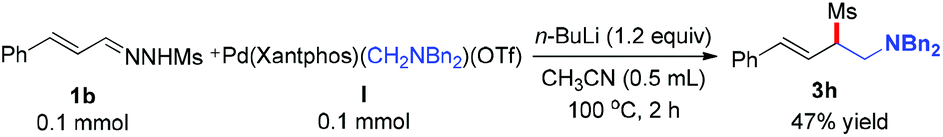DOI:
10.1039/C5QO00381D
(Research Article)
Org. Chem. Front., 2016,
3, 259-267
Palladium-catalysed coupling reaction of aminals with N-sulfonyl hydrazones to give allylic sulfones†
Received
18th November 2015
, Accepted 20th December 2015
First published on 22nd December 2015
Abstract
Palladium-catalysed cross-coupling of aminals with N-sulfonyl hydrazones has been established via C–N bond activation under base-free conditions, in which one C–C bond and one C–S bond were simultaneously generated. It was successfully applied in the construction of a variety of aminomethyl substituted allylic sulfones. Preliminary mechanistic studies indicated that the unique electrophilic cyclopalladated complex was involved in the catalytic cycle.
 Hanmin Huang | Hanmin Huang was born in Hubei, China, and completed his M.S. degree at Huazhong University of Science & Technology. He obtained his Ph.D. degree in 2003 at Dalian Institute of Chemical Physics, Chinese Academy of Sciences, under the supervision of Professor Huilin Chen and Professor Zhuo Zheng. He then moved to Nagoya University and worked as a JSPS postdoctoral research fellow with Professor Masato Kitamura. In April 2008, he initiated his independent research in Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, as a full Professor financed by the “Hundred Talent Program of CAS”. His current research interests are focused on the development of transition-metal catalyzed new reactions and understanding their mechanism. |
Introduction
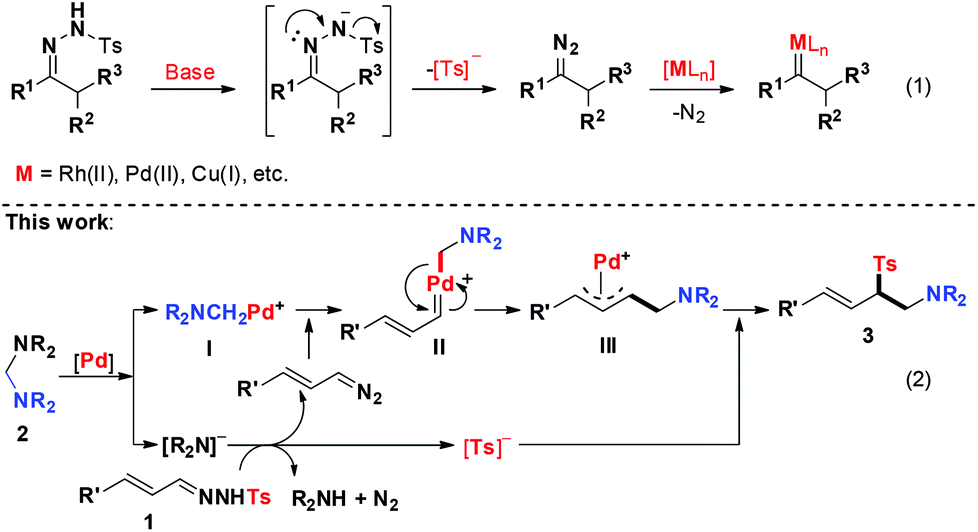
Transition-metal catalysed insertion of metal carbenes into the X–H (X = N, O, S, B, Si, etc.) bond is a powerful tool for constructing carbon–heteroatom bonds, which has been extensively explored and considerable progress has been achieved.1 In contrast, the selective insertion of metal carbenes into the C–X bond via the stepwise ylide formation mechanism remains largely elusive since the selective control of the migration of two distinct carbon moieties attached to a heteroatom is challenging.2 To circumvent this problem, we recently developed a novel palladium-catalysed protocol for the formal insertion of carbenoids into aminals via C–N bond activation, which led to the generation of α,β-diaminoacidesters with quaternary carbon-centers. Mechanistic studies disclosed that the C–N bond of aminals was selectively cleaved by the palladium catalyst to generate the cyclopalladated complex which was then trapped by diazoacetate to form the Pd–carbene complex to facilitate C–C and C–N bond formation.3
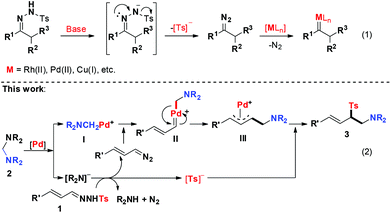
The N-sulfonyl hydrazones are easily synthesized from simple starting materials and have been widely utilized in the past few decades.4 In the presence of a strong base, the unstable diazo compound could be generated in situ and one molecule of Ts− was released from an N-sulfonyl hydrazone. In this regard, N-sulfonyl hydrazone could act as a surrogate of the diazo compound (Scheme 1, eqn (1)). Indeed, the first example of employing N-sulfonyl hydrazone as a diazo compound precursor in the palladium-catalysed cross-coupling reaction with aryl halides was discovered by Barluenga in 2007.5 This pioneering work opened a new way for the development of metal–carbene chemistry. Since then, a number of transition-metal catalysed cross-coupling reactions with N-sulfonyl hydrazones as carbene precursors have been developed.6,7 However, a stoichiometric amount of an external base was generally required in these reactions.
 |
| | Scheme 1 A new strategy for the synthesis of aminomethyl substituted allylic sulfones. | |
Inspired by these results and on the basis of our recent work in Pd-catalyzed C–N bond activation chemistry,3,8 we hypothesized that the N-sulfonyl hydrazone would act as a useful surrogate of the diazo intermediate to react with an aminal under palladium catalysis, since the released R2N− can act as a strong base to facilitate the production of the diazo intermediate, which could be further reacted with the cyclopalladated complex I (Scheme 1, eqn (2)). Herein, we report palladium-catalysed C–C and C–S bond formation reactions between aminals and N-sulfonyl hydrazones via C–N bond activation, which provides an unusual and reliable approach to aminomethyl allylic sulfones. Notably, the allylic sulfone motif exists in numerous bioactive compounds, such as antibacterial agents and herbicides.9
Results and discussion
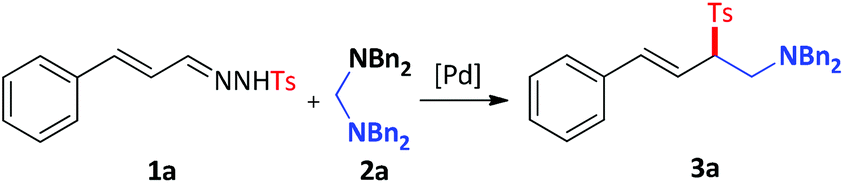
Initially, we started our investigation by employing N-tosylhydrazone 1a and N,N,N′,N′-tetrabenzylmethanediamine 2a as the model substrates (Table 1, entry 1). As might be expected, the reaction took place and gave the desired product 3a in 39% isolated yield in the presence of a catalytic amount of Pd(PPh3)4 in 1,4-dioxane at 80 °C for 12 h. With the initial result in hand, we proceeded to optimize the reaction conditions. In the beginning, the yield of 3a couldn't be improved by using different palladium catalysts (Table 1, entries 2–5). To our delight, when the cationic palladium-catalyst, Pd(DPEphos)(CH3CN)2(OTf)2, was introduced into the reaction system, the yield of 3a was promoted to 50% (Table 1, entry 6). Further screening of solvents (see the ESI†) demonstrated that strong polar organic solvents and protic solvents were failed to give satisfactory results and n-Bu2O proved to be the best solvent, giving 3a in 56% yield (entries 6–10). Since N-tosylhydrazone 1a might be decomposed fast under high temperature, the reaction temperature was tested, and we found that the yield of 3a was reduced at 60 °C (Table 1, entry 11) even after prolonged the reaction time (see ESI†). However, the desired product could be obtained in a better yield through slightly elevating the temperature (Table 1, entry 12). To further improve the efficiency of the reaction, the impact of the additive was investigated (see the ESI†). 3 Å MS emerged as the choice to give 3a in good yield (Table 1, entry 13). It's worth noting that the reaction could occur faster under higher concentration (Table 1, entry 14). Last but not least, we examined the effect of reaction time and found 10 hours was the best choice for this transformation, providing the desired product in 68% isolated yield (Table 1, entry 15).
Table 1 Optimization of reaction conditionsa
|
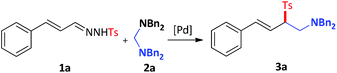
|
| Entry |
Pd |
Solvent |
Additive |
Yield (%) |
|
Reaction conditions: 1a (0.4 mmol), 2a (0.8 mmol), [Pd] (2.5 mol%), solvent (2.0 mL), 80 °C, 12 h, isolated yield.
60 °C.
100 °C.
3 Å MS (20 mg).
n-Bu2O (1.0 mL).
