
Gold-catalyzed tandem synthesis of bioactive spiro-dipyrroloquinolines and its application in the one-step synthesis of incargranine B aglycone and seneciobipyrrolidine (I)†
Can-Liang
Ma‡
a,
Xiao-Hua
Li‡
b,
Xiao-Long
Yu
b,
Xiao-Long
Zhu
b,
Yong-Zhou
Hu
a,
Xiao-Wu
Dong
*a,
Bin
Tan
*b and
Xin-Yuan
Liu
*b
aZhejiang Province Key Laboratory of Anti-Cancer Drug Research, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, 310058, P. R. China. E-mail: dongxw@zju.edu.cn; Tel: (+86)571-88981051
bDepartment of Chemistry, South University of Science and Technology of China, Shenzhen, 518055, P. R. China. E-mail: tanb@sustc.edu.cn; liuxy3@sustc.edu.cn; Tel: (+86)755-88018304
First published on 6th January 2016
Abstract
The Au-catalyzed tandem process of aminoalkynes was explored, providing simple and efficient access to richly functionalized dipyrroloquinoline frameworks with good to excellent yields. The reaction exhibits great efficiency and high atom economy in multiple-bond formation for constructing bioactive azaspiro polycyclic molecules with densely multiple stereogenic centers including quaternary carbons, and shows a broad substrate scope and synthetically important functional group tolerance, which have been illustrated in the first one-step synthesis of incargranine B aglycone and seneciobipyrrolidine (I).
 Xin-Yuan Liu | Dr Xin-Yuan Liu received his Ph.D. degree from the University of Hong Kong (HKU) in 2010 and then worked as a Post-doctoral Fellow in HKU and in the Scripps Research Institute from 2010 to 2012. Dr Liu joined the South University of Science and Technology of China as Associate Professor in 2012. His research interests are focused on the design of homogeneous and heterogeneous catalysis, their applications in asymmetric functionalization of unactivated alkenes and alkynes, and the application of chemistry in medicinal chemistry. He has published more than forty research papers in international journals, including Nat. Commun., J. Am. Chem. Soc., Angew., Chem. Comm. and so on. Citation times for his research papers have been up to 1300. |
Polycyclic azaheterocycles are a component omnipresent in a wide range of naturally occurring and biologically active molecules.1 In particular, spiroheterocycles with multiple stereogenic centers are key subunits found in a large number of natural alkaloids like cylindricine A, ansalactam A, haplophytine and grandilodine A with a great diversity of important biological properties (Fig. 1).2 Despite the significant efforts in the development of efficient strategies for the synthesis of spiroheterocyclic alkaloids, the one-step and stereocontrolled construction of these molecular scaffolds with high atom economy, preferably by using simple catalytic systems starting from readily available acyclic starting materials, has been much less explored and it remains a very important and formidable synthetic challenge because of their structural complexity/diversity.
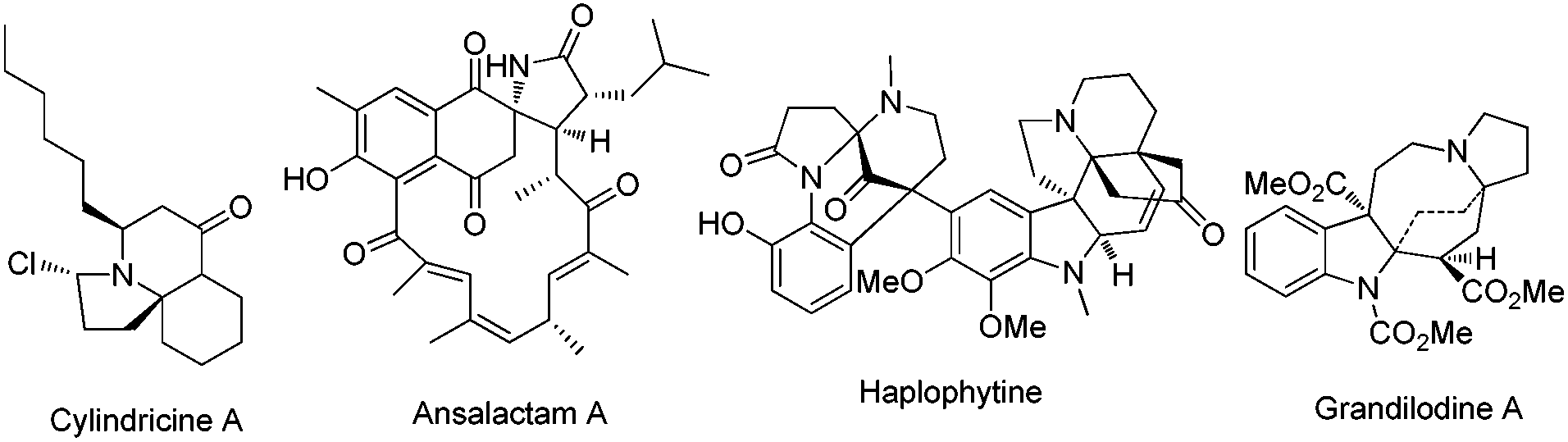 | ||
| Fig. 1 Natural products and pharmaceuticals containing an azaspiro moiety. | ||
We and Xu have independently reported the synthesis of substituted azaheterocycles via transition-metal-catalyzed tandem cyclization of aminoalkynes with some electrophiles or nucleophiles (Scheme 1a).3,4 In these previously reported reactions,3,4 the transformation might be realized using aminoalkynes for the generation of an activated enamine intermediate through metal-catalyzed hydroamination to spur further transformations. On the other hand, the aza-Diels–Alder reaction of 2-azadienes and electron-rich olefins to access azaheterocycles represents one of the most important developments in modern synthetic chemistry and has a broad range of applications in the synthesis of natural products and bioactive compounds,5,6 owing to their bond-forming efficiency, atom economy, excellent stereoselectivity, and product structural diversity/complexity. We wondered if the synthetic potential of the aminoalkyne reactivity as enamine precursors in the presence of transition-metal-catalysis could be further harnessed to simultaneously serve as both dienophile and 2-azadiene through the aza-Diels–Alder reaction to directly access different types of polycyclic azaheterocycles (Scheme 1b). Interestingly, this proposed reaction would overcome one of the main limitations of the aza-Diels–Alder reaction involving the extremely unstable enamine/iminium ion reagents.7 In this scenario, several major challenges have to be overcome to accomplish the desired reaction. First, the difficulty in controlling both regioselectivity and diastereoselectivity owing to concomitant generation of two or three stereocenters with such a strategy has to be substantially overcome. The other is that the rapid enamine/iminium equilibrium may lead to a number of reaction pathways and combination cascades such as aldol, Mannich and Claisen to give a mixture of products. Herein, we present the gold-catalyzed8 one-pot tandem reaction for the efficient formation of multiple bonds, and thus provide facile access to richly functionalized spiro-dipyrroloquinoline frameworks bearing two quaternary stereogenic centers with potential anticancer activity (Scheme 1b). Significantly, this highly convenient and economical methodology was successfully applied to the first one-step synthesis of incargranine B aglycone and (±)-seneciobipyrrolidine (I) in good yield on a gram-scale (Scheme 1c).
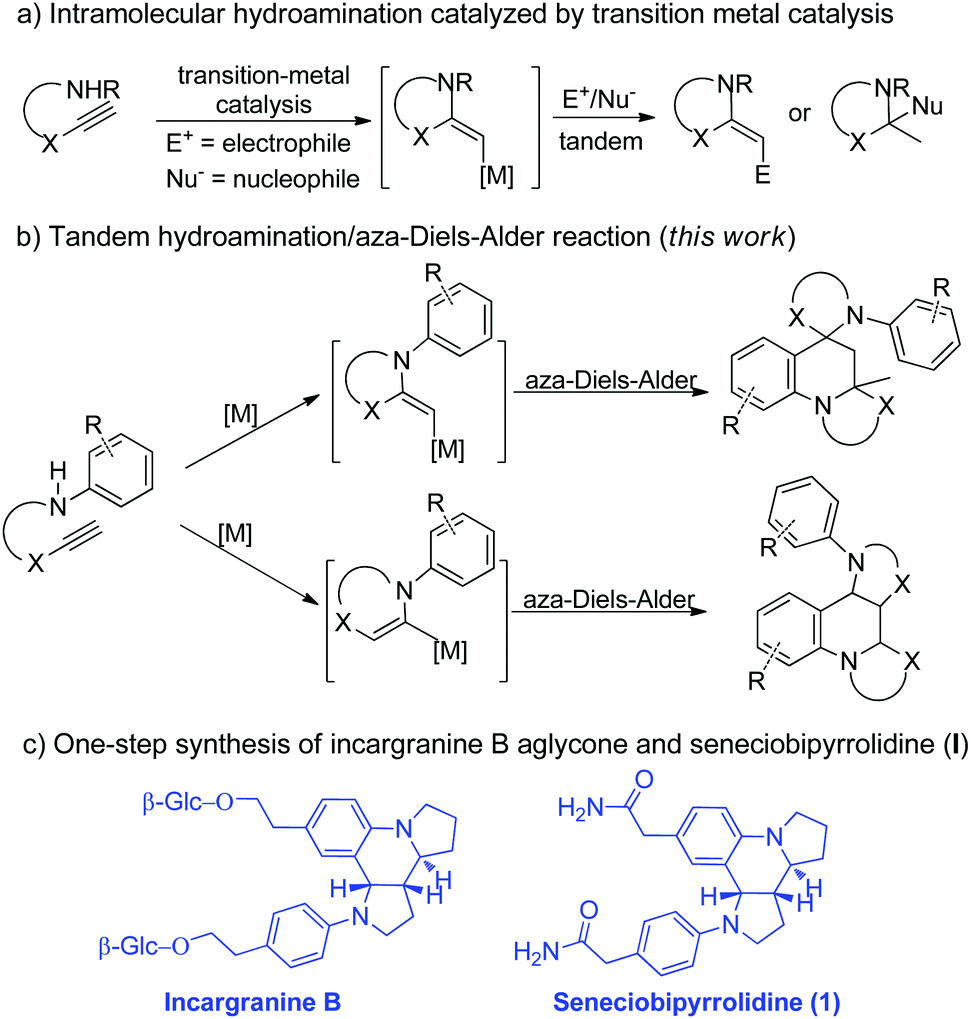 | ||
| Scheme 1 A tandem synthesis of complex bioactive polycyclic azaheterocycles. | ||
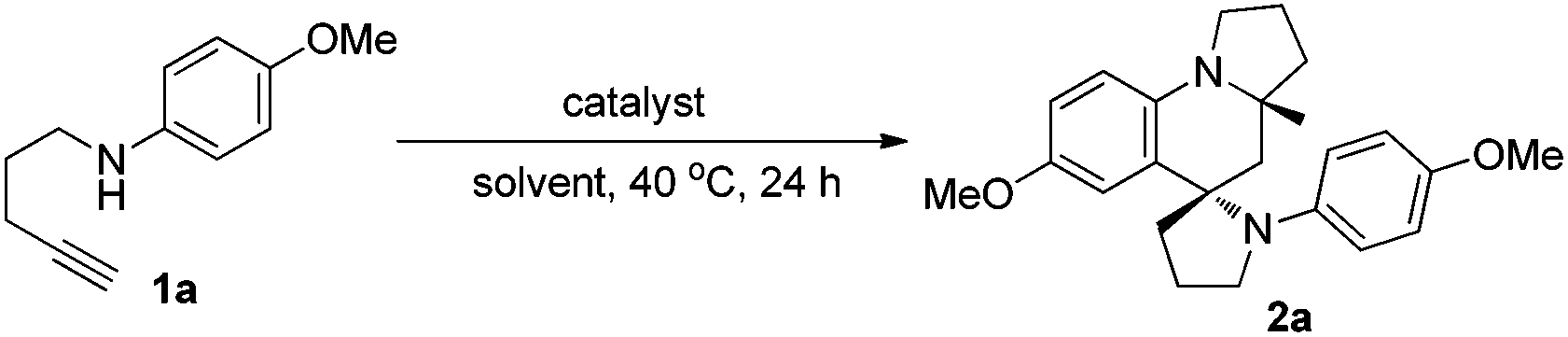
Our investigation started with the use of 1,4-aminoalkyne 1a as the model substrate for identifying a suitable catalytic system, based on our previous reports that such substrates can easily undergo hydroamination under transition-metal-catalysis to give enamine/iminium ion intermediates.3,4 Thus, we initially examined the reaction of 1a in the presence of 5 mol% of PPh3AuCl/AgSbF6 (mol ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in ethanol (EtOH) at 40 °C for 24 h. To our delight, the desired product 2a could be obtained with an excellent all-trans diastereoselectivity, albeit with only 10% yield, demonstrating that a gold catalyst can selectively catalyse such tandem reactions (Table 1, entry 1). To improve the product yield, a panel of gold(I) complexes with different ancillary ligands was screened for the activity and diastereo-induction in the tandem reaction. However, using gold complexes bearing different auxiliary ligands including NHC9 and (tBu)2(o-diphenyl)P10 resulted in the formation of product 2a in only moderate yield (entries 2 and 3). Next, we screened various coinage metal salts like AgSbF6, AuCl and KAuCl4 for this tandem reaction, and found that KAuCl4 was most beneficial for the reaction to exclusively provide the desired product 2a in 71% yield as a single diastereoisomer and with up to almost complete chemoselectivity (entries 4–6). Further screening of solvents showed that a protic solvent EtOH gave the best result, while aprotic solvents toluene, THF and dioxane gave low product yields (entries 6–10). We then investigated the effect of additives and found that the use of 5 Å MS or the absence of MS gave the desired product in relatively lower yield as compared with the use of 4 Å MS (entries 11 and 12).
:1) in ethanol (EtOH) at 40 °C for 24 h. To our delight, the desired product 2a could be obtained with an excellent all-trans diastereoselectivity, albeit with only 10% yield, demonstrating that a gold catalyst can selectively catalyse such tandem reactions (Table 1, entry 1). To improve the product yield, a panel of gold(I) complexes with different ancillary ligands was screened for the activity and diastereo-induction in the tandem reaction. However, using gold complexes bearing different auxiliary ligands including NHC9 and (tBu)2(o-diphenyl)P10 resulted in the formation of product 2a in only moderate yield (entries 2 and 3). Next, we screened various coinage metal salts like AgSbF6, AuCl and KAuCl4 for this tandem reaction, and found that KAuCl4 was most beneficial for the reaction to exclusively provide the desired product 2a in 71% yield as a single diastereoisomer and with up to almost complete chemoselectivity (entries 4–6). Further screening of solvents showed that a protic solvent EtOH gave the best result, while aprotic solvents toluene, THF and dioxane gave low product yields (entries 6–10). We then investigated the effect of additives and found that the use of 5 Å MS or the absence of MS gave the desired product in relatively lower yield as compared with the use of 4 Å MS (entries 11 and 12).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Additive | Yieldb [%] |
| a Reaction conditions: 1a (0.2 mmol), catalyst (5 mol%), solvent (1 mL), at 40 °C under argon for 24 h. b Determined by 1H NMR spectroscopy using CH2Br2 as an internal standard. c Au(L)Cl = (tBu)2(o-diphenyl)PAuCl. d NHC = N,N′-bis(2,6-diisopropylphenyl)-imidazol-2-ylidene. | ||||
| 1 | PPh3AuCl/AgSbF6 | EtOH | 4 Å MS | 10 |
| 2 | Au(L)Clc/AgSbF6 | EtOH | 4 Å MS | 48 |
| 3 | (NHC)AuCld/AgSbF6 | EtOH | 4 Å MS | 26 |
| 4 | AgSbF6 | EtOH | 4 Å MS | 12 |
| 5 | AuCl | EtOH | 4 Å MS | 9 |
| 6 | KAuCl4 | EtOH | 4 Å MS | 71 |
| 7 | KAuCl4 | DCE | 4 Å MS | 11 |
| 8 | KAuCl4 | Toluene | 4 Å MS | 8 |
| 9 | KAuCl4 | THF | 4 Å MS | 9 |
| 10 | KAuCl4 | Dioxane | 4 Å MS | 6 |
| 11 | KAuCl4 | EtOH | 5 Å MS | 50 |
| 12 | KAuCl4 | EtOH | — | 64 |
With this set of optimized reaction conditions, the scope of this gold(III)-catalyzed tandem reaction is demonstrated with a variety of differently substituted 1,4-aminoalkynes. Gratifyingly, good to excellent yields for the tandem reaction were generally obtained for most of the substrates under the mild reaction conditions. As can be seen in Scheme 2, the reaction provided the azaspiro polycycles 2 in 62–77% yields regardless of the substrates 1 with either electron-donating substituents, such as OMe (1a), Me (1c), and OPh (1d), or synthetically attractive electron-withdrawing groups, such as F (1e), Cl (1f), and Br (1g) at the para position of the aryl group. Notably, F, Cl and Br substituents can be tolerated in this reaction, thereby, facilitating further modifications at halogenated positions (2e–2g). 3,5-Dimethyl-N-(pent-4-yn-1-yl)aniline (1h) bearing dimethyl groups gave the corresponding product 2h in 90% yield as well. Interestingly, when 3-methyl-N-(pent-4-yn-1-yl)aniline (1i) was used in the reaction, the 6-position C–H bond with less steric hindrance was selectively activated to afford the corresponding product 2i in 69% yield as a single diastereoisomer, with no 2-position C–H bond activated product 2i′, thus exhibiting not only excellent diastereoselectivity but also excellent regioselectivity for such a tandem reaction. Notably, the 1,4-aminoalkynes with R2 being phenyl, cyclohexyl or cyclopentyl group underwent this Au(III)-catalyzed tandem cyclization to furnish spirocyclic products 2j–2l in 40–85% yields with the rapidly concomitant installation of three or two new spiro rings. The relative configuration of 2g was determined by X-ray crystallographic analysis (Fig. 2a).11 The relative configuration of all other products was determined with reference to 2g.
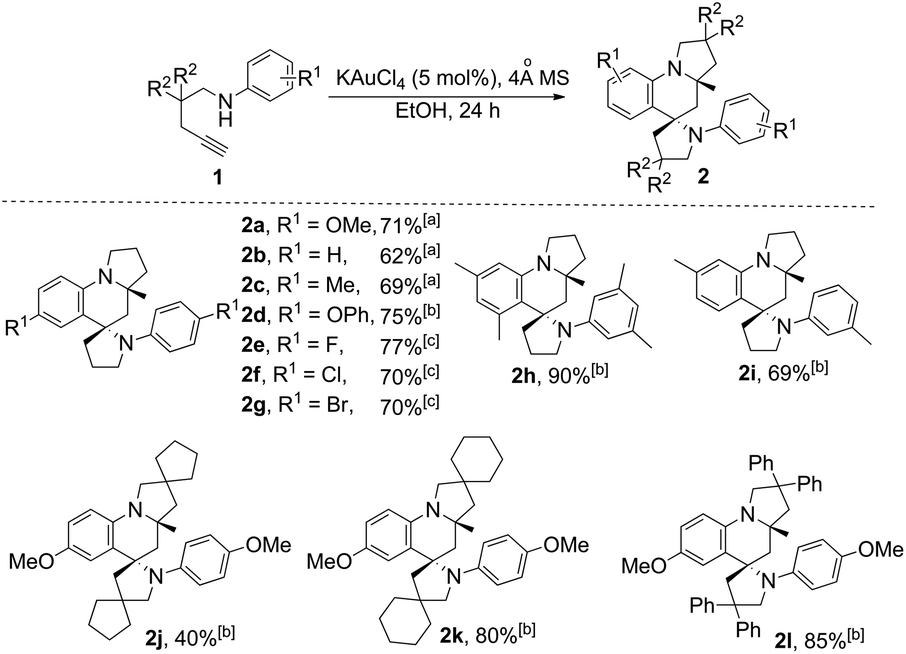 | ||
| Scheme 2 KAuCl4-catalyzed synthesis of azaspiro polycycles. Reaction conditions: 1 (1.0 mmol), KAuCl4 (5 mol%), 4 Å MS (100 mg), EtOH (1 mL) for 24 h. [a] At 40 °C. [b] At rt. [c] At 60 °C. | ||
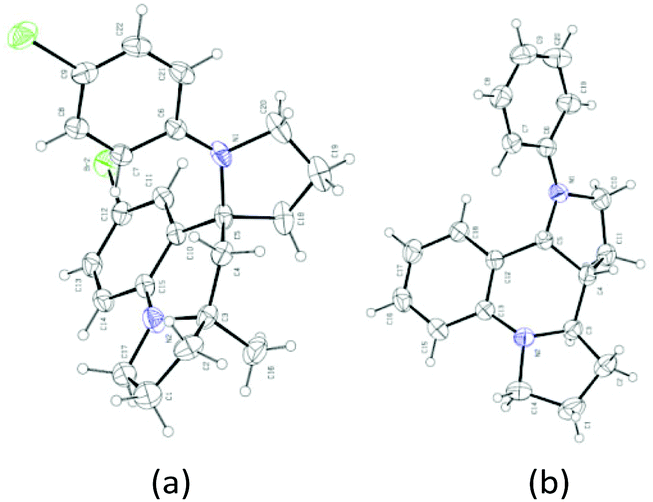 | ||
| Fig. 2 X-ray structures of 2g (a) and 4′ (b). | ||
When 1,4-aminoalkynes with an ortho-substituent (e.g., 2-Me) or strong electron-withdrawing substituents (e.g., 3,5-dichloro) were used, unfortunately, the reaction did not give the corresponding azaspiro polycycles under the conditions described above, and this could be attributed to the weak activation of the KAuCl4 catalyst for the subsequent aza-Diels–Alder reaction of the sterically more congested or electron-poor 2-azadiene intermediate.8b To further expand the scope of such useful tandem reactions, we tested the use of the more reactive Au(I) cation derived from the reaction of (tBu)2(o-diphenyl)PAuCl10 with an equimolar amount of AgN(Tf)2 as the catalyst. After a careful survey of the reaction conditions, we were pleased to find that the corresponding products 2m–2o were afforded in 53–95% yields, when 1,4-aminoalkynes 1m–1o bearing an ortho-substituent (e.g., 2-Me) and strong electron-withdrawing substituents (e.g., 3,5-dichloro, 4-CF3) were treated in the presence of 5 mol% of (t-Bu)2(o-biphenyl)PAuCl/AgN(Tf)2 (mol ratio = 1:1) with 4 Å MS in DCE (Scheme 3).
 | ||
| Scheme 3 Au(I)-catalyzed synthesis of azaspiro polycycles. Reaction conditions: 1 (1.0 mmol), (tBu)2(o-diphenyl)PAuCl/AgN(Tf)2 (5 mol%), 4 Å MS (100 mg), DCE (1 mL). [b] At rt for 11 h. [c] At 75 °C for 48 h. | ||
The azaspiro polycyclic molecules easily constructed from the current reaction from simple starting materials have a similar core structure with a wide variety of biologically active natural products (Fig. 1).2 The structure resemblance encouraged us to evaluate the biological activity of our products. Our preliminary studies revealed that 2j exhibited significantly high cytotoxicities against the A549 (human lung carcinoma) cell line (IC50 = 5.37 μM) and the MGC80-3 (human gastric adenocarcinoma) cell line (IC50 = 10.76 μM), suggesting a potential application of this class of complex polycyclic azaheterocylic molecules in anti-cancer studies.
The current protocol was also applied to achieve the one-step synthesis of two alkaloids encompassing a dipyrroloquinoline12 ring framework. To achieve such a dipyrroloquinoline ring framework, we selected 1,3-aminoalkyne 3 as the model substrate and were delighted to find that the tricyclic azaheterocyles 4 and 4′ as a mixture of two diastereomers (45:34 dr) were obtained in 79% yield with the use of (tBu)2(o-diphenyl)PAu(CH3CN)SbF6 (5 mol%) as a catalyst in DCM at room temperature (Scheme 4, eqn (1)). It should be noted that only two diastereoisomers of these products with three stereocenters were selectively obtained, favoring 2,3-cis-substituted diastereoselectivity and easily separated by simple flash chromatography. The relative configurations of 4′ were determined by X-ray crystallographic analysis (Fig. 2b).11 It is well-known that incargranine B was isolated from Incarvillea mairei var. grandiflora in 2010 by Zhang and co-workers,13a yet only one total synthesis of this molecule requiring six steps with only 50% combined yield in the key step has been recently reported by Lawrence and co-workers.13b For the purpose of accessing incargranine B aglycone 6 and 6′, we conducted the reaction of 5 under the standard conditions, and observed that the desired product was afforded in almost quantitative yield as a 56:40 mixture of diastereomers (Scheme 4, eqn (2)). This protocol could be scaled up to a gram-scale for the synthesis of incargranine B aglycone without a decrease in product yield. It should be noted that the use of the combination of the Au(I) catalyst and diphenyl phosphate as the catalyst resulted in improving the diastereoselectivity to 70:26 (6:6′). Most importantly, seneciobipyrrolidine (I) has been more recently isolated from Senecio scandens Buch.-Ham ex D. Don by Tan and co-workers, the latter is a plant used in folk medicine for the treatment of inflammation and bacterial infection in China.14 To synthesize (±)-seneciobipyrrolidine (I) for the first time, we carried out the tandem reaction of 7 with 5 mol% of the Au(I) catalyst alone, or the combination of the Au(I) catalyst and diphenyl phosphate, respectively. To our delight, the desired products 8 and 8′ were afforded in 84% and 85% yields as a 51:33 and 74:11 mixture of diastereomers; the analytical data were in agreement with those reported for natural seneciobipyrrolidine (I) (Scheme 4, eqn (3)).14
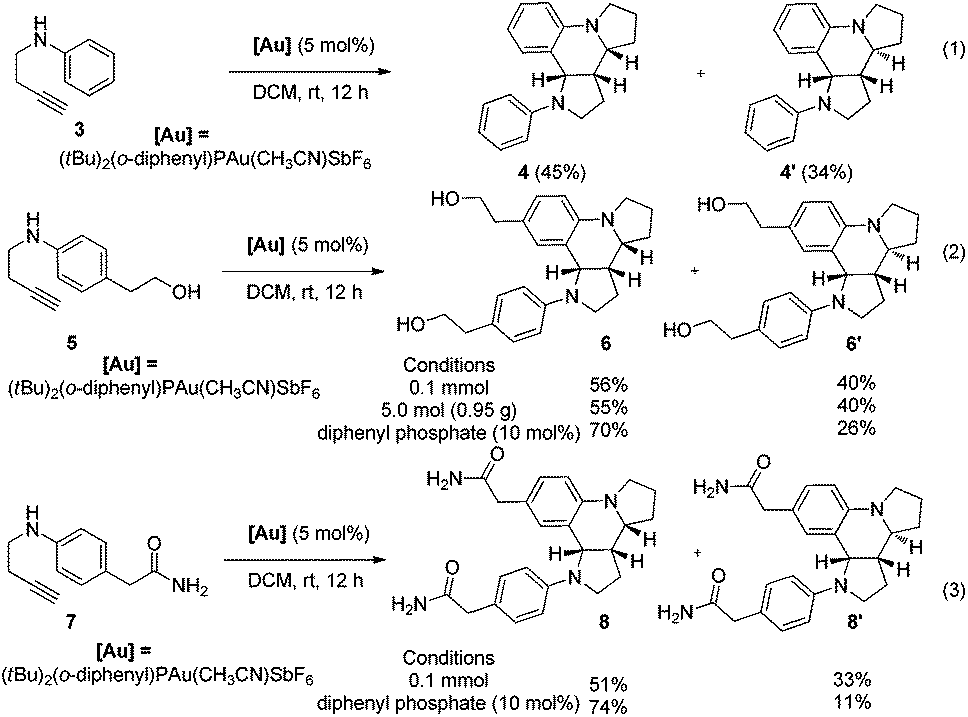 | ||
| Scheme 4 One-step synthesis of tricyclic azaheterocyles. | ||
Furthermore, we examined 1,3-aminoalkyne bearing internal alkyne (4-methoxy-N-(pent-3-yn-1-yl)aniline 1p) as the reaction substrate using KAuCl4 as the catalyst, to be of interest, the reaction gave compound 2a with good diastereoselectivity, albeit with moderate yield (38%). This could be attributed to the lower reaction activity of the internal alkyne (Scheme 5).
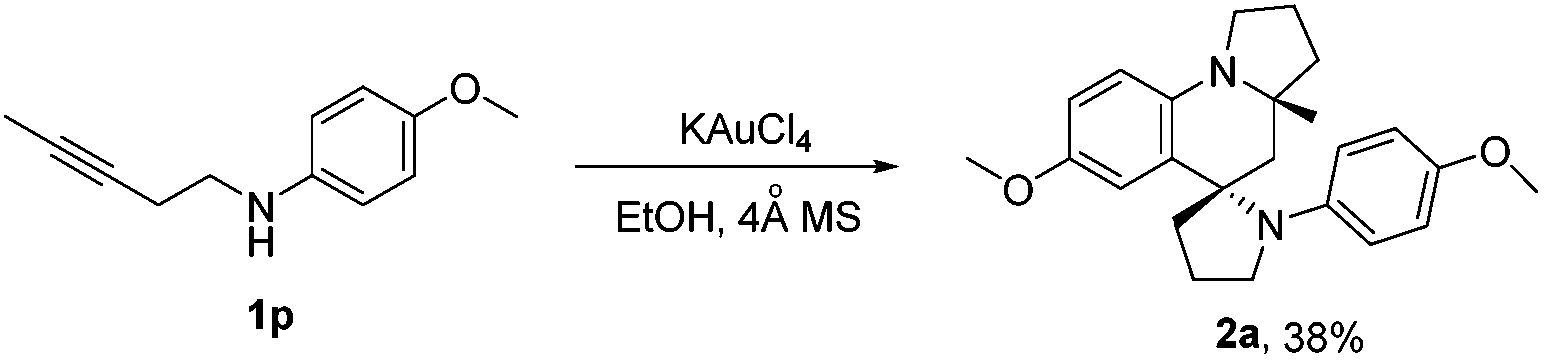 | ||
| Scheme 5 KAuCl4-catalyzed synthesis of azaspiro polycycles. Reaction conditions: 1p (1.0 mmol), KAuCl4 (5 mol%), 4 Å MS (100 mg), EtOH (1 mL) for 24 h at 40 °C. | ||
On the basis of the established reactivity of gold–alkyne complexes toward nucleophiles,3,8,15 a reaction mechanism for the Au-catalyzed tandem reaction of aminoalkynes is proposed (Scheme 6a and b). Taking 1,4-aminoalkyne 1 as an example, coordination of the triple bond of 1 to a gold catalyst16 and the subsequent nucleophilic attack of a nitrogen atom to the gold-coordinated alkyne A affords the gold–enamine intermediate B that is protonated to give enamine intermediate C. Once enamine C is formed, the second catalytic cycle “Povarov reaction” likely proceeds in a stepwise manner17 initiated by the Mannich reaction rather than via a concerted aza-Diels–Alder mechanism due to the polarized nature of the enamine double bond under the current reaction system. This hypothesis is supported by the finding that the seven-membered ring product 10 was obtained in 40% yield via the intramolecular trap of the iminium intermediate generated after the Mannich reaction by the hydroxyl nucleophile when 1,3-aminoalkyne 9 bearing an ortho-hydroxyl group was treated with a catalytic amount of (tBu)2(o-diphenyl)PAu(CH3CN)SbF6 for 0.5 h under otherwise identical conditions. