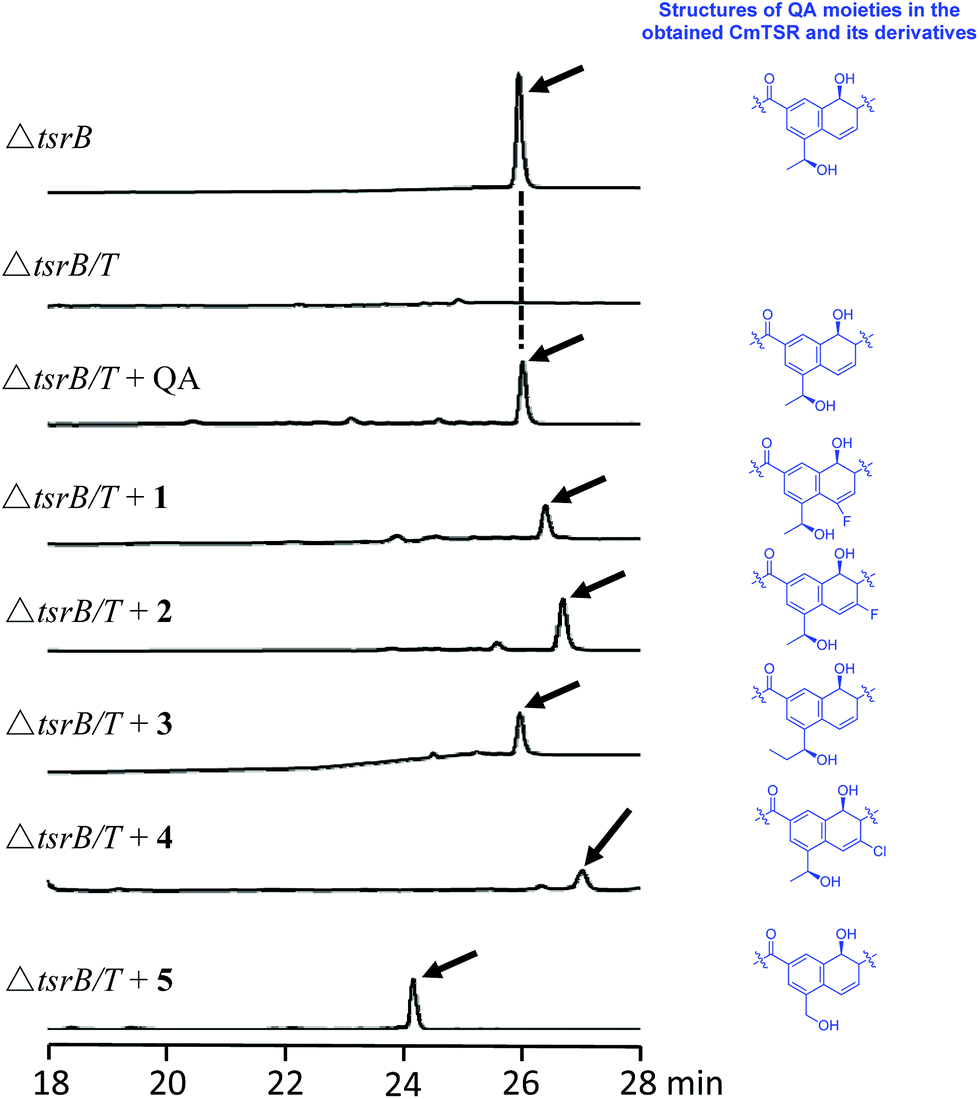 | ||
| Fig. 2 HPLC analysis of the fermentation cultures of the single mutant strain ΔtsrB, and double mutant strain ΔtsrB/T, in the absence or presence of the exogenous QA and its analogs, and the resulting CmTSR products are indicated by the arrows respectively. | ||
To generate CmTSR derivatives with regioselectively modified QA moieties, various QA analogs (1–7; Scheme 1) were synthesized using the robust protocol that we had previously developed.17 QA analogs (1–5) were fed to ΔtsrB/T during the fermentation process. Using this approach, five new CmTSR derivatives, 5′-fluoro-CmTSR (∼4 mg L−1), 6′-fluoro-CmTSR (∼10 mg L−1), 6′-chloro-CmTSR (<0.05 mg L−1), 12′-methyl-CmTSR (∼3.5 mg L−1), and 12′-de-methyl-CmTSR (2 mg L−1), were efficiently obtained (Fig. 2, Scheme 1). These derivatives were purified, and their chemical structures were further elucidated via1H, 13C, 19F and 2D NMR analyses (ESI†) in which data were compared with the corresponding data for parent compounds (TSR, CmTSR) and previously characterized TSR derivatives with various QA moieties (5′-fluoro-TSR, 6′-fluoro-TSR, and 12′-methyl-TSR). These comparisons indicated that in the tested CmTSR derivatives, the C-terminal amide of the parent compound TSR had been replaced by a methyl-esterified structure; in addition, there were regioselective modifications of the QA moieties that corresponded to ester analogs of the quinolone ketones fed to the mutant strain.
