
Regiospecific synthesis of distally chlorinated ketones via C–C bond cleavage of cycloalkanols†
Xuefeng
Fan‡
a,
Huijun
Zhao‡
a,
Jiajia
Yu
a,
Xiaoguang
Bao
*b and
Chen
Zhu
*ac
aKey Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, 199 Ren-Ai Road, Suzhou, Jiangsu 215123, China. E-mail: chzhu@suda.edu.cn
bCollege of Chemistry, Chemical Engineering and Materials Science, Soochow University, 199 Ren-Ai Road, Suzhou, Jiangsu 215123, China. E-mail: xgbao@suda.edu.cn
cKey Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
First published on 10th December 2015
Abstract
A variety of distally Csp3-chlorinated ketones are synthesized with good regioselectivities via silver-catalyzed ring opening of cycloalkanols under mild reaction conditions. The reaction uses only routine, inexpensive reagents and can be scaled up to gram quantities. A novel radical-mediated C–C bond cleavage/C–Cl bond formation is proposed.
Introduction
Since the ketone motif is ubiquitous in organic compounds, the convenient construction of diverse ketone derivatives is of upmost importance in organic synthesis. Halogen-substituted ketones are privileged precursors for further manipulations, and easily afford numerous ketone derivatives, e.g., via nucleophilic substitutions1–4 or cross-couplings.5 As a prominent representative, chlorine-substituted ketones frequently serve as key synthetic building blocks for the synthesis of pharmaceutically active molecules.6 In addition, bromine- and iodine-substituted ketones are often derived from the corresponding chlorinated ketones by the Finkelstein reaction with bromide or iodide salts.7 Therefore, the development of an efficient, direct approach to chlorinated ketones is highly desirable.Electrophilic chlorination of a ketone enolate provides ready access to α-chlorinated ketones.8 However, the straightforward introduction of a chlorine atom into the remote (β, γ, δ, etc.) positions of a ketone is more challenging. While there is no generally direct chlorination procedure for distally chlorinated ketones, a circuitous route relies on the Friedel–Craft reaction,9 although stoichiometric strong Lewis acids and moisture sensitive acyl chlorides are required and the substrate scope is limited to electron-rich aryl ketones (Scheme 1A). Recently, Li et al. developed an elegant modification of the Hunsdiecker reaction10 to produce a variety of alkyl chlorides under mild reaction conditions via silver-catalyzed decarboxylative chlorination (Scheme 1B).11 But this was not a specific method to prepare distally chlorinated ketones, and thus only a few examples of chlorinated ketones were included.
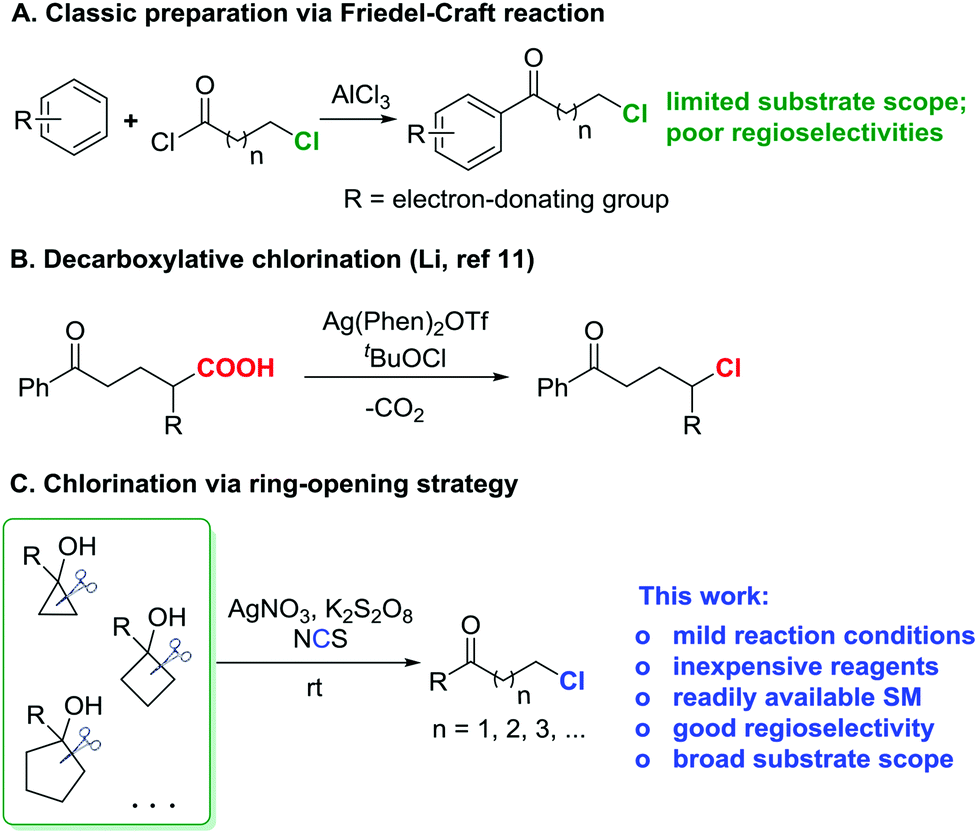 | ||
| Scheme 1 Synthesis of distally chlorinated ketones: classic preparation (A); decarboxylative chlorination (B); ring-opening chlorination (C). | ||
Recently, we reported silver-catalyzed ring openings of cyclopropanols and cyclobutanols to generate β- and γ-fluorinated ketones.12a We also disclosed manganese-catalyzed oxidative azidation of cyclobutanols to produce γ-azido ketones.12b Cycloalkanols proved to be versatile precursors for the regioselective synthesis of distally functionalized ketones. To enlarge the utility of this “radical clock” strategy,13 we herein disclose our new findings for the efficient synthesis of distally chlorinated ketones (Scheme 1C). A wide range of chlorinated ketones are furnished under mild reaction conditions. Moreover, many merits are affiliated with this transformation: (a) only routine, inexpensive reagents are employed; (b) the cycloalkanol starting materials are conveniently available in one step via addition of Grignard reagents to cycloketones or the Kulinkovich reaction;14 and (c) a novel sequence of C–C bond cleavage and subsequent regioselective C–Cl bond formation is involved.
Results and discussion
Computational study
At the outset, computational studies at the UB3LYP/6-311++G(d,p)//UB3LYP/6-31+G(d,p) level of theory were employed to probe the feasibility of the radical-mediated ring openings (Fig. 1). Computational findings suggest that cyclopropoxy (1a) and cyclobutoxy (2a) radicals readily undergo a nearly barrierless ring-opening to terminal carbon-centered radicals 1b and 2b, respectively. Moreover, the formation of 1b and 2b is significantly exothermic by 14.9 and 23.6 kcal mol−1, respectively, whereas, the predicted energy barriers of ring-opening for cyclopentoxy (3a) and cyclohexoxy (4a) radicals are 4.9 and 6.0 kcal mol−1, respectively. The exothermicity for the formation of radicals 3b and 4b is computed to be 9.4 and 5.5 kcal mol−1, which are less exothermic than the generation of 1b and 2b. Therefore, one can conclude that the ring-opening of O-centered three- and four-membered rings occur more readily than the corresponding five- and six-membered rings due, at least partially, to the larger ring tension for smaller rings.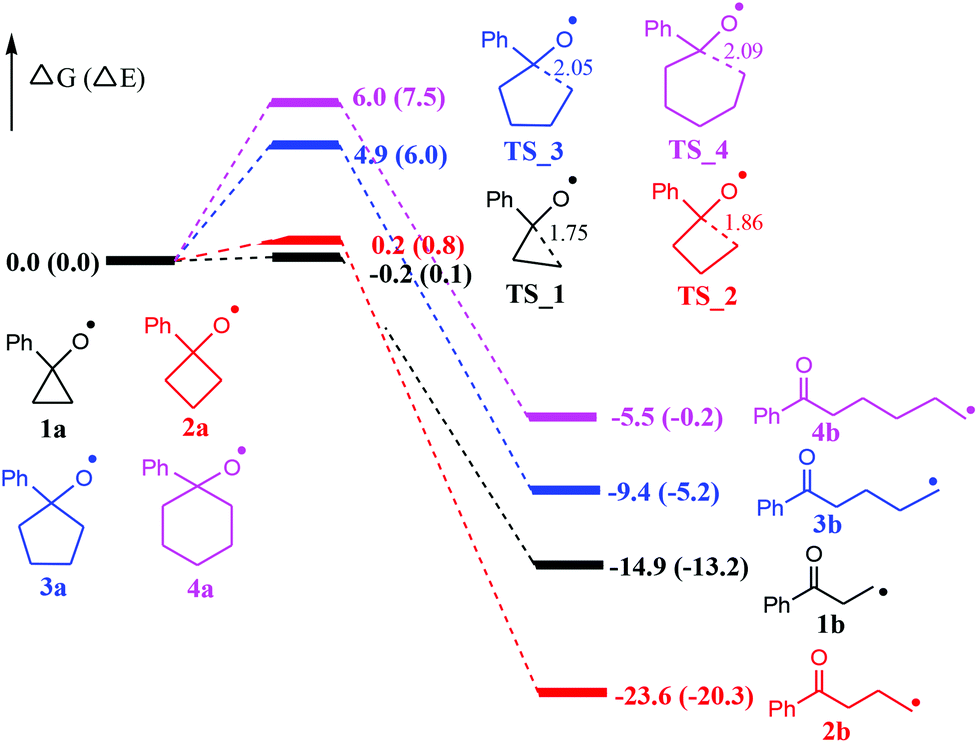 | ||
| Fig. 1 Energy profiles (in kcal mol−1) for the radical-mediated ring openings. Bond lengths are shown in Å. | ||
Experimental study
Given the theoretical understanding, we started the experimental studies with the use of 1-phenylcyclobutanol 5a as the model substrate. The reaction parameters were systematically surveyed (Table 1). The silver-catalyzed chlorination of 5a smoothly proceeded using NCS as the chlorine source and K2S2O8 as the oxidant. No chloride 6a was obtained without silver catalysis (entry 1). Among the various silver salts examined, AgNO3 exhibited a better catalytic ability (entries 2–6). The biphasic solution of DCE/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) facilitated the reaction, giving a higher yield than other mixed solvents (entries 6–12). The use of 1.2 equiv. of K2S2O8 gave the optimal yield (entry 14). The consumption of K2S2O8 could be reduced to a catalytic amount without a considerable decrease of yield (entry 15). However, the use of a catalytic amount of K2S2O8 significantly decreased the conversion rate (entries 15 and 16).
:1) facilitated the reaction, giving a higher yield than other mixed solvents (entries 6–12). The use of 1.2 equiv. of K2S2O8 gave the optimal yield (entry 14). The consumption of K2S2O8 could be reduced to a catalytic amount without a considerable decrease of yield (entry 15). However, the use of a catalytic amount of K2S2O8 significantly decreased the conversion rate (entries 15 and 16).
|
|
||||
|---|---|---|---|---|
| Entry | Silver salt | Solvent | K2S2O8 equiv. | Yieldb (%) |
| a 5a (0.20 mmol), Ag salt (0.04 mmol, 20 mol%), NCS (0.30 mmol, 1.5 equiv.), and K2S2O8 in solvent/H2O (0.5 mL/0.5 mL, 0.2 M), 25 °C. b Isolated yields. c 24 h. d 48 h. | ||||
| 1 | None | DCE/H2O | 2.5 | 0 |
| 2 | AgF | DCE/H2O | 2.5 | 64 |
| 3 | AgOAc | DCE/H2O | 2.5 | 61 |
| 4 | AgOTf | DCE/H2O | 2.5 | 70 |
| 5 | AgBF4 | DCE/H2O | 2.5 | 57 |
| 6 | AgNO3 | DCE/H2O | 2.5 | 72 |
| 7 | AgNO3 | DCM/H2O | 2.5 | 70 |
| 8 | AgNO3 | Toluene/H2O | 2.5 | 53 |
| 9 | AgNO3 | THF/H2O | 2.5 | 22 |
| 10 | AgNO3 | CH3CN/H2O | 2.5 | <10 |
| 11 | AgNO3 | Dioxane/H2O | 2.5 | <10 |
| 12 | AgNO3 | MeOH/H2O | 2.5 | 39 |
| 13 | AgNO3 | DCE/H2O | 2.0 | 71 |
| 14 | AgNO3 | DCE/H2O | 1.2 | 76 |
| 15c | AgNO3 | DCE/H2O | 0.5 | 71 |
| 16d | AgNO3 | DCE/H2O | 0.2 | 47 |
To evaluate the generality of the protocol, we first investigated the chlorination of cyclobutanols. A wide range of cyclobutanols were smoothly converted to the desired γ-chlorinated ketones, while exhibiting good functionality tolerance (Scheme 2). The steric effect seemed to have little influence on the reaction. In the cases of electron-rich substrates, regardless of the arene substitution at o-, m-, or p-positions, all reactions were completed within a few hours (6b–6h). Similar yields of 6i–6k were produced by different halogenated aryl cyclobutanols. Notably, the presence of aryl halide in the products reserves an option for further functionalization. When strong electron-withdrawing groups (CF3, OCF3, etc.) were present, the reaction could be promoted by the addition of acetic acid as the co-solvent (6l and 6m). The reason is so far unclear.15 Not only aryl, but also alkyl cyclobutanols were suitable substrates and furnished the expected products in acceptable yields (6n and 6o). Heteroaryl substrates were compatible with the reaction conditions (6p and 6q). The example of 6r was noteworthy, since the alkene moiety did not suffer from the potential alkyl radical-triggered cyclization. Though less reactive, the substrates with steric hindrance on the cyclobutyl ring were readily chlorinated in synthetically useful yields (6s–6u).
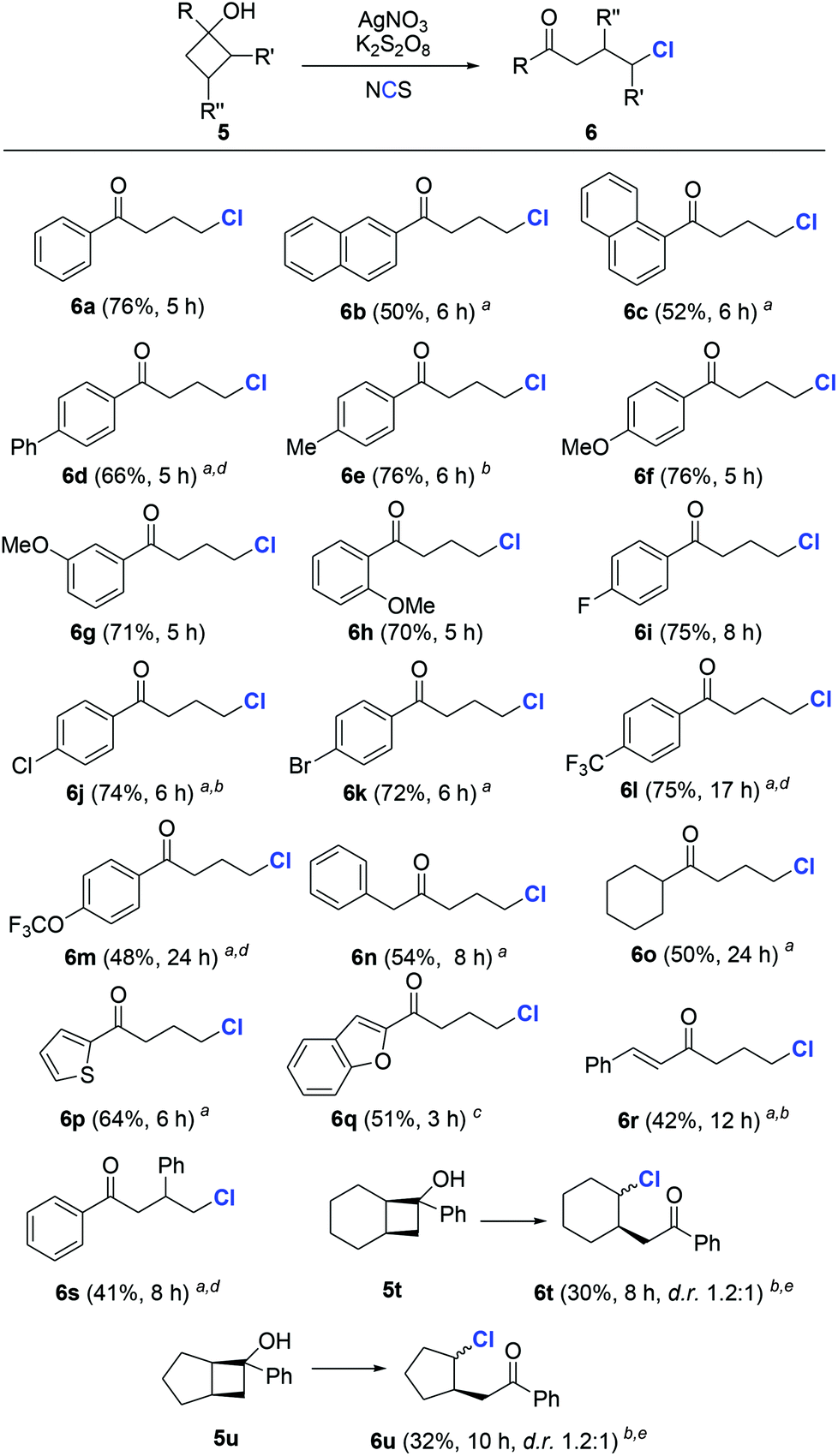 | ||
| Scheme 2 Synthesis of γ-chlorinated ketones from cyclobutanols. Typical conditions: 5 (0.20 mmol), AgNO3 (0.04 mmol, 20 mol%), NCS (0.30 mmol, 1.5 equiv.), and K2S2O8 (0.24 mmol, 1.2 equiv.) in DCE/H2O (0.5 mL/0.5 mL, 0.2 M), 25 °C. Isolated yields. a30 mol% AgNO3. b2.5 equiv. K2S2O8. c0.5 equiv. K2S2O8. d0.2 mL of AcOH as a co-solvent. e50 mol% AgNO3. | ||
Encouraged by these results, a variety of cyclopropanols were subsequently examined under the preceding reaction conditions (Scheme 3). Both electron-rich and -deficient aryl cyclopropanols afforded β-chlorinated ketones in moderate to good yields (8a–8e). Likewise, the alkyl and alkenyl cyclopropanols with diverse substituents were also amenable to transformation into the corresponding β-chlorinated ketones (8f–8l). Surprisingly, the chlorination predominantly occurred at the less substituted carbon when polysubstituted cyclopropanols were utilized (8m and 8n). It was important to point out that the isolated yields were compromised, in some cases, due to the formation of arylvinylketone byproducts.
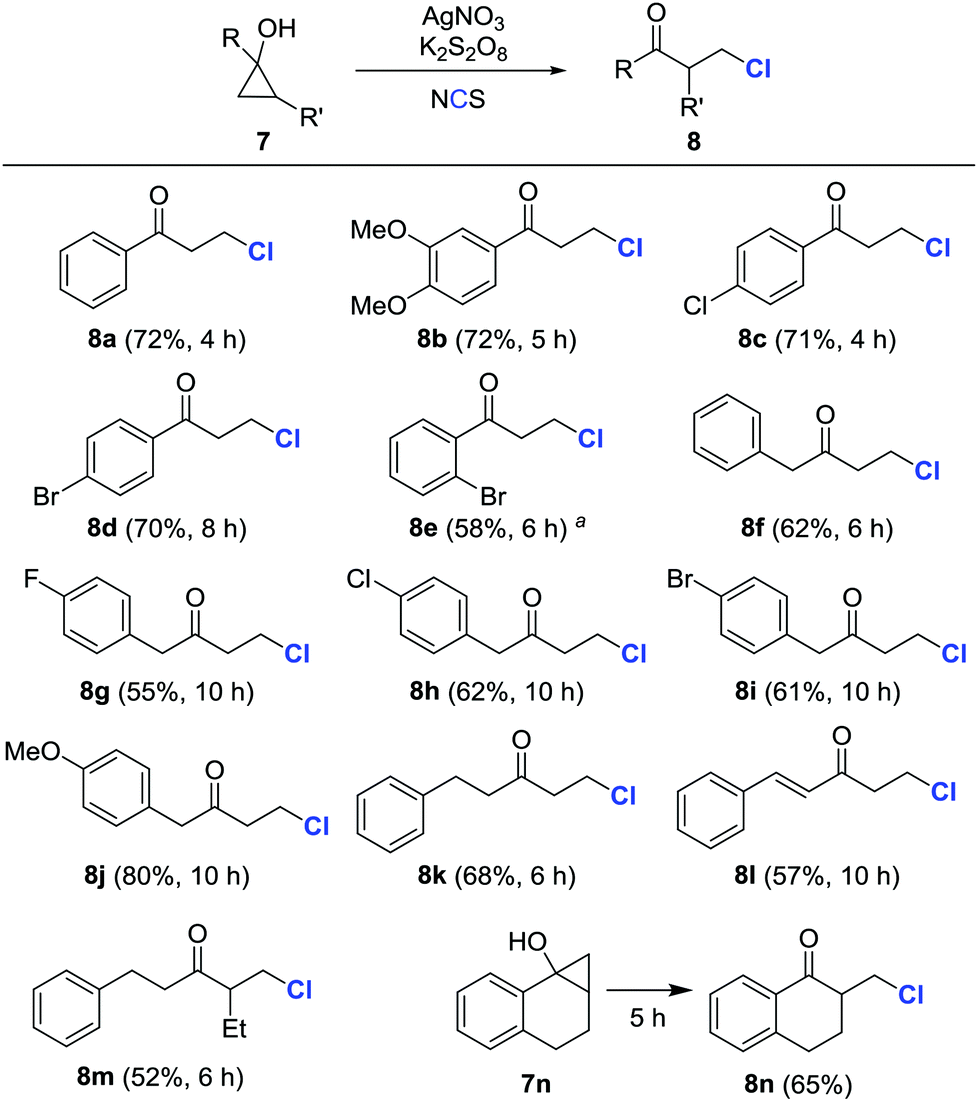 | ||
| Scheme 3 Synthesis of β-chlorinated ketones from cyclopropanols. Typical conditions: 7 (0.20 mmol), AgNO3 (0.04 mmol, 20 mol%), NCS (0.30 mmol, 1.5 equiv.), and K2S2O8 (0.24 mmol, 1.2 equiv.) in DCE/H2O (0.5 mL/0.5 mL, 0.2 M), 25 °C. Isolated yields. a0.5 equiv. K2S2O8. | ||
According to the computational studies in Fig. 1, the alkoxy radical-mediated opening of less strained five- or six-membered rings is feasible,16 but less favorable than three- or four-membered rings. Indeed, when a series of cyclopentanols (9a–9e), cyclohexanols (11a–11c), and cycloheptanol (13) was employed, modest yields of the desired, distally chlorinated ketones were achieved, but only by increasing the amount of silver catalyst (Scheme 4). For cycloalkanols with lower reactivity, the material balance consisted mostly of recovered starting materials, or the byproduct olefin formed via dehydration of tertiary alcohols. Interestingly, no competitive 1,5-hydrogen abstraction took place in the cases of 11 and 13. This platform provides a direct and regioselective approach to distally chlorinated carbonyl compounds which are sometimes difficult to obtain via other methods.
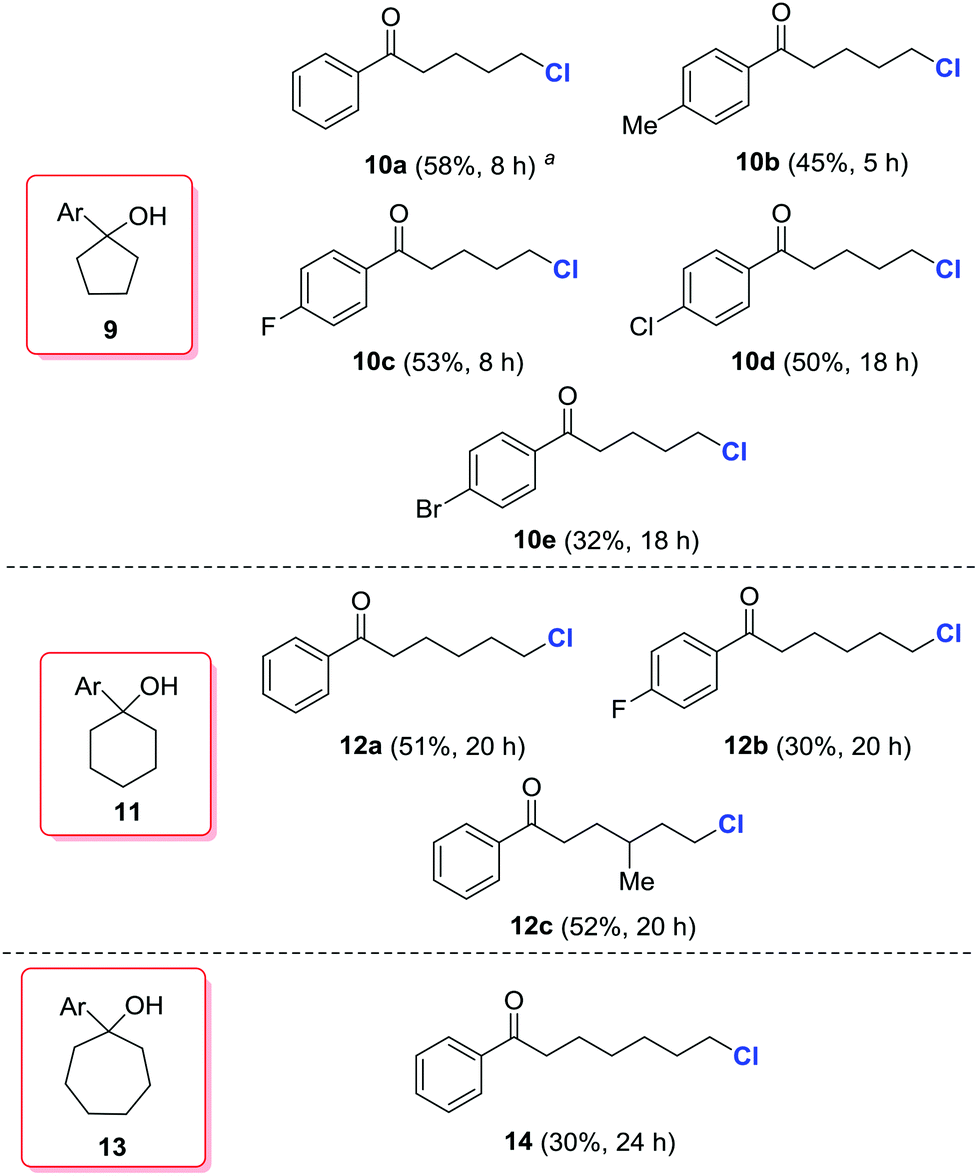 | ||
| Scheme 4 Synthesis of δ-, ε-, and ζ-chlorinated ketones. Typical conditions: cycloalkanol (0.20 mmol), AgNO3 (0.1 mmol, 0.5 equiv.), NCS (0.30 mmol, 1.5 equiv.), and K2S2O8 (0.50 mmol, 2.5 equiv.) in DCE/H2O (0.5 mL/0.5 mL, 0.2 M), 25 °C. Isolated yields. a30 mol% AgNO3. | ||
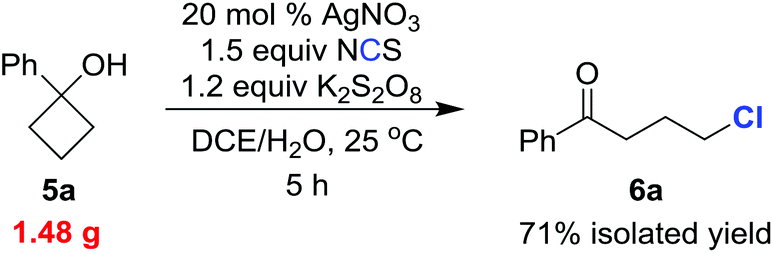
To test the practicality of our methodology, we carried out the chlorination on a gram scale (eqn (1)). Reaction of 1.48 g (10 mmol) of 5a under the standard reaction conditions gave the chlorinated ketone 6a (1.31 g) within 5 h in 71% isolated yield.
 | (1) |
This protocol can also be applied to cycloalkanol brominations under similar reaction conditions. The desired brominated ketones 15a–15e were readily furnished in synthetically useful yields using NBS in lieu of NCS (Scheme 5).
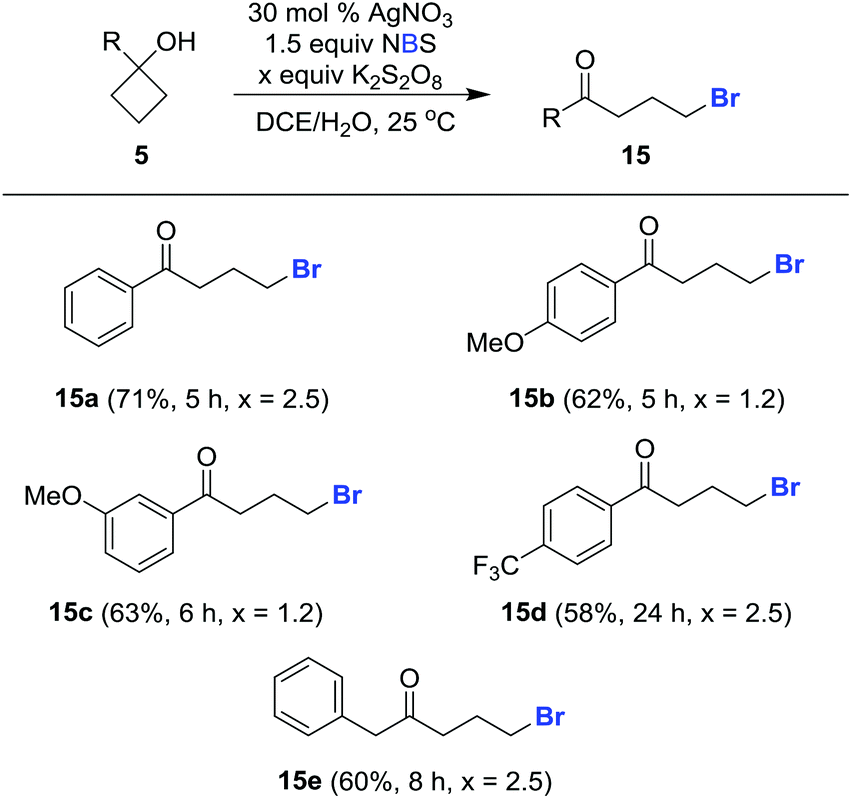 | ||
| Scheme 5 Synthesis of brominated ketones. Typical conditions: cyclobutanol (0.20 mmol), AgNO3 (0.06 mmol, 0.3 equiv.), NBS (0.30 mmol, 1.5 equiv.), and K2S2O8 (1.2 or 2.5 equiv.) in DCE/H2O (0.5 mL/0.5 mL, 0.2 M), 25 °C. Isolated yields. | ||
Proposed mechanism
A set of control experiments was conducted to elucidate the mechanistic pathway. As shown in the entry 1 of Table 1, the reaction did not proceed in the absence of a silver catalyst. Also, the reaction did not work if K2S2O8 was removed from the standard reaction conditions. In addition, the reaction was significantly suppressed in the presence of the anti-oxidant/radical traps TEMPO or BHT. This evidence suggests that the reaction involves a radical pathway. Indeed, when the reaction of 5a took place in the absence of NCS, 1-tetralone, which was generated via a radical-mediated intramolecular cyclization, was obtained.17Based on the experimental observations, a proposed mechanism is outlined in Fig. 2. Initially, cycloalkanol 5 is oxidized to the cycloalkoxy radical a by the metastable AgII species, which is generated by the interaction of AgNO3 and K2S2O8 (path a).18 The alkyl radical b, the open-chain tautomer of a, is then intercepted by NCS to eventually afford the chlorinated ketone 6. The released imidyl radical c might also be involved in the generation of the radical a from 5via hydrogen abstraction (BDE: t-alkanol O–H 105 kcal mol−1versus imide N–H 107 kcal mol−1) (path b).19
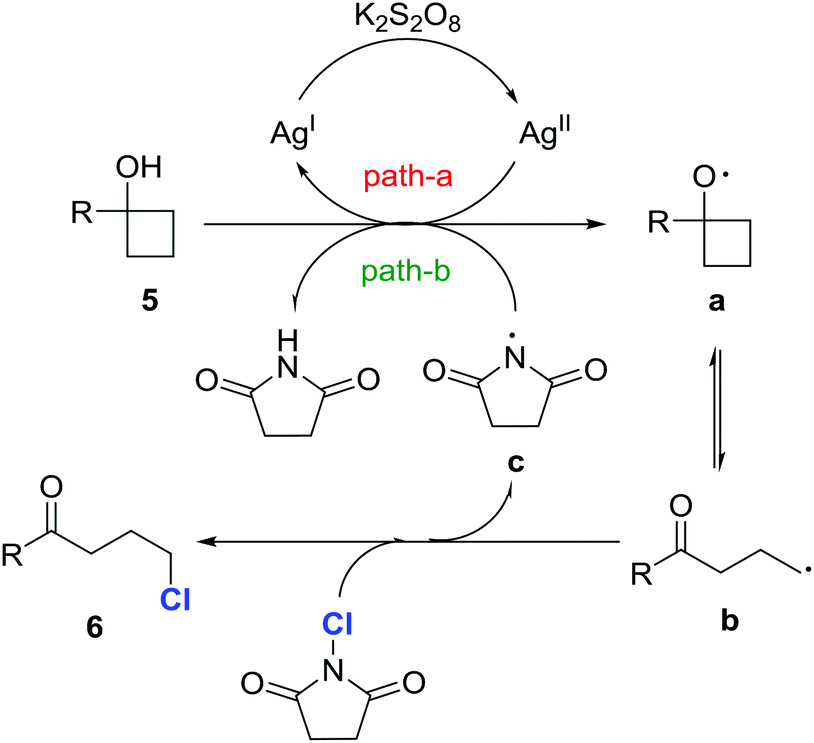 | ||
| Fig. 2 A plausible mechanism. | ||
Conclusions
A novel, synthetically useful protocol for the production of distally chlorinated ketones has been established. A wide range of β, γ, δ, and even more remotely chlorinated ketones are efficiently synthesized under mild reaction conditions. The reaction is carried out with only routine, inexpensive reagents and can be readily scaled up to gram amounts. This protocol can be further applied to the distal bromination of cycloalkanols. Further applications of this “radical clock” strategy are ongoing in our laboratory.Experimental section
Cyclobutanol 5 (0.2 mmol, 1.0 equiv.), AgNO3 (0.04 mmol, 0.2 equiv.), K2S2O8 (0.24 mmol, 1.2 equiv.), and NCS (0.3 mmol, 1.5 equiv.) were loaded in a flask which was subjected to evacuation/flushing with nitrogen three times. A mixed solvent of DCE/H2O (0.5/0.5 mL, 0.2 M) was added into the mixture via a syringe and the mixture was then stirred at 25 °C until the starting material was consumed as determined by TLC. The mixture was extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with brine, dried over Na2SO4, filtered, concentrated, and purified by flash chromatography on silica gel (ethyl acetate/PE) to give the halogenated ketone 6.Competing interest
The authors declare no competing financial interests.Acknowledgements
C. Z. is grateful for the financial support from Soochow University, the National Natural Science Foundation of China (Grant no. 21402134), the Natural Science Foundation of Jiangsu (Grant no. BK20140306), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). X. B. is supported by the Natural Science Foundation of China (NSFC 21302133). Professor John R. Falck of the University of Texas Southwestern Medical Center is thanked for kindly proofreading the manuscript.Notes and references
- For examples of forming C–C bonds, see: (a) S. Danishefsky and B. H. Migdalof, J. Am. Chem. Soc., 1969, 91, 2806 CrossRef CAS; (b) A. E. Ahmed, P. E. Hanna and V. R. Grund, J. Med. Chem., 1976, 19, 1174 CrossRef; (c) D. Barlocco and G. Cignarella, Synthesis, 1985, 876 CrossRef CAS; (d) S. Basurto, S. Garcia, A. G. Neo, T. Torroba, C. F. Marcos, D. Miguel, J. Barbera, M. B. Ros and M. R. de la Fuente, Chem. – Eur. J., 2005, 11, 5362 CrossRef CAS PubMed; (e) F. Zhu, P. Xu, F. Zhou, C. Wang and J. Zhou, Org. Lett., 2015, 17, 972 CrossRef CAS PubMed.
- For examples of forming C–N bonds, see: (a) P. Sulmon, N. D. Kimpe and N. Schamp, J. Org. Chem., 1988, 53, 4462 CrossRef CAS; (b) T. G. Archibald and K. Baum, J. Org. Chem., 1990, 55, 3565 CrossRef; (c) R. M. Luond, J. Walker and R. W. Neier, J. Org. Chem., 1992, 57, 5013 CrossRef; (d) H. Karoui, C. Nsanzumuhire, F. L. Moigne and P. Tordo, J. Org. Chem., 1999, 64, 1471 CrossRef CAS PubMed; (e) F. Lehmann, E. A. Currier, R. Olsson, U. Hacksell and K. Luthman, Bioorg. Med. Chem., 2005, 13, 3057 CrossRef CAS PubMed.
- For examples of forming C–O bonds, see: (a) N. D. Kimpe, P. Sulmon, L. Moens and N. Schamp, J. Org. Chem., 1986, 51, 3839 CrossRef; (b) S. Chang, D. P. Fernando and S. King, Org. Process Res. Dev., 2001, 5, 141 CrossRef CAS; (c) J.-L. Clement, J.-P. Finet, C. Frejaville and P. Tordo, Org. Biomol. Chem., 2003, 1, 1591 RSC; (d) A. Foroumadi, A. Samzadeh-Kermani, S. Emami, G. Dehghan, M. Sorkhi, F. Arabsorkhi, M. R. Heidari, M. Abdollahi and A. Shafiee, Bioorg. Med. Chem. Lett., 2007, 17, 6764 CrossRef CAS PubMed; (e) R. Willand-Charnley, B. W. Puffer and P. H. Dussault, J. Am. Chem. Soc., 2014, 136, 5821 CrossRef CAS PubMed.
- For examples of forming C–S and C–P bonds, see: (a) A. E. Lukach, D. G. Morris, A. N. Santiago and R. A. Rossi, J. Org. Chem., 1995, 60, 1000 CrossRef CAS; (b) T. L. Fevig, W. G. Phillips and P. H. Lau, J. Org. Chem., 2001, 66, 2493 CrossRef CAS PubMed; (c) S. R. S. S. Kotti, X. Xu, G. Li and A. D. Headley, Tetrahedron Lett., 2004, 45, 1427 CrossRef CAS; (d) J. Xiang, M. Ipek, V. Suri, M. Tam, Y. Xing, N. Huang, Y. Zhang, J. Tobin, T. S. Mansour and J. McKew, Bioorg. Med. Chem., 2007, 15, 4396 CrossRef CAS PubMed; (e) A. Aguilar-Aguilar, L. S. Liebeskind and E. Pena-Cabrera, J. Org. Chem., 2007, 72, 8539 CrossRef CAS PubMed.
- For examples, see: (a) C. Liu, C. He, W. Shi, M. Chen and A. Lei, Org. Lett., 2007, 9, 5601 CrossRef CAS PubMed; (b) N. A. Strotman, S. Sommer and G. C. Fu, Angew. Chem., Int. Ed., 2007, 46, 3556 CrossRef CAS PubMed; (c) P. M. Lundin, J. Esquivias and G. C. Fu, Angew. Chem., Int. Ed., 2009, 48, 154 CrossRef CAS PubMed; (d) S. Lou and G. C. Fu, J. Am. Chem. Soc., 2010, 132, 1264 CrossRef CAS PubMed; (e) Z. Shen, K. K. K. Goh, C. H. A. Wong, Y. Yang, Y. Lai, H. Cheong and T.-P. Loh, Chem. Commun., 2011, 47, 4778 RSC; (f) Q. Jiang, T. Guo, Q. Wang, P. Wu and Z. Yu, Adv. Synth. Catal., 2013, 355, 1874 CrossRef CAS.
- For examples, see: (a) M. A. Iorio, T. Paszkowska Reymer and V. Frigeni, J. Med. Chem., 1987, 30, 1906 CrossRef CAS PubMed; (b) I. van Winjngaarden, C. G. Kruse, J. A. M. van der Heyden and M. Th. M. Tulip, J. Med. Chem., 1988, 31, 1934 CrossRef; (c) C.-A. Chen, Y. Jiang, K. Lu, I. Daniewska, C. G. Mazza, L. Negron, C. Forray, T. Parola, B. Li, L. G. Hegde, T. D. Wolinksy, D. A. Craig, R. Kong, J. M. Wetzel, K. Andersen and M. R. Marzabadi, J. Med. Chem., 2007, 50, 3883 CrossRef CAS PubMed.
- For examples, see: (a) P. Traxler, U. Trinks, E. Buchdunger, H. Mett, T. Meyer, M. Muller, U. Regenass, J. Rosel and N. Lydon, J. Med. Chem., 1995, 38, 2441 CrossRef CAS PubMed; (b) R. Giovannini, T. Stüdemann, A. Devasagayaraj, G. Dussin and P. Knochel, J. Org. Chem., 1999, 64, 3544 CrossRef CAS PubMed; (c) J. Zhou and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 14726 CrossRef CAS PubMed; (d) J. Chen, Z. Sun, Y. Zhang, X. Zeng, C. Qing, J. Liu, L. Li and H. Zhang, Bioorg. Med. Chem. Lett., 2009, 19, 5496 CrossRef CAS PubMed; (e) T. K. Olszewski, C. Bomont, P. Coutrot and C. Grison, J. Organomet. Chem., 2010, 695, 2354 CrossRef CAS.
- For examples, see: (a) D. Yang, Y.-L. Yan and B. Liu, J. Org. Chem., 2002, 67, 7429 CrossRef CAS PubMed; (b) R. Frantz, L. Hintermann, M. Perseghini, D. Broggini and A. Togni, Org. Lett., 2003, 5, 1709 CrossRef CAS PubMed; (c) C. Wang and J. Tunge, Chem. Commun., 2004, 2694 RSC; (d) M. Marigo, S. Bachmann, N. Halland, A. Braunton and K. A. Jørgensen, Angew. Chem., Int. Ed., 2004, 43, 5507 CrossRef CAS PubMed; (e) M. Marigo, N. Kumaragurubaran and K. A. Jørgensen, Chem. – Eur. J., 2004, 10, 2133 CrossRef CAS PubMed; (f) F. Grein, A. C. Chen, D. Edwards and C. M. Crudden, J. Org. Chem., 2006, 71, 861 CrossRef CAS PubMed; (g) K. Shibatomi, Y. Soga, A. Narayama, I. Fujisawa and S. Iwasa, J. Am. Chem. Soc., 2012, 134, 9836 CrossRef CAS PubMed.
- For examples, see: (a) W. J. Close, J. Am. Chem. Soc., 1957, 79, 1455 CrossRef CAS; (b) T. Noguchi, M. Hasegawa, K. Tomisawa and M. Mitsukuchi, Bioorg. Med. Chem., 2003, 11, 4729 CrossRef CAS PubMed; (c) K. Lukin, M. C. Hsu, G. Chambournier, B. Kotecki, C. J. Venkatramani and M. R. Leanna, Org. Process Res. Dev., 2007, 11, 578 CrossRef CAS; (d) F. Bertolini, S. Crotti, V. D. Bussolo, F. Macchia and M. Pineschi, J. Org. Chem., 2008, 73, 8998 CrossRef CAS PubMed; (e) F. Yu, J.-N. Zhou, X.-C. Zhang, Y.-Z. Sui, F.-F. Wu, L.-J. Xie, A. S. C. Chan and J. Wu, Chem. – Eur. J., 2011, 17, 14234 CrossRef CAS PubMed; (f) O. Pablo, D. Guijarro and M. Yus, J. Org. Chem., 2013, 78, 9181 CrossRef CAS PubMed.
- For seminal work, see: (a) A. Borodine, Ann., 1861, 119, 121 Search PubMed; (b) H. Hunsdiecker and C. Hunsdiecker, Ber., 1942, 75, 291 Search PubMed. For reviews, see: (c) R. G. Johnson and R. K. Ingham, Chem. Rev., 1956, 56, 219 CrossRef CAS; (d) C. V. Wilson, Org. React., 1957, 332 CAS.
- (a) Z. Wang, L. Zhu, F. Yin, Z. Su, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 4258 CrossRef CAS PubMed. Also see other leading examples of silver-catalyzed decarboxylative functionalization of alkanoic acids: (b) F. Yin, Z. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 10401 CrossRef CAS PubMed; (c) X. Liu, Z. Wang, X. Cheng and C. Li, J. Am. Chem. Soc., 2012, 134, 14330 CrossRef CAS PubMed; (d) C. Liu, X. Wang, Z. Li, L. Cui and C. Li, J. Am. Chem. Soc., 2015, 137, 9820 CrossRef CAS PubMed.
- (a) H. Zhao, X. Fan, J. Yu and C. Zhu, J. Am. Chem. Soc., 2015, 137, 3490 CrossRef CAS PubMed; (b) R. Ren, H. Zhao, L. Huan and C. Zhu, Angew. Chem., Int. Ed., 2015, 54, 12692 CrossRef CAS PubMed.
- For examples of ring openings of cycloalkanols via one-electron oxidation, see: (a) K. Meyer and J. Rocek, J. Am. Chem. Soc., 1972, 94, 1209 CrossRef CAS; (b) S. Chiba, Z. Cao, S. Bialy and K. Narasaka, Chem. Lett., 2006, 35, 18 CrossRef CAS; (c) J. Jiao, L. X. Nguyen, D. R. Patterson and R. A. Flowers II, Org. Lett., 2007, 9, 1323 CrossRef CAS PubMed; (d) B. M. Casey, C. A. Eakin and R. A. Flowers II, Tetrahedron Lett., 2009, 50, 1264 CrossRef CAS PubMed; (e) Y.-F. Wang and S. Chiba, J. Am. Chem. Soc., 2009, 131, 12570 CrossRef CAS PubMed; (f) Y.-F. Wang, K. K. Toh, E. P. J. Ng and S. Chiba, J. Am. Chem. Soc., 2011, 133, 6411 CrossRef CAS PubMed; (g) A. Ilangovan, S. Saravanakumar and S. Malayappasamy, Org. Lett., 2013, 15, 4968 CrossRef CAS PubMed; (h) M. S. Bloom, D. D. Bume, C. R. Pitts and T. Lectka, Chem. – Eur. J., 2015, 21, 8060 CrossRef PubMed; (i) S. Ren, C. Feng and T.-P. Loh, Org. Biomol. Chem., 2015, 13, 5105 RSC; (j) D. G. Kananovich, Y. A. Konik, D. M. Zubrytski, I. Järving and M. Lopp, Chem. Commun., 2015, 51, 8349 RSC; (k) Y. Li, Z. Ye, T. M. Bellman, T. Chi and M. Dai, Org. Lett., 2015, 17, 2186 CrossRef CAS PubMed; (l) Z. Ye and M. Dai, Org. Lett., 2015, 17, 2190 CrossRef CAS PubMed.
- For similar examples of using AcOH as the co-solvent in silver-catalyzed oxidative reactions, see: (a) L. Zhu, H. Chen, Z. Wang and C. Li, Org. Chem. Front., 2014, 1, 1299 RSC; (b) C. Zhang, Z. Li, L. Zhu, L. Yu, Z. Wang and C. Li, J. Am. Chem. Soc., 2013, 135, 14082 CrossRef CAS PubMed; (c) Z. Li, Z. Wang, L. Zhu, X. Tan and C. Li, J. Am. Chem. Soc., 2014, 136, 16439 CrossRef CAS PubMed.
- (a) O. G. Kulinkovich, S. V. Sviridov, D. A. Vasilevski and T. S. Zh. Pritytskaya, Org. Khim., 1989, 25, 2244 CAS; (b) O. G. Kulinkovich, Eur. J. Org. Chem., 2004, 4517 CrossRef CAS.
- For a recent example of the radical-mediated opening of less strained rings, see: C. R. Pitts, M. S. Bloom, D. D. Bume, Q. A. Zhang and T. Lectka, Chem. Sci., 2015, 6, 5225 RSC.
- J. Yu, H. Zhao, S. Liang, X. Bao and C. Zhu, Org. Biomol. Chem., 2015, 13, 7924 CAS.
- (a) J. M. Anderson and J. K. Kochi, J. Am. Chem. Soc., 1970, 92, 1651 CrossRef CAS; (b) J. M. Anderson and J. K. Kochi, J. Org. Chem., 1970, 35, 986 CrossRef CAS.
- (a) F. G. Bordwell and W. Z. Liu, J. Am. Chem. Soc., 1996, 118, 10819 CrossRef CAS; (b) M. J. Bausch, B. David, V. Prasad, L. H. Wang and A. Vaughn, J. Phys. Org. Chem., 1992, 5, 1 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00368g |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2016 |