10 h.
|
| 1 |
Pd(PPh3)4 |
1,4-Dioxane |
— |
39 |
| 2 |
Pd(OAc)2 |
1,4-Dioxane |
— |
35 |
| 3 |
Pd(XantPhos)Cl2 |
1,4-Dioxane |
— |
40 |
| 4 |
Pd(DPEPhos)Cl2 |
1,4-Dioxane |
— |
28 |
| 5 |
Pd(XantPhos)(CH3CN)2(OTf)2 |
1,4-Dioxane |
— |
34 |
| 6 |
Pd(DPEPhos)(CH3CN)2(OTf)2 |
1,4-Dioxane |
— |
50 |
| 7 |
Pd(DPEPhos)(CH3CN)2(OTf)2 |
DMF |
— |
0 |
| 8 |
Pd(DPEPhos)(CH3CN)2(OTf)2 |
DMSO |
— |
0 |
| 9 |
Pd(DPEPhos)(CH3CN)2(OTf)2 |
MeOH |
— |
12 |
| 10 |
Pd(DPEPhos)(CH3CN)2(OTf)2 |
n-Bu2O |
— |
56 |
| 11b |
Pd(DPEPhos)(CH3CN)2(OTf)2 |
n-Bu2O |
— |
26 |
| 12c |
Pd(DPEPhos)(CH3CN)2(OTf)2 |
n-Bu2O |
— |
60 |
| 13c,d |
Pd(DPEPhos)(CH3CN)2(OTf)2 |
n-Bu2O |
3 Å MS |
63 |
| 14c,d,e |
Pd(DPEPhos)(CH3CN)2(OTf)2 |
n-Bu2O |
3 Å MS |
63 |
| 15c,d,e,f |
Pd(DPEPhos)(CH3CN)2(OTf)2 |
n-Bu2O |
3 Å MS |
68 |
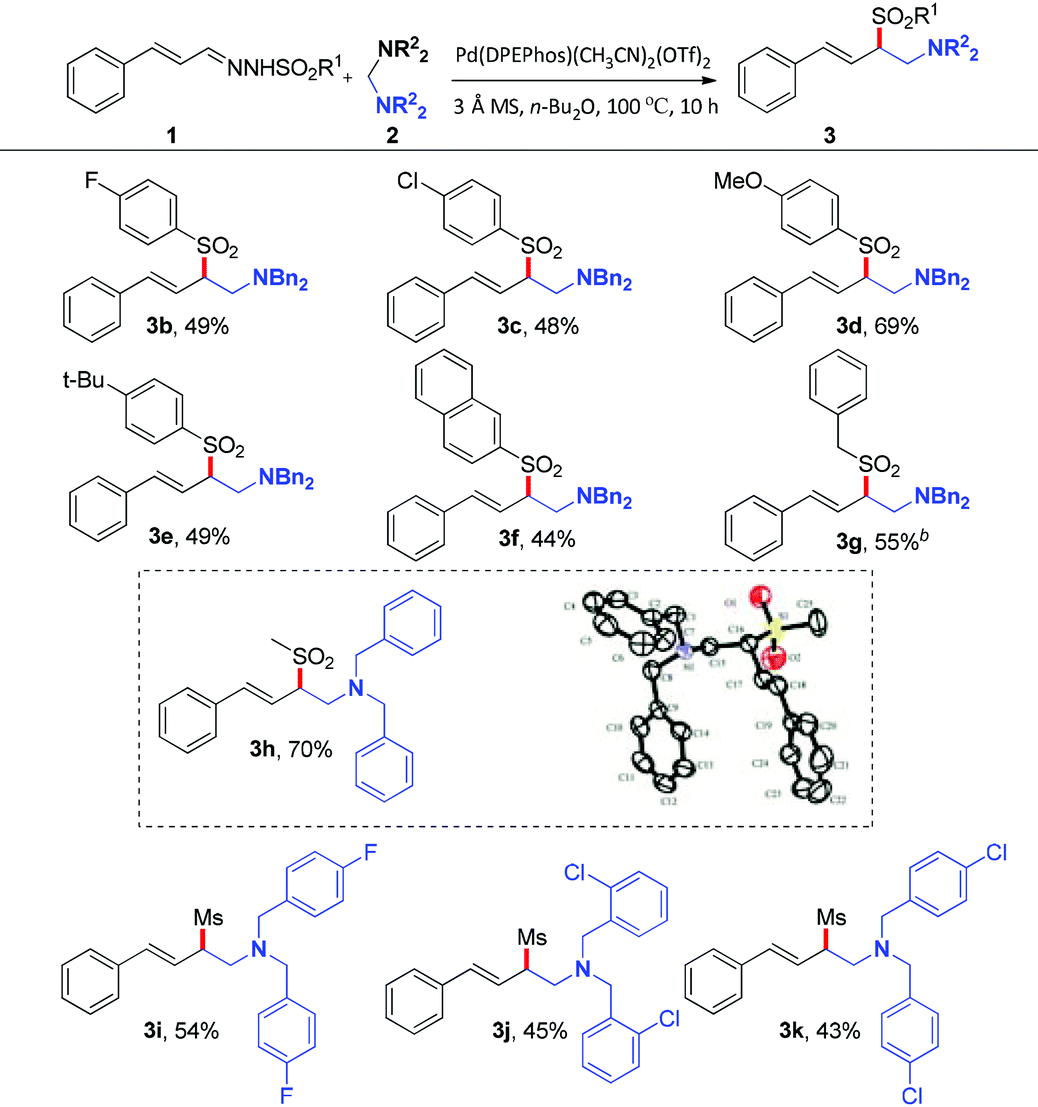
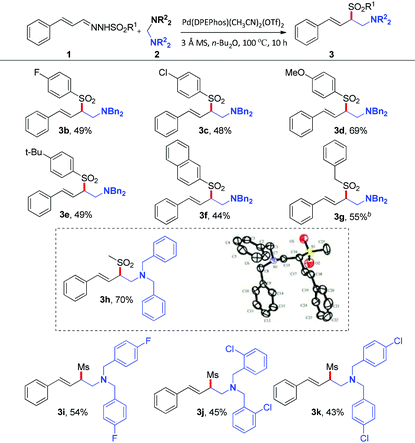
With the optimized reaction conditions in hand, the substrate scope was next investigated by employing various N-sulfonyl hydrazones and aminals. Firstly, the effect of the sulfonyl group was investigated. As illustrated in Table 2, hydrazones bearing electron-donating or electron-withdrawing groups on the phenyl ring of sulfonyl groups can be transformed into corresponding products smoothly. As might be expected, N-sulfonyl hydrazones with electron-donating groups underwent the reaction to give the desired products (3d, 3e) in 69% and 49% yields, respectively. On the contrary, relatively lower yields were obtained when substrates bear electron-withdrawing groups (3b, 3c). The reason should be assigned to the nucleophilicity of the corresponding sulfinate anions. It's interesting to note that the reaction of the N-mesylhydrazone 1h with 2a proceeded smoothly, giving the desired product 3h in 70% yield. Besides, the solid state structure of 3h was unambiguously characterized by single-crystal X-ray crystallographic analysis (see the ESI†).10 As for the aminal component, fluoro and chloro functional groups attached to the phenyl ring of the aminal can tolerate current reaction conditions to give the corresponding products in moderate yields (43–54% yields, 3i–3k).
Table 2 Substrate scope of N-sulfonyl hydrazones and aminalsa
|
Reaction conditions: 1 (0.4 mmol), 2 (0.8 mmol), Pd(DPEphos)(CH3CN)2(OTf)2 (2.5 mol%), 3 Å MS (20 mg), n-Bu2O (1.0 mL), 100 °C, 10 h, isolated yield.
60 °C, 24 h.
|
|
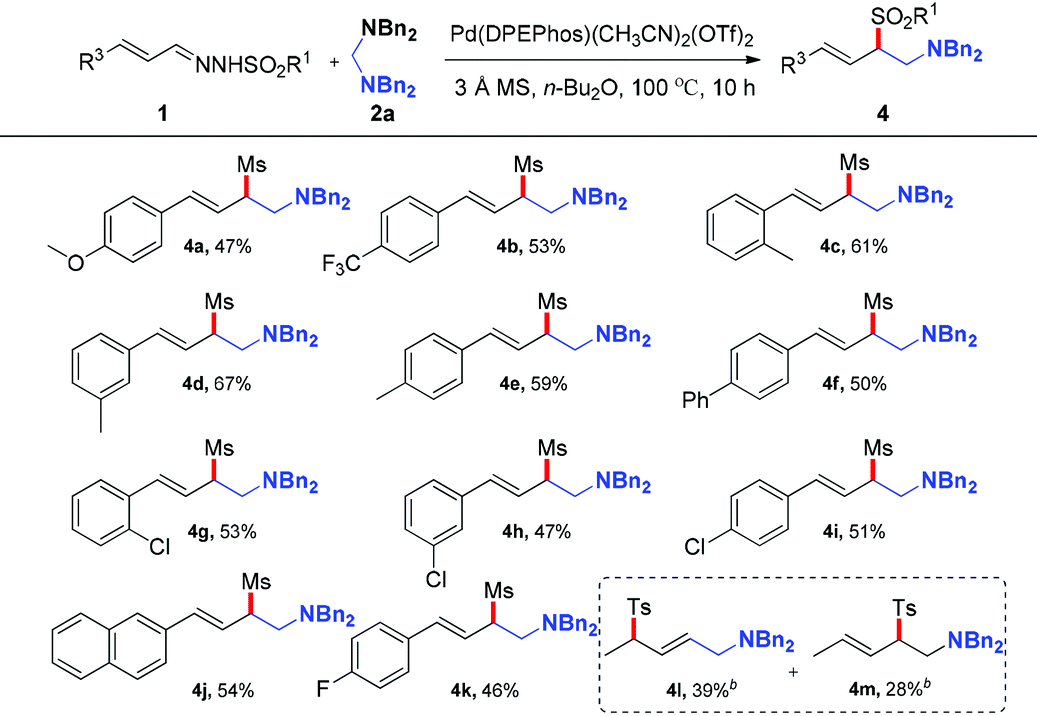
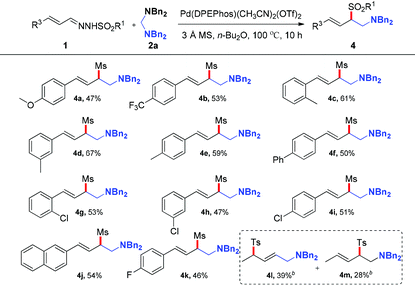
After investigating the effect of the sulfonyl group and aminals, various types of N-mesylhydrazones derived from cinnamaldehydes, which bear a variety of substituents on the phenyl ring, were also investigated in the reaction with 2a. As shown in Table 3, the reaction proceeded smoothly to give the products in moderate yields in the presence of both electron-rich and -deficient aromatic systems. Generally, hydrazones with electron-donating groups on the phenyl ring provided higher yields than those with electron-withdrawing groups. Typical functional groups such as methoxyl (4a), methyl (4c–4e), fluoride (4k) and chloride (4g–4i) were well tolerated under the optimized reaction conditions. In addition, 2-naphthyl-substituted hydrazone was also compatible with this transformation, generating the corresponding product 4j in 54% yield. After the exploration of the reaction scope of aryl hydrazones, we turned our attention to more challenging crotonaldehyde derived hydrazones. N′-((E)-but-2-enylidene)-4-methylbenzenesulfonohydrazide proceeded smoothly, producing the corresponding adducts (4l, 4m) in 39% and 28% yields with lower regioselectivity, respectively.
Table 3 Substrate scope of N-sulfonyl hydrazonesa
|
Reaction conditions: 1 (0.4 mmol), 2a (0.8 mmol), Pd(DPEphos)(CH3CN)2(OTf)2 (2.5 mol%), 3 Å MS (20 mg), n-Bu2O (1.0 mL), 100 °C, 10 h, isolated yield.
Pd(Xantphos)(CH3CN)2(OTf)2 (2.5 mol%), 1,4-dioxane (1.0 mL).
|
|
To gain insight into the mechanism of this reaction, a stoichiometric reaction of cyclopalladated complex I with N′-((E)-3-phenylallylidene)methanesulfonohydrazide 1b was conducted in CH3CN at 100 °C for 2 h in the presence of n-BuLi. This transformation provided the desired product 3h in 47% yield, suggesting that metal complex I was most likely to be involved in the catalytic cycle (Scheme 2).
 |
| | Scheme 2 Plausible mechanistic study. | |
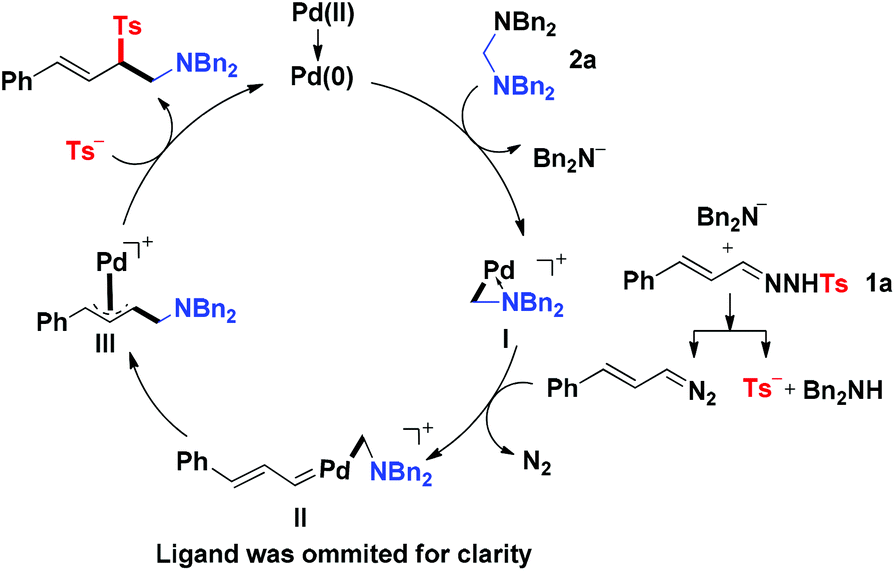
On the basis of the results described above and previous reports,3,6 a plausible mechanism of this novel reaction was proposed as the following catalytic cycle (Fig. 1). The catalyst precursor Pd(II) species were reduced to Pd(0) species, which reacted with aminal 2a through oxidative addition to produce the Pd(II) complex I and release one molecule of R2N−. The R2N− acted as a strong base to react with N-tosylhydrazone 1 to produce (E)-(3-diazoprop-1-en-1-yl)benzene together with TS−. The (E)-(3-diazoprop-1-en-1-yl)benzene was then trapped by the cyclopalladated complex I to generate the carbene intermediate II, which underwent subsequently migratory insertion of the aminomethyl group to form π-allylpalladium intermediate III. The intermediate III was then attacked by Ts− to produce the desired product and regenerate the active Pd(0) for the next catalytic cycle.
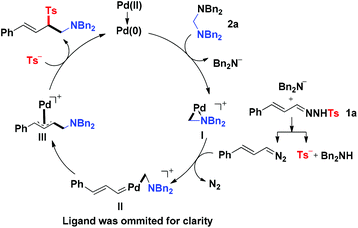 |
| | Fig. 1 Plausible reaction mechanism. | |
Conclusions
In summary, we have established an unprecedented palladium-catalysed cross-coupling of aminals with N-sulfonyl hydrazones under external base-free conditions, in which one C–C bond and one C–S bond were simultaneously constructed. It was successfully applied in the construction of a variety of aminomethyl substituted allylic sulfones under mild conditions. A series of aminals and N-sulfonyl hydrazones with different substituents were smoothly transformed to the corresponding products. Asymmetric catalysis and reactions of aminals with other types of N-sulfonyl hydrazones are currently in progress in our group.
Experimental section
General
All non-aqueous reactions and manipulations were performed by using standard Schlenk techniques. All solvents before use were dried and degassed by standard methods and stored under a nitrogen atmosphere. All reactions were monitored by TLC with silica gel-coated plates. NMR spectra were recorded on a BRUKER Avance III 400 MHz spectrometer. Chemical shifts were reported in parts per million (ppm) downfield from TMS with the solvent resonance as the internal standard. Coupling constants (J) were reported in Hz and referred to apparent peak multiplications. High resolution mass spectra (HRMS) were recorded on a Bruker MicroTOF-QII mass spectrometer (ESI). Cinnamaldehyde, crotonaldehyde, and solvents were purchased from Aldrich. Sulfonyl chlorides were purchased from Alfa Aesar. Aminals used here were known compounds and synthesized according to the reported methods.11a,11b 4-Fluorobenzenesulfonohydrazide, 4-tert-butylbenzenesulfonohydrazide and N-sulfonyl hydrazones (1c–1o) were synthesized according to the literature procedure: 4-fluorobenzenesulfonohydrazide,11c 4-tert-butylbenzenesulfonohydrazide,11c1c,11d1d, and11d1e–1o.11eN′-((1E,2E)-But-2-en-1-ylidene)-4-methylbenzenesulfonohydrazide was known compounds and was prepared according to ref. 11e. N-Sulfohydrazides used for the preparation of hydrazones were synthesized according to the literature.11f,g Substituted cinnamaldehydes used for the preparation of N-mesylhydrazones were known compounds and synthesized according to the literature.11h
General procedure for the reaction of aminals with N-sulfonyl hydrazones
N-Sulfonyl hydrazones 1 (0.4 mmol), aminals 2 (0.8 mmol), Pd(DPEPhos)(CH3CN)2(OTf)2 (0.01 mmol, 2.5 mol%), 3 Å MS (20.0 mg), and n-Bu2O (1.0 mL) were added to a 25 mL flame-dried Young-type tube under a nitrogen atmosphere. The mixture was degassed by the freeze–thaw method, and stirred at 100 °C for 10 h. After cooling to room temperature, the solvent was evaporated under reduced pressure and the residue was purified by flash chromatography on silica gel (petroleum ether/ethyl acetate = 50/1–5/1) to afford the products.
4-Fluoro-N′-((E)-3-phenylallylidene)benzenesulfonohydrazide (1c).
The title compound was prepared according to the general procedure and purified by recrystallization to give white needles (4.1 g, 72% yield). 1H NMR (400 MHz, DMSO-d6) δ 6.88 (dd, J1 = 16.0 Hz, J2 = 9.2 Hz, 1H), 7.00 (d, J = 16.0 Hz, 1H), 7.29–7.38 (m, 3H), 7.45–7.50 (m, 2H), 7.57 (d, J = 7.2 Hz, 2H), 7.75 (d, J = 8.4 Hz, 1H), 7.88–7.92 (m, 2H), 11.51 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 116.4, 116.6, 124.6, 127.1, 128.7, 128.9, 130.1, 130.2, 135.4, 135.6, 139.5, 149.7, 163.2, 165.7; 19F NMR (376 MHz, DMSO-d6) δ −105.8; HRMS (ESI) calcd for C15H13FN2O2SNa [M + Na]: 327.0574, found: 327.0567.
4-tert-Butyl-N′-((E)-3-phenylallylidene)benzenesulfonohydrazide (1d).
The title compound was prepared according to the general procedure and purified by recrystallization to give white needles (5.0 g, 81% yield). 1H NMR (400 MHz, DMSO-d6) δ 1.27 (s, 9H), 6.88 (dd, J1 = 16.0 Hz, J2 = 9.2 Hz, 1H), 6.97 (d, J = 16.0 Hz, 1H), 7.27–7.36 (m, 3H), 7.55 (d, J = 7.2 Hz, 2H), 7.64 (d, J = 8.4 Hz, 2H), 7.75(d, J = 8.8 Hz, 1H), 7.80 (d, J = 10.4 Hz, 2H), 11.48 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 35.9, 40.1, 130.0, 131.3, 132.2, 132.3, 134.0, 134.1, 140.9, 141.6, 144.4, 154.4, 161.2; HRMS (ESI) calcd for C19H23N2O2S [M + H]: 343.1475, found: 343.1457.
N′-((E)-3-(4-Methoxyphenyl)allylidene)methanesulfonohydrazide (1e).
The title compound was prepared according to the general procedure and purified by recrystallization to give white needles (3.3 g, 92% yield). 1H NMR (400 MHz, DMSO-d6) δ 3.03 (s, 3H), 3.77 (s, 3H), 6.85 (dd, J1 = 16.0 Hz, J2 = 8.8 Hz, 1H), 6.93–6.97 (m, 3H), 7.54 (d, J = 8.8 Hz, 2H), 7.78 (d, J = 9.2 Hz, 1H), 10.87 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 38.4, 55.2, 114.3, 122.5, 128.4, 128.6, 138.8, 149.4, 159.9; HRMS (ESI) calcd for C11H15N2O3S [M + H]: 255.0798, found: 255.0792.
N′-((E)-3-(4-(Trifluoromethyl)phenyl)allylidene)methanesulfonohydrazide (1f).
The title compound was prepared according to the general procedure and purified by recrystallization to give white needles (2.8 g, 95% yield). 1H NMR (400 MHz, DMSO-d6) δ 3.04 (s, 3H), 7.11–7.13 (m, 2H), 7.73 (d, J = 8.4 Hz, 2H), 7.80–7.83 (m, 3H), 11.13 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 38.6, 122.8, 125.5, 125.6, 125.6, 127.6, 127.6, 128.3, 128.7, 136.9, 139.8, 148.1; 19F NMR (376 MHz, DMSO-d6) δ −61.0; HRMS (ESI) calcd for C11H11F3N2O2SNa [M + Na]: 315.0386, found: 315.0378.
N′-((E)-3-o-Tolylallylidene)methanesulfonohydrazide (1g).
The title compound was prepared according to the general procedure and purified by recrystallization to give white needles (2.3 g, 72% yield). 1H NMR (400 MHz, DMSO-d6) δ 2.37 (s, 3H), 3.03 (s, 3H), 6.88 (dd, J1 = 16.0 Hz, J2 = 9.2 Hz, 1H), 7.20–7.25 (m, 4H), 7.65–7.67 (m, 1H), 7.85 (d, J = 9.6 Hz, 1H), 10.98 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 19.4, 38.5, 125.4, 125.8, 126.3, 128.7, 130.5, 134.4, 136.0, 136.4, 149.2; HRMS (ESI) calcd for C11H15N2O2S [M + H]: 239.0849, found: 239.0840.
N′-((E)-3-m-Tolylallylidene)methanesulfonohydrazide (1h).
The title compound was prepared according to the general procedure and purified by recrystallization to give white needles (155.0 mg, 47% yield). 1H NMR (400 MHz, DMSO-d6) δ 2.32 (s, 3H), 3.02 (s, 3H), 6.90–7.01 (m, 2H), 7.15 (d, J = 7.6 Hz, 1H), 7.25–7.29 (m, 1H), 7.38–7.41 (m, 2H), 7.79 (d, J = 8.0 Hz, 1H), 10.95 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 20.9, 38.5, 124.2, 124.7, 127.6, 128.7, 129.6, 135.6, 137.9, 139.0, 148.9; HRMS (ESI) calcd for C11H14N2O2SNa [M + Na]: 261.0668, found: 261.0665.
N′-((E)-3-p-Tolylallylidene)methanesulfonohydrazide (1i).
The title compound was prepared according to the general procedure and purified by recrystallization to give white needles (2.0 g, 56% yield). 1H NMR (400 MHz, DMSO-d6) δ 2.31 (s, 3H), 3.02 (s, 3H), 6.86–7.00 (m, 2H), 7.20 (d, J = 8.0 Hz, 2H), 7.49 (d, J = 8.0 Hz, 2H), 7.78 (d, J = 8.8 Hz, 1H), 10.92 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 20.9, 38.4, 123.8, 127.0, 129.4, 133.0, 138.5, 138.9, 149.1; HRMS (ESI) calcd for C11H14N2O2SNa [M + Na]: 261.0668, found: 261.0673.
N′-((E)-3-(Biphenyl-4-yl)allylidene)methanesulfonohydrazide (1j).
The title compound was prepared according to the general procedure and purified by recrystallization to give white needles (2.6 g, 87% yield). 1H NMR (400 MHz, DMSO-d6) δ 3.03 (s, 3H), 6.98–7.10 (m, 2H), 7.36–7.40 (m, 2H), 7.46–7.50 (m, 2H), 7.72 (d, J = 8.4 Hz, 6H), 7.82 (d, J = 8.4 Hz, 1H), 10.99 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 38.5, 124.9, 126.5, 127.0, 127.7, 129.0, 134.9, 138.4, 139.4, 140.3, 148.9; HRMS (ESI) calcd for C16H16N2O2SNa [M + Na]: 323.0825, found: 323.0818.
N′-((E)-3-(2-Chlorophenyl)allylidene)methanesulfonohydrazide (1k).
The title compound was prepared according to the general procedure and purified by recrystallization to give white needles (1.8 g, 84% yield). 1H NMR (400 MHz, DMSO-d6) δ 3.04 (s, 3H), 7.07 (dd, J1 = 16.0 Hz, J2 = 9.2 Hz, 1H), 7.28 (d, J = 16.0 Hz, 1H), 7.33–7.38 (m, 2H), 7.49–7.51 (m, 1H), 7.85–7.90 (m, 2H), 11.10 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 38.6, 127.3, 127.6, 127.9, 129.8, 130.2, 132.4, 133.3, 133.5, 148.4; HRMS (ESI) calcd for C10H11ClN2O2SNa [M + Na]: 281.0122, found: 281.0108.
N′-((E)-3-(3-Chlorophenyl)allylidene)methanesulfonohydrazide (1l).
The title compound was prepared according to the general procedure and purified by recrystallization to give white needles (2.7 g, 89% yield) 1H NMR (400 MHz, DMSO-d6) δ 3.03 (s, 3H), 6.98–7.09 (m, 2H), 7.36–7.43 (m, 2H), 7.59 (d, J = 7.2 Hz, 1H), 7.69 (s, 1H), 7.78 (d, J = 8.4 Hz, 1H), 11.06 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 38.6, 125.5, 126.5, 126.7, 128.4, 130.6, 133.6, 137.2, 138.0, 148.3; HRMS (ESI) calcd for C10H11ClN2O2SNa [M + Na]: 281.0122, found: 281.0110.
N′-((E)-3-(4-Chlorophenyl)allylidene)methanesulfonohydrazide (1m).
The title compound was prepared according to the general procedure and purified by recrystallization to give white needles (2.8 g, 95% yield). 1H NMR (400 MHz, DMSO-d6) δ 3.03 (s, 3H), 6.95–7.05 (m, 2H), 7.45 (d, J = 8.4 Hz, 2H), 7.64 (d, J = 8.8 Hz, 2H), 7.76–7.78 (m, 1H), 11.02 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 38.5, 125.7, 128.7, 128.8, 133.2, 134.7, 137.4, 148.5; HRMS (ESI) calcd for C10H11ClN2O2SNa [M + Na]: 281.0122, found: 281.0114.
N′-((E)-3-(Naphthalen-2-yl)allylidene)methanesulfonohydrazide (1n).
The title compound was prepared according to the general procedure and purified by recrystallization to give white needles (2.9 g, 92% yield). 1H NMR (400 MHz, DMSO-d6) δ 3.05 (s, 3H), 7.08–7.21 (m, 2H), 7.50–7.55 (m, 2H), 7.83–7.92 (m, 5H), 8.03 (s, 1H), 11.02 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 38.5, 123.6, 125.3, 126.6, 127.6, 128.1, 128.4, 133.0, 133.4, 138.9, 148.9; HRMS (ESI) calcd for C14H15N2O2S [M + H]: 275.0849, found: 275.0835.
N′-((E)-3-(4-Fluorophenyl)allylidene)methanesulfonohydrazide (1o).
The title compound was prepared according to the general procedure and purified by recrystallization to give white needles (2.1 g, 82% yield). 1H NMR (400 MHz, DMSO-d6) δ 3.02 (s, 3H), 6.90–7.04 (m, 2H), 7.20–7.24 (m, 2H), 7.64–7.68 (m, 2H), 7.78 (d, J = 8.8 Hz, 1H), 10.97 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 38.5, 115.6, 115.8, 124.7, 124.7, 129.1, 129.2, 132.4, 132.4, 137.7, 148.8, 161.1, 163.5; 19F NMR (376 MHz, DMSO-d6) δ −112.3; HRMS (ESI) calcd for C10H11FN2O2SNa [M + Na]: 265.0417, found: 265.0407.
(E)-N,N-Dibenzyl-4-phenyl-2-tosylbut-3-en-1-amine (3a).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (130.5 mg, 68% yield). 1H NMR (400 MHz, CDCl3) δ 2.40 (s, 3H), 2.97–3.03 (m, 1H), 3.20 (dd, J1 = 13.2 Hz, J2 = 3.2 Hz, 1H), 3.45 (d, J = 13.6 Hz, 2H), 3.70–3.79 (m, 3H), 5.73 (dd, J1 = 16.0 Hz, J2 = 9.6 Hz, 1H), 6.19 (d, J = 16.0 Hz, 1H), 7.20–7.36 (m, 17H), 7.58 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 21.7, 51.7, 59.1, 67.9, 121.6, 126.6, 127.2, 128.2, 128.3, 128.6, 129.0, 129.0, 129.5, 134.7, 136.2, 137.9, 138.8, 144.5; HRMS (ESI) calcd for C31H32NO2S [M + H]: 482.2148, found: 482.2151.
(E)-N,N-Dibenzyl-2-(4-fluorophenylsulfonyl)-4-phenylbut-3-en-1-amine (3b).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (94.0 mg, 49% yield). 1H NMR (400 MHz, CDCl3) δ 2.97–3.03 (m, 1H), 3.22 (dd, J1 = 13.2 Hz, J2 = 3.6 Hz, 1H), 3.48 (d, J = 13.6 Hz, 2H), 3.70–3.76 (m, 3H), 5.70 (dd, J1 = 16.0 Hz, J2 = 9.6 Hz, 1H), 6.16 (d, J = 16.0 Hz, 1H), 7.08–7.12 (m, 2H), 7.21–7.24 (m, 11H), 7.28–7.37 (m, 4H), 7.70 (dd, J1 = 8.8 Hz, J2 = 5.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 51.5, 59.2, 68.1, 116.0, 116.2, 121.2, 126.6, 127.2, 128.3, 128.4, 128.7, 129.0, 131.8, 131.9, 135.9, 138.2, 138.7; 19F NMR (376 MHz, DMSO-d6) δ −103.6; HRMS (ESI) calcd for C30H29FNO2S [M + H]: 486.1898, found: 486.1886.
(E)-N,N-Dibenzyl-2-(4-chlorophenylsulfonyl)-4-phenylbut-3-en-1-amine (3c).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (96.4 mg, 48% yield). 1H NMR (400 MHz, CDCl3) δ 2.96–3.02 (m, 1H), 3.20 (dd, J1 = 13.2 Hz, J2 = 3.6 Hz, 1H), 3.48 (d, J = 13.6 Hz, 2H), 3.71–3.76 (m, 3H), 5.70 (dd, J1 = 16.0 Hz, J2 = 9.6 Hz, 1H), 6.19 (d, J = 16.0 Hz, 1H), 7.23–7.26 (m, 12H), 7.31–7.35 (m, 3H), 7.41 (d, J = 8.8 Hz, 2H), 7.61 (d, J = 8.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 51.5, 59.2, 68.0, 121.0, 126.6, 127.3, 128.3, 128.4, 128.7, 129.0, 129.1, 130.5, 135.9, 136.1, 138.3, 138.6, 140.4; HRMS (ESI) calcd for C30H29ClNO2S [M + H]: 502.1602, found: 502.1597.
(E)-N,N-Dibenzyl-2-(4-methoxyphenylsulfonyl)-4-phenylbut-3-en-1-amine (3d).
The title compound was prepared according to the general procedure and purified by column chromatography to give a white solid (137.2 mg, 69% yield). 1H NMR (400 MHz, CDCl3) δ 2.97–3.03 (m, 1H), 3.20 (dd, J1 = 13.2 Hz, J2 = 3.6 Hz, 1H), 3.45 (d, J = 13.6 Hz, 2H), 3.71–3.78 (m, 3H), 3.84 (s, 3H), 5.74 (dd, J1 = 16.0 Hz, J2 = 9.6 Hz, 1H), 6.19 (d, J = 15.6 Hz, 1H), 6.89 (d, J = 9.2 Hz, 2H), 7.20–7.25 (m, 12H), 7.29–7.36 (m, 3H), 7.62 (d, J = 8.8 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 51.7, 55.6, 59.1, 68.1, 114.0, 121.7, 126.6, 127.2, 128.2, 128.3, 128.6, 129.0, 129.2, 131.2, 136.2, 137.8, 138.8, 163.6; HRMS (ESI) calcd for C31H32NO3S [M + H]: 498.2097, found: 498.2093.
(E)-N,N-Dibenzyl-2-(4-tert-butylphenylsulfonyl)-4-phenylbut-3-en-1-amine (3e).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (103.0 mg, 49% yield). 1H NMR (400 MHz, CDCl3) δ 1.31 (s, 9H), 2.99–3.05 (m, 1H), 3.21 (dd, J1 = 13.2 Hz, J2 = 3.2 Hz, 1H), 3.45 (d, J = 13.6 Hz, 2H), 3.72–3.82 (m, 3H), 5.72 (dd, J1 = 16.0 Hz, J2 = 9.6 Hz, 1H), 6.14 (d, J = 16.0 Hz, 1H), 7.18–7.25 (m, 11H), 7.28–7.35 (m, 4H), 7.44 (d, J = 8.4 Hz, 2H), 7.60–7.62 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 31.1, 35.2, 51.6, 59.0, 68.0, 121.6, 125.8, 126.6, 127.2, 128.2, 128.3, 128.6, 128.9, 129.0, 134.5, 136.2, 137.9, 138.8, 157.5; HRMS (ESI) calcd for C34H38NO2S [M + H]: 524.2618, found: 524.2610.
(E)-N,N-Dibenzyl-2-(naphthalen-2-ylsulfonyl)-4-phenylbut-3-en-1-amine (3f).
The title compound was prepared according to the general procedure and purified by column chromatography to give a white solid (92.0 mg, 44% yield). 1H NMR (400 MHz, CDCl3) δ 3.02–3.07 (m, 1H), 3.25 (dd, J1 = 13.2 Hz, J2 = 3.2 Hz, 1H), 3.47 (d, J = 13.6 Hz, 2H), 3.73 (d, J = 13.2 Hz, 2H), 3.82–3.88 (m, 1H), 5.78 (dd, J1 = 15.6 Hz, J2 = 9.2 Hz, 1H), 6.20 (d, J = 16.0 Hz, 1H), 7.15–7.20 (m, 12H), 7.25–7.33 (m, 3H), 7.57–7.61 (m, 1H), 7.64–7.68 (m, 2H), 7.86–7.90 (m, 3H), 8.28 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 51.8, 59.1, 68.0, 121.3, 123.7, 126.6, 127.2, 127.5, 127.9, 128.2, 128.6, 128.9, 128.9, 129.2, 129.5, 130.9, 132.0, 134.7, 135.3, 136.1, 138.2, 138.7; HRMS (ESI) calcd for C34H32NO2S [M + H]: 518.2148, found: 518.2134.
(E)-N,N-Dibenzyl-2-(benzylsulfonyl)-4-phenylbut-3-en-1-amine (3g).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (106.0 mg, 55% yield). 1H NMR (400 MHz, CDCl3) δ 3.00–3.05 (m, 1H), 3.21 (dd, J1 = 13.2 Hz, J2 = 3.6 Hz, 1H), 3.46 (d, J = 13.6 Hz, 2H), 3.67–3.73 (m, 3H), 4.11 (d, J = 14.0 Hz, 1H), 4.21 (d, J = 14.0 Hz, 1H), 5.93 (dd, J1 = 16.0 Hz, J2 = 9.6 Hz, 1H), 6.45 (d, J = 16.0 Hz, 1H), 7.19–7.26 (m, 9H), 7.29–7.43 (m, 11H); 13C NMR (100 MHz, CDCl3) δ 51.1, 57.5, 59.1, 64.6, 121.8, 126.8, 127.2, 127.3, 128.3, 128.6, 128.8, 128.9, 129.0, 130.9, 135.8, 138.1, 138.7; HRMS (ESI) calcd for C31H32NO2S [M + H]: 482.2148, found: 482.2148.
(E)-N,N-Dibenzyl-2-(methylsulfonyl)-4-phenylbut-3-en-1-amine (3h).
The title compound was prepared according to the general procedure and purified by column chromatography to give a white solid (113.5 mg, 70% yield). 1H NMR (400 MHz, CDCl3) δ 2.75 (s, 3H), 3.00–3.06 (m, 1H), 3.27 (dd, J1 = 13.2 Hz, J2 = 4.0 Hz, 1H), 3.53 (d, J = 13.6 Hz, 2H), 3.70–3.76 (m, 1H), 3.81 (d, J = 13.6 Hz, 2H), 5.94 (dd, J1 = 16.0 Hz, J2 = 9.6 Hz, 1H), 6.58 (d, J = 16.0 Hz, 1H), 7.24–7.33 (m, 11H), 7.38–7.39 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 39.1, 51.2, 59.2, 67.0, 121.7, 126.8, 127.3, 128.4, 128.6, 128.8, 129.0, 135.7, 138.0, 138.7; HRMS (ESI) calcd for C25H28NO2S [M + H]: 406.1835, found: 406.1842.
(E)-N,N-Bis(4-fluorobenzyl)-2-(methylsulfonyl)-4-phenylbut-3-en-1-amine (3i).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (95.0 mg, 54% yield). 1H NMR (400 MHz, CDCl3) δ 2.78 (s, 3H), 2.97–3.03 (m, 1H), 3.27 (dd, J1 = 13.6 Hz, J2 = 3.6 Hz, 1H), 3.48 (d, J = 13.6 Hz, 2H), 3.71–3.78 (m, 3H), 5.92 (dd, J1 = 16.0 Hz, J2 = 9.6 Hz, 1H), 6.59 (d, J = 15.6 Hz, 1H), 6.92–6.96 (m, 4H), 7.22–7.26 (m, 4H), 7.33–7.42 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 39.1, 50.7, 58.2, 66.9, 115.1, 115.3, 121.8, 126.7, 128.8, 128.9, 130.4, 130.5, 134.2, 134.2, 135.5, 137.9, 160.9, 163.3; 19F NMR (376 MHz, CDCl3) δ −115.2; HRMS (ESI) calcd for C25H26F2NO2S [M + H]: 442.1647, found: 442.1650.
(E)-N,N-Bis(2-chlorobenzyl)-2-(methylsulfonyl)-4-phenylbut-3-en-1-amine (3j).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (85.0 mg, 45% yield). 1H NMR (400 MHz, CDCl3) δ 2.77 (s, 3H), 3.03–3.08 (m, 1H), 3.42 (dd, J1 = 13.2 Hz, J2 = 3.2 Hz, 1H), 3.76–3.84 (m, 5H), 5.94 (dd, J1 = 16.0 Hz, J2 = 10.0 Hz, 1H), 6.57 (d, J = 16.0 Hz, 1H), 7.10–7.18 (m, 4H), 7.25–7.38 (m, 7H), 7.46 (dd, J1 = 7.2 Hz, J2 = 1.6 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 39.1, 51.9, 56.4, 67.0, 121.4, 126.7, 126.8, 128.5, 128.6, 128.7, 129.6, 131.2, 134.3, 135.7, 135.9, 138.3; HRMS (ESI) calcd for C25H26Cl2NO2S [M + H]: 474.1056, found: 474.1047.
(E)-N,N-Bis(4-chlorobenzyl)-2-(methylsulfonyl)-4-phenylbut-3-en-1-amine (3k).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (81.0 mg, 43% yield). 1H NMR (400 MHz, CDCl3) δ 2.78 (s, 3H), 2.96–3.02 (m, 1H), 3.27 (dd, J1 = 13.2 Hz, J2 = 3.6 Hz, 1H), 3.48 (d, J = 13.6 Hz, 2H), 3.70–3.78 (m, 3H), 5.90 (dd, J1 = 15.6 Hz, J2 = 9.6 Hz, 1H), 6.58 (d, J = 16.0 Hz, 1H), 7.19–7.26 (m, 8H), 7.34–7.43 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 39.1, 50.8, 58.3, 66.9, 121.8, 126.7, 128.6, 128.9, 130.2, 133.1, 135.4, 137.0, 138.0; HRMS (ESI) calcd for C25H26Cl2NO2S [M + H]: 474.1056, found: 474.1060.
(E)-N,N-Dibenzyl-4-(4-methoxyphenyl)-2-(methylsulfonyl)but-3-en-1-amine (4a).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (82.0 mg, 47% yield). 1H NMR (400 MHz, CDCl3) δ 2.75 (s, 3H), 2.98–3.04 (m, 1H), 3.26 (dd, J1 = 13.2 Hz, J2 = 3.6 Hz, 1H), 3.52 (d, J = 13.6 Hz, 2H), 3.69–3.75 (m, 1H), 3.80 (d, J = 13.6 Hz, 2H), 3.85 (s, 3H), 5.79 (dd, J1 = 15.6 Hz, J2 = 9.6 Hz, 1H), 6.53 (d, J = 15.6 Hz, 1H), 6.92 (d, J = 8.8 Hz, 2H), 7.23–7.33 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 39.1, 51.1, 55.4, 59.1, 67.1, 114.1, 119.3, 127.3, 128.1, 128.4, 128.5, 129.0, 137.4, 138.7, 160.0; HRMS (ESI) calcd for C26H30NO3S [M + H]: 436.1941, found: 436.1942.
(E)-N,N-Dibenzyl-2-(methylsulfonyl)-4-(4-(trifluoromethyl)phenyl)but-3-en-1-amine (4b).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (100.0 mg, 53% yield). 1H NMR (400 MHz, CDCl3) δ 2.75 (s, 3H), 3.02–3.07 (m, 1H), 3.28 (dd, J1 = 13.6 Hz, J2 = 4.0 Hz, 1H), 3.54 (d, J = 13.2 Hz, 1H), 3.69–3.75 (m, 1H), 3.81 (d, J = 13.6 Hz, 2H), 6.01 (dd, J1 = 15.6 Hz, J2 = 9.6 Hz, 1H), 6.58 (d, J = 16.0 Hz, 1H), 7.26–7.30 (m, 10H), 7.46 (d, J = 8.4 Hz, 2H), 7.65 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 39.2, 51.3, 59.2, 66.9, 115.6, 115.9, 121.3, 121.3, 127.4, 128.3, 128.4, 128.4, 129.0, 131.9, 136.7, 138.6, 161.7, 164.1; 19F NMR (376 MHz, CDCl3) δ −112.6; HRMS (ESI) calcd for C26H27F3NO2S [M + H]: 474.1709, found: 474.1700.
(E)-N,N-Dibenzyl-2-(methylsulfonyl)-4-o-tolylbut-3-en-1-amine (4c).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (101.0 mg, 61% yield). 1H NMR (400 MHz, CDCl3) δ 2.39 (s, 3H), 2.75 (s, 3H), 3.00–3.06 (m, 1H), 3.27 (dd, J1 = 13.2 Hz, J2 = 3.6 Hz, 1H), 3.53 (d, J = 13.6 Hz, 2H), 3.70–3.81 (m, 3H), 5.92 (dd, J1 = 15.6 Hz, J2 = 9.6 Hz, 1H), 6.56 (d, J = 15.6 Hz, 1H), 7.14–7.20 (m, 3H), 7.24–7.32 (m, 11H); 13C NMR (100 MHz, CDCl3) δ 19.8, 39.1, 51.4, 59.2, 67.3, 123.1, 126.0, 126.3, 127.3, 128.4, 128.5, 129.0, 130.5, 134.8, 135.6, 135.9, 138.7; HRMS (ESI) calcd for C26H30NO2S [M + H]: 420.1992, found: 420.1985.
(E)-N,N-Dibenzyl-2-(methylsulfonyl)-4-m-tolylbut-3-en-1-amine (4d).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (112.0 mg, 67% yield). 1H NMR (400 MHz, CDCl3) δ 2.37 (s, 3H), 2.76 (s, 3H), 3.01–3.06 (m, 1H), 3.28 (dd, J1 = 13.2 Hz, J2 = 3.6 Hz, 1H), 3.54 (d, J = 13.6 Hz, 2H), 3.71–3.81 (m, 3H), 5.84 (dd, J1 = 15.6 Hz, J2 = 9.6 Hz, 1H), 6.86 (d, J = 15.6 Hz, 1H), 7.20–7.28 (m, 9H), 7.28–7.33 (m, 4H), 7.41–7.43 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 21.4, 39.1, 51.1, 59.2, 67.0, 121.6, 124.1, 127.3, 127.4, 128.4, 128.7, 129.0, 129.4, 135.6, 138.0, 138.4, 138.7; HRMS (ESI) calcd for C26H30NO2S [M + H]: 420.1992, found: 420.1982.
(E)-N,N-Dibenzyl-2-(methylsulfonyl)-4-p-tolylbut-3-en-1-amine (4e).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (99.0 mg, 59% yield). 1H NMR (400 MHz, CDCl3) δ 2.38 (s, 3H), 2.74 (s, 3H), 2.99–3.05 (m, 1H), 3.26 (dd, J1 = 13.2 Hz, J2 = 3.6 Hz, 1H), 3.52 (d, J = 13.2 Hz, 2H), 3.70–3.80 (m, 3H), 5.89 (dd, J1 = 15.6 Hz, J2 = 9.6 Hz, 1H), 6.55 (d, J = 16.0 Hz, 1H), 7.18–7.25 (m, 2H), 7.27–7.32 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 21.3, 39.1, 51.1, 59.1, 67.0, 120.7, 126.7, 127.3, 128.4, 129.0, 129.5, 133.0, 137.9, 138.7, 138.7; HRMS (ESI) calcd for C26H30NO2S [M + H]:420.1992, found: 420.1990.
(E)-N,N-Dibenzyl-4-(biphenyl-4-yl)-2-(methylsulfonyl)but-3-en-1-amine (4f).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (95.0 mg, 50% yield). 1H NMR (400 MHz, CDCl3) δ 2.77 (s, 3H), 3.02–3.08 (m, 1H), 3.29 (dd, J1 = 13.6 Hz, J2 = 4.0 Hz, 1H), 3.54 (d, J = 13.6 Hz, 2H), 3.73–3.82 (m, 3H), 5.98 (dd, J1 = 16.0 Hz, J2 = 9.6 Hz, 1H), 6.62 (d, J = 16.0 Hz, 1H), 7.25–7.40 (m, 11H), 7.45–7.49 (m, 4H), 7.62–7.65 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 39.2, 51.2, 59.2, 67.1, 121.7, 127.0, 127.2, 127.3, 127.4, 127.6, 128.4, 128.9, 129.0, 134.7, 137.5, 138.7, 140.4, 141.4; HRMS (ESI) calcd for C31H32NO2S [M + H]: 482.2148, found: 482.2129.
(E)-N,N-Dibenzyl-4-(2-chlorophenyl)-2-(methylsulfonyl)but-3-en-1-amine (4g).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (94.0 mg, 53% yield). 1H NMR (400 MHz, CDCl3) δ 2.78 (s, 3H), 3.01–3.07 (m, 1H), 3.29 (dd, J1 = 13.2 Hz, J2 = 3.6 Hz, 1H), 3.54 (d, J = 13.6 Hz, 2H), 3.70–3.76 (m, 1H), 3.81 (d, J = 13.6 Hz, 2H), 5.87 (dd, J1 = 15.6 Hz, J2 = 9.6 Hz, 1H), 7.04 (d, J = 15.6 Hz, 1H), 7.22–7.26 (m, 4H), 7.27–7.32 (m, 8H), 7.40–7.47 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 39.3, 51.0, 59.4, 67.0, 125.1, 127.1, 127.2, 127.3, 128.4, 129.1, 129.6, 129.8, 133.2, 133.9, 134.1, 138.6; HRMS (ESI) calcd for C25H27ClNO2S [M + H]: 440.1446, found: 440.1433.
(E)-N,N-Dibenzyl-4-(3-chlorophenyl)-2-(methylsulfonyl)but-3-en-1-amine (4h).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (82.0 mg, 47% yield). 1H NMR (400 MHz, CDCl3) δ 2.74 (s, 3H), 3.00–3.05 (m, 1H), 3.27 (dd, J1 = 13.2 Hz, J2 = 3.6 Hz, 1H), 3.53 (d, J = 13.6 Hz, 2H), 3.65–3.71 (m, 1H), 3.80 (d, J = 13.6 Hz, 2H), 5.89 (dd, J1 = 16.0 Hz, J2 = 9.6 Hz, 1H), 6.49 (d, J = 16.0 Hz, 1H), 7.21–7.23 (m, 1H), 7.25–7.31 (m, 12H), 7.34 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 39.2, 51.3, 59.4, 66.8, 123.3, 125.0, 126.6, 127.4, 128.4, 128.5, 129.1, 130.0, 134.8, 136.4, 137.5, 138.6; HRMS (ESI) calcd for C25H27ClNO2S [M + H]: 440.1446, found: 440.1445.
(E)-N,N-Dibenzyl-4-(4-chlorophenyl)-2-(methylsulfonyl)but-3-en-1-amine (4i).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (89.0 mg, 51% yield). 1H NMR (400 MHz, CDCl3) δ 2.75 (s, 3H), 2.99–3.05 (m, 1H), 3.26 (dd, J1 = 13.2 Hz, J2 = 4.0 Hz, 1H), 3.53 (d, J = 13.6 Hz, 2H), 3.67–3.80 (m, 3H), 5.89 (dd, J1 = 16.0 Hz, J2 = 9.6 Hz, 1H), 6.51 (d, J = 15.6 Hz, 1H), 7.22–7.27 (m, 3H), 7.28–7.30 (m, 9H), 7.34–7.36 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 39.2, 51.3, 59.3, 66.9, 122.3, 127.4, 127.9, 128.4, 129.0, 129.0, 134.2, 134.4, 136.6, 138.6; HRMS (ESI) calcd for C25H27ClNO2S [M + H]: 440.1446, found: 440.1442.
(E)-N,N-Dibenzyl-2-(methylsulfonyl)-4-(naphthalen-2-yl)but-3-en-1-amine (4j).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (98.0 mg, 54% yield). 1H NMR (400 MHz, CDCl3) δ 2.78 (s, 3H), 3.05–3.11 (m, 1H), 3.30 (dd, J1 = 13.6 Hz, J2 = 4.0 Hz, 1H), 3.54 (d, J = 13.6 Hz, 2H), 3.76–3.83 (m, 3H), 6.05 (dd, J1 = 16.0 Hz, J2 = 9.6 Hz, 1H), 6.74 (d, J = 16.0 Hz, 1H), 7.22–7.29 (m, 6H), 7.31–7.33 (m, 4H), 7.48–7.57 (m, 3H), 7.75 (s, 1H), 7.84–7.86 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 39.2, 51.2, 59.2, 67.1, 122.1, 123.5, 126.5, 126.6, 127.2, 127.3, 127.8, 128.2, 128.4, 128.5, 129.0, 133.2, 133.4, 133.5, 138.0, 138.7; HRMS (ESI) calcd for C29H30NO2S [M + H]: 456.1992, found: 456.1997.
(E)-N,N-Dibenzyl-4-(4-fluorophenyl)-2-(methylsulfonyl)but-3-en-1-amine (4k).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (77.0 mg, 46% yield). 1H NMR (400 MHz, CDCl3) δ 2.75 (s, 3H), 2.99–3.04 (m, 1H), 3.27 (dd, J1 = 13.2 Hz, J2 = 3.6 Hz, 1H), 3.53 (d, J = 13.6 Hz, 2H), 3.67–3.73 (m, 1H), 3.80 (d, J = 13.6 Hz, 2H), 5.83 (dd, J1 = 16.0 Hz, J2 = 9.6 Hz, 1H), 6.53 (d, J = 16.0 Hz, 1H), 7.05–7.09 (m, 2H), 7.24–7.27 (m, 3H), 7.28–7.35 (m, 9H); 13C NMR (100 MHz, CDCl3) δ 39.2, 51.3, 59.2, 66.9, 115.6, 115.9, 121.3, 121.3, 127.4, 128.3, 128.4, 128.4, 129.0, 131.9, 136.7, 138.6, 161.7, 164.1; 19F NMR (376 MHz, CDCl3) δ −112.6; HRMS (ESI) calcd for C25H27FNO2S [M + H]: 424.1741, found: 424.1732.
(E)-N,N-Dibenzyl-4-tosylpent-2-en-1-amine (4l).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (65.0 mg, 39% yield). 1H NMR (400 MHz, CDCl3) δ 1.43 (d, J = 6.8 Hz, 3H), 2.22 (s, 3H), 2.89–3.04 (m, 2H), 3.43 (q, J = 24.8 Hz, 4H), 3.65–3.72 (m, 1H), 5.48–5.66 (m, 2H), 7.15 (d, J = 8.0 Hz, 2H), 7.21–7.23 (m, 2H), 7.24–7.31 (m, 8H), 7.66 (d, J = 8.4 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 13.4, 21.4, 55.2, 58.0, 63.6, 125.8, 126.9, 128.2, 128.7, 129.0, 129.5, 134.1, 139.3, 144.5; HRMS (ESI) calcd for C26H30NO2S [M + H]: 420.1992, found: 420.1988.
(E)-N,N-Dibenzyl-2-tosylpent-3-en-1-amine (4m).
The title compound was prepared according to the general procedure and purified by column chromatography to give a colorless oil (47.0 mg, 28% yield). 1H NMR (400 MHz, CDCl3) δ 1.68 (dd, J1 = 6.4 Hz, J2 = 1.2 Hz, 3H), 2.42 (s, 3H), 2.82–2.88 (m, 1H), 3.07 (dd, J1 = 12.8 Hz, J2 = 3.2 Hz, 1H), 3.43 (d, J = 13.6 Hz, 2H), 3.55–3.61 (m, 1H), 3.67 (d, J = 13.6 Hz, 2H), 5.07–5.13 (m, 1H), 5.34–5.43 (m, 1H), 7.20–7.28 (m, 12H), 7.57 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 18.2, 21.6, 51.6, 58.8, 67.7, 123.1, 127.1, 128.2, 128.9, 129.0, 129.3, 135.0, 138.8, 144.3; HRMS (ESI) calcd for C26H30NO2S [M + H]: 420.1992, found: 420.1975.
Acknowledgements
The project was supported by the National Natural Science Foundation of China (21222203, 21372231 and 21133011) and Chinese Academy of Sciences.
Notes and references
- For leading reviews, see:
(a) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861 CrossRef CAS PubMed;
(b) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed;
(c) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS;
(d) S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365 CrossRef CAS;
(e) X. Zhao, Y. Zhang and J. Wang, Chem. Commun., 2012, 48, 10162 RSC;
(f) D. Gillingham and N. Fei, Chem. Soc. Rev., 2013, 42, 4918 RSC;
(g) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS PubMed.
-
(a)
J. Wang, in Comprehensive Organic Chemistry III, ed. D. M. P. Mingos and R. H. Crabtree, Elsevier, Oxford, 2007, pp. 151–178 Search PubMed;
(b) A. Padwa, L. S. Beall, C. K. Eidell and K. J. Worsencroft, J. Org. Chem., 2001, 66, 2414 CrossRef CAS PubMed;
(c) J. S. Clark and M. D. Middleton, Org. Lett., 2002, 4, 765 CrossRef CAS PubMed;
(d) C.-Y. Zhou, W.-Y. Yu, P. W. H. Chan and C.-M. Che, J. Org. Chem., 2004, 69, 7072 CrossRef CAS PubMed;
(e) J. A. Vanecko and F. G. West, Org. Lett., 2005, 7, 2949 CrossRef CAS PubMed.
- G. Qin, L. Li, J. Li and H. Huang, J. Am. Chem. Soc., 2015, 137, 12490 CrossRef CAS PubMed.
- For a leading review, see:
(a) J. R. Fulton, V. K. Aggarwal and J. d. Vicente, Eur. J. Org. Chem., 2005, 1479 CrossRef CAS. For selected references, see:
(b) W. R. Bamford and T. S. Stevens, J. Chem. Soc., 1952, 4735 RSC;
(c) K. Inamoto, M. Katsuno, T. Yoshino, I. Suzuki, K. Hiroya and T. Sakamoto, Chem. Lett., 2004, 1026 CrossRef CAS;
(d) F. Hu, J. Yang, Y. Xia, C. Ma, H. Xia, Y. Zhang and J. Wang, Org. Chem. Front., 2015, 2, 1450 RSC.
-
(a) J. Barluenga, P. Moriel, C. Valdés and F. Aznar, Angew. Chem., Int. Ed., 2007, 46, 5587 CrossRef CAS PubMed;
(b) J. Barluenga, M. T. Gamasa, P. Moriel, F. Aznar and C. Valdés, Chem. – Eur. J., 2008, 14, 4792 CrossRef CAS;
(c) J. Barluenga, M. Escribano, P. Moriel, F. Aznar and C. Valdés, Chem. – Eur. J., 2009, 15, 13291 CrossRef CAS PubMed.
- For leading review on using N-sulfonyl hydrazones as carbene precursors, see:
(a) J. Barluenga and C. Valdés, Angew. Chem., Int. Ed., 2011, 50, 7486 CrossRef CAS PubMed. For selected references, see:
(b) L. Zhou, F. Ye, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2010, 132, 13590 CrossRef CAS;
(c) Q. Xiao, Y. Xia, H. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2011, 50, 1114 CrossRef CAS;
(d) F. Ye, X. Ma, Q. Xiao, H. Li, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2012, 134, 5742 CrossRef CAS;
(e) P.-X. Zhou, Y.-Y. Ye, L.-B. Zhao, J.-Y. Hou, X. Kang, D.-Q. Chen, Q. Tang, J.-Y. Zhang, Q.-X. Huang, L. Zheng, J.-W. Ma, P.-F. Xu and Y.-M. Liang, Chem. – Eur. J., 2014, 20, 16093 CrossRef CAS PubMed.
- For leading reviews on metal carbenes, see:
(a) Y. Zhang and J. Wang, Eur. J. Org. Chem., 2011, 1015 CrossRef;
(b) X. Zhao, Y. Zhang and J. Wang, Chem. Commun., 2012, 48, 10162 RSC;
(c) Y. Xia, Y. Zhang and J. Wang, ACS Catal., 2013, 3, 2586 CrossRef CAS;
(d) Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236 CrossRef CAS PubMed;
(e) Z. Liu and J. Wang, J. Org. Chem., 2013, 78, 10024 CrossRef CAS PubMed.
-
(a) Y. Xie, J. Hu, Y. Wang, C. Xia and H. Huang, J. Am. Chem. Soc., 2012, 134, 20613 CrossRef CAS PubMed;
(b) Y. Xie, J. Hu, P. Xie, B. Qian and H. Huang, J. Am. Chem. Soc., 2013, 135, 18327 CrossRef CAS PubMed;
(c) J. Hu, Y. Xie and H. Huang, Angew. Chem., Int. Ed., 2014, 53, 7272 CrossRef CAS;
(d) J. Hu, Y. Xie and H. Huang, J. Mol. Catal., 2014, 28, 197 CAS;
(e) G. Zhang, B. Gao and H. Huang, Angew. Chem., Int. Ed., 2015, 54, 7657 CrossRef CAS PubMed;
(f) C. Qiao, J. Hu and H. Huang, J. Mol. Catal., 2015, 29, 103 CAS.
-
(a) F. Reck, F. Zhou, M. Girardot, G. Kern, C. J. Eyermann, N. J. Hales, R. R. Ramsay and M. B. Gravestock, J. Med. Chem., 2005, 48, 499 CrossRef CAS PubMed;
(b)
N. Neamati, G. W. Kabalka, B. Venkataiah and R. Dayam, W. O. Patent, 081966, 2007 Search PubMed;
(c)
M. Muehlebach, W. Lutz, J. Wenger, J. Finney, C. J. Mathews and D. Fawke, W. O. Patent, 110308, 2008 Search PubMed.
- CCDC 1437594 contains the supplementary crystallographic data for this paper.
-
(a) H. Heaney, G. Papageorgiou and R. F. Wilkins, Tetrahedron, 1997, 53, 2941 CrossRef CAS;
(b) T. Rosenau, A. Potthast and P. Kosma, Tetrahedron, 2004, 60, 301 CrossRef CAS;
(c) A. G. Myers, B. Zheng and M. Movassaghi, J. Org. Chem., 1997, 62, 7507 CrossRef CAS PubMed;
(d) M. J. Genin, D. A. Allwine, D. J. Anderson, M. R. Barbachyn, D. E. Emmert, S. A. Garmon, D. R. Graber, K. C. Grega, J. B. Hester, D. K. Hutchinson, J. Morris, R. J. Reischer, C. W. Ford, G. E. Zurenko, J. C. Hamel, R. D. Schaadt, D. Stapert and B. H. Yagi, J. Med. Chem., 2000, 43, 953 CrossRef CAS PubMed;
(e) V. K. Aggarwal, E. Alonso, I. Bae, G. Hynd, K. M. Lydon, M. J. Palmer, M. Patel, M. Porcelloni, J. Richardson, R. A. Stenson, J. R. Studley, J. Vassé and C. L. Winn, J. Am. Chem. Soc., 2003, 125, 10926 CrossRef CAS;
(f) F.-L. Yang, X.-T. Ma and S.-K. Tian, Chem. – Eur. J., 2012, 18, 1582 CrossRef CAS PubMed;
(g) E. L. Luzina and A. V. Popov, J. Fluorine Chem., 2013, 41 CrossRef CAS;
(h) G. Battistuzzi, S. Cacchi and G. Fabrizi, Org. Lett., 2003, 5, 777 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra of substrates and products. CCDC 1437594. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00381d |
|
| This journal is © the Partner Organisations 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.