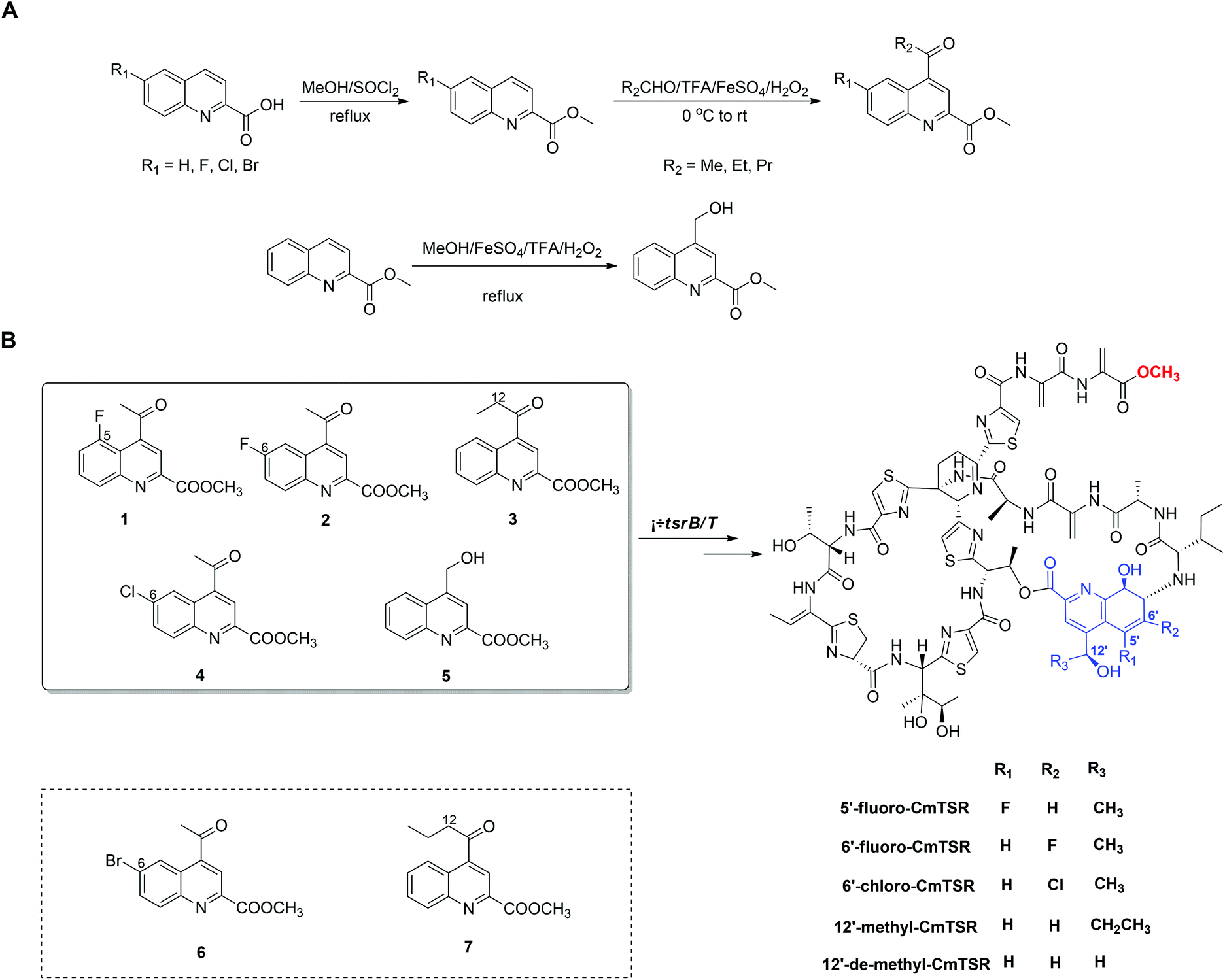 | ||
| Scheme 1 Chemical synthesis routes of the QA analogs (A) and the mutational biosynthesis strategy for generating concurrently modified CmTSR derivatives (B), in which the successfully incorporated QA analogs are shown in a black rectangle while the unsuccessfully incorporated ones are shown in a dashed rectangle. | ||
To detect differences among the antibacterial activities of these derivatives, nine clinically isolated pathogens, including methicillin-resistant Staphylococcus aureus (MRSA), penicillin-resistant Streptococcus pneumoniae (PRSP), and vancomycin-resistant Enterococcus faecium (VRE), were randomly chosen and used to test the derivatives’ minimum inhibitory concentrations (MICs) (Table 1 and S6†). The experimental results suggested that most of these derivatives exhibited greater activity than the parent compound CmTSR, and the following order of potency was observed: 5′-fluoro-CmTSR > 6′-fluoro-CmTSR > 12′-methyl-CmTSR ≥ 12′-de-methyl-CmTSR ≥ CmTSR > 6′-chloro-CmTSR. These observations were consistent with the findings from our prior in silico molecular modeling.14 The steric effect caused by a methyl group at the 12′C position and the electronic effect caused by fluorination at 5′C or 6′C increased the binding affinities between TSR compounds and target biomacromolecules. In contrast, the inductive and increased steric effects resulting from the introduction of a large chlorine atom into the CmTSR QA moiety may have destroyed the interaction between QA and A1067 of the 23S rRNA, leading to a marked reduction in the activity of 6′-chloro-CmTSR. Moreover, the doubly modified CmTSR derivatives were more potent than the corresponding singly modified molecules (TSR variants with either C-terminus or QA moiety-derived modifications); this result was consistent with the expectation of synergistic effects produced by these double modifications of TSR and revealed additional details regarding the complex SAR of TSR's side ring and tail.
| PRSP | MRSA | VRE | CD | |
|---|---|---|---|---|
| TSR | 0.001–0.008 | 0.032–0.064 | 0.032–0.064 | 0.025–0.05 |
| CmTSR | 0.000125–0.002 | 0.008 | 0.008–0.016 | 0.008–0.0125 |
| 5′-F-CmTSR | <0.000125 | 0.00025–0.0005 | 0.00025–0.0005 | 0.000125–0.0005 |
| 6′-F-CmTSR | <0.000125 | 0.0005–0.001 | 0.0005–0.001 | 0.001–0.002 |
| 12′-Me-CmTSR | <0.000125 | 0.002–0.004 | 0.008 | 0.004–0.0125 |
| 12′-de-Me-CmTSR | 0.000125–0.001 | 0.004–0.008 | 0.008–0.016 | 0.004–0.016 |
| 6′-Cl-CmTSR | 0.25–0.5 | 1.0–2.0 | 1.0 | 0.5–1.0 |
| VAN | 0.25 | 0.5–1.0 | >256 | 0.2–0.4 |
Although mutational biosynthesis has exhibited great power with respect to expanding the molecular diversity and utility of polyketide and non-ribosomal peptide natural products,18 to date, there exist only a few successful examples of mutational biosynthesis in RiPP engineering. These successes have featured the incorporation of non-amino acid building blocks, such as QA in TSR biosynthesis13a and methyl indolic acid (MIA) in nosiheptide (NOS) biosynthesis.19 Research has demonstrated that the biosynthesis of the QA moiety, which is independent of post-translational modifications to the TSR precursor peptide, is mediated by four enzymes in addition to TsrT (TsrA, TsrE, TsrD, and TsrU; Fig. 1 and S2†).13a As chemical modules, synthetic QA analogs fed into ΔtsrB/T can replace the functions of four gene modules in TSR biosynthesis (compounds 1–4 for TsrT, TsrA, TsrE, and TsrD; compound 5 for TsrA, TsrE, TsrD, and TsrU). This unusual molecular engineering strategy for generating RiPP antibiotics, which is based on mutational biosynthesis and knocking out genes that encode tailoring enzymes, greatly facilitates the expansion of TSR diversity and overcomes the limitations imposed by the substrate specificities of TsrT, TsrA, TsrE, TsrD, and TsrU. Notably, a natural 12′-de-methyl-siomycin (SIO) derivative known as siomycin D has been isolated from the SIO-producing strain Streptomyces sioyaensis (Fig. S1†).20 However, no such analogs had been found in TSR-producing strains until we conducted the aforementioned molecular engineering study; this phenomenon can most likely be attributed to the relatively strict recognition of building blocks in the TSR biosynthetic system. Overly modified QA analogs could not be utilized by the microbial cell factory; for instance, compounds 6 and 7 were not incorporated into the CmTSR architecture. Thus, our understanding of the tolerance of the TSR biosynthetic system was enhanced; in particular, we determined that the enzymes responsible for QA incorporation and the closure of TSR's side ring could tolerate fluoro- and chloro-substitution at the 6C position of QA and that the extension or shortening of a methyl group on the 12C atom of QA could also be recognized by the biosynthetic system. However, the substitution of larger halogen atoms (e.g., Br) at the 6C position of QA or the extension of an ethyl group at the 12C position of QA could not be tolerated. In fact, when compound 5 was fed to ΔtsrB/T to produce a chlorine-substituted CmTSR derivative at the 6C position of QA, a markedly reduced yield was obtained (Fig. 2).
Recent developments in drug delivery systems21 have accelerated the clinical use of thiopeptide antibiotics with large molecular weights and poor water solubilities. However, to date, LFF571, a semi-synthesized molecule generated from the natural product GE2270A via C-terminal modifications, is the only thiopeptide antibiotic undergoing phase II clinical testing for the treatment of moderate Clostridium difficile (CD) infections (CDI; NCT01232595).22 Progress towards the discovery of more potent derivatives and the development of robust large-scale production methods will renew interest in the future clinical use of molecules in the thiopeptide family. With the exception of 6′-chloro-CmTSR, all of the newly obtained CmTSR derivatives in this study exhibited greater antibacterial activity than the parent compound TSR. To the best of our knowledge, 5′-fluoro-CmTSR even exhibited greater antibacterial activity than all known natural thiopeptide antibiotics. To tap into more potential applications of our examined derivatives and compare their antibacterial activities with that of LFF571, more than ten clinically isolated CD strains were randomly selected for the determination of the MICs of these CmTSR derivatives (Table 1 and S6). The clinically utilized first-line antibiotic vancomycin (VAN) was chosen as the control drug. The experimental results indicated that the newly engineered CmTSRs exhibited more potent activities than LFF571 and VAN with respect to eliminating CD strains, suggesting that CmTSRs could serve as drug leads with great potential in the future development of clinically employed anti-infective agents.
Conclusions
In conclusion, based on the proposed synergistic effect of doubly modified TSR molecules, we constructed a new mutant strain, Streptomyces laurentii ΔtsrB/T, that has lost the ability to produce TSR. The exogenous chemical feeding of QA analogs restored the antibiotic production for this microbial cell factory and resulted in the generation of five new CmTSR derivatives with greatly increased antibacterial activities relative to the parent compound. Thus, our understanding of the tolerance for QA incorporation in the TSR biosynthetic system was enhanced. The strategy described in this paper could inspire studies in synthetic biology by encouraging the consideration of chemical modules as an approach for replacing multiple gene functions in the generation of unnatural natural products; in addition, the newly obtained CmTSR derivatives may serve as drug leads in pharmaceutical chemistry and as chemical probes in chemical biology.Acknowledgements
This work was supported in part by grants from NSFC (31430005 and 91413101), STCSM (13XD1404500 and 14JC1407700), “973 program” (2012CB721100), and MST (2012AA02A706) of China. This research was also funded in part by the Youth Innovation Foundation Fellowship, Chinese Academy of Sciences (2014228 to Dr Shoufeng Wang). We thank Prof. Heinz G. Floss, University of Washington, for providing the TSR-producing strain S. laurentii ATCC 31255.Notes and references
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Göransson, C. W. Gruber, D. H. Ha, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Müller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Sussmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108 RSC.
- M. C. Bagley, J. W. Dale, E. A. Merritt and X. Xiong, Chem. Rev., 2005, 105, 685 CrossRef CAS PubMed.
- S. K. Radhakrishnan, U. G. Bhat, D. E. Hughes, I.-C. Wang, R. H. Costa and A. L. Gartel, Cancer Res., 2006, 66, 9731 CrossRef CAS PubMed.
- M. N. Aminake, S. Schoof, L. Sologub, M. Leubner, M. Kirschner, H.-D. Arndt and G. Pradel, Antimicrob. Agents Chemother., 2011, 5, 1338 CrossRef PubMed.
- M. Ueno, S. Furukawa, F. Abe, M. Ushioda, K. Fujine, S. Johki, H. Hatori and H. Ueda, J. Antibiot., 2004, 57, 590 CrossRef CAS PubMed.
- N. S. Hegde, D. A. Sanders, R. Rodriguez and S. Balasubramanian, Nat. Chem., 2011, 3, 725 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, B. S. Safina, M. Zak, A. A. Estrada and S. H. Lee, Angew. Chem., Int. Ed., 2004, 43, 5087 CrossRef CAS PubMed; (b) K. C. Nicolaou, M. Zak, B. S. Safina, S. H. Lee and A. A. Estrada, Angew. Chem., Int. Ed., 2004, 43, 5092 CrossRef CAS PubMed; (c) K. C. Nicolaou, B. S. Safina, M. Zak, S. H. Lee, M. Nevalainen, M. Bella, A. A. Estrada, C. Funke, F. J. Zécri and S. Bulat, J. Am. Chem. Soc., 2005, 127, 11159 CrossRef CAS PubMed.
- (a) S. Schoof, S. Baumann, B. Ellinger and H.-D. Arndt, ChemBioChem, 2009, 10, 242 CrossRef CAS PubMed; (b) C. L. Myers, P. C. Hang, G. Ng, J. Yuen and J. F. Honek, Bioorg. Med. Chem., 2010, 18, 4231 CrossRef CAS PubMed; (c) S. Schoof, G. Pradel, M. N. Aminake, B. Ellinger, S. Baumann, M. Potowski, Y. Najajreh, M. Kirschner and H.-D. Arndt, Angew. Chem., Int. Ed., 2010, 49, 3317 CrossRef CAS PubMed.
- Q. Zhang and W. Liu, Nat. Prod. Rep., 2013, 30, 218 RSC.
- T. Young, P. Dorrestein and C. Walsh, Chem. Biol., 2012, 19, 1600 CrossRef CAS PubMed.
- (a) M. D. Tianero, M. S. Donia, T. S. Young, P. G. Schultz and E. W. Schmidt, J. Am. Chem. Soc., 2012, 134, 418 CrossRef CAS PubMed; (b) F. J. Piscotta, J. M. Tharp, W. R. Liu and A. J. Link, Chem. Commun., 2015, 51, 409 RSC.
- (a) C. Li, F. Zhang and W. Kelly, Mol. BioSyst., 2011, 7, 82 RSC; (b) C. Li, F. Zhang and W. Kelly, Chem. Commun., 2012, 48, 558 RSC.
- (a) L. Duan, S. Wang, R. Liao and W. Liu, Chem. Biol., 2012, 19, 443 CrossRef CAS PubMed; (b) H. Guo, J. Wang, Y. Li, Y. Yu, Q. Zheng, J. Wu and W. Liu, Chem. Sci., 2014, 5, 240 RSC.
- S. Wang, Q. Zheng, J. Wang, Z. Zhao, Q. Li, Y. Yu, R. Wang and W. Liu, Org. Chem. Front., 2015, 2, 106 RSC.
- Q. Zheng, Q. Wang, S. Wang, J. Wu, Q. Gao and W. Liu, Chem. Biol., 2015, 22, 1002 CrossRef CAS PubMed.
- R. Liao and W. Liu, J. Am. Chem. Soc., 2011, 133, 2852 CrossRef CAS PubMed.
- Q. Zheng, S. Wang and W. Liu, Tetrahedron, 2014, 70, 7686 CrossRef CAS.
- (a) S. Weist and R. D. Süssmuth, Appl. Microbiol. Biotechnol., 2005, 68, 141 CrossRef CAS PubMed; (b) Y. Yan, J. Chen, L. Zhang, Q. Zheng, Y. Han, H. Zhang, D. Zhang, T. Awakawa, I. Abe and W. Liu, Angew. Chem., Int. Ed., 2013, 52, 12308 CrossRef CAS PubMed.
- Q. Zhang, Y. Li, D. Chen, Y. Yu, L. Duan, B. Shen and W. Liu, Nat. Chem. Biol., 2011, 7, 154 CrossRef CAS PubMed.
- K. Tokura, K. Tori, Y. Yoshimura, K. Okabe, H. Otsuka, K. Matsushita, F. Inagaki and T. Miyazawa, J. Antibiot., 1980, 33, 1563 CrossRef CAS PubMed.
- (a) M. Wang and A. Gartel, Mol. Cancer Ther., 2011, 10, 2287 CrossRef CAS PubMed; (b) S. Lehar, T. Pillow, M. Xu, L. Staben, K. Kajihara, R. Vandlen, L. DePalatis, H. Raab, W. Hazenbos, J. Morisaki, J. Kim, S. Park, M. Darwish, B.-C. Lee, H. Hernandez, K. Loyet, P. Lupardus, R. Fong, D. Yan, C. Chalouni, E. Luis, Y. Khalfin, E. Plise, J. Cheong, J. Lyssikatos, M. Strandh, K. Koefoed, P. Andersen, J. Flygare, M. Tan, E. Brown and S. Mariathasan, Nature, 2015, 527, 323 CrossRef CAS PubMed.
- (a) M. J. LaMarche, J. A. Leeds, A. Amaral, J. T. Brewer, S. M. Bushell, G. Deng, J. M. Dewhurst, J. Ding, J. Dzink-Fox, G. Gamber, A. Jain, K. Lee, L. Lee, T. Lister, D. McKenney, S. Mullin, C. Osborne, D. Palestrant, M. A. Patane, E. M. Rann, M. Sachdeva, J. Shao, S. Tiamfook, A. Trzasko, L. Whitehead, A. Yifru, D. Yu, W. Yan and Q. Zhu, J. Med. Chem., 2012, 55, 2376 CrossRef CAS PubMed; (b) A. Jarrad, T. Karoli, M. Blaskovich, D. Lyras and M. Cooper, J. Med. Chem., 2015, 58, 5164 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00433k |
| ‡ These authors equally contributed to this work. |
| This journal is © the Partner Organisations 2016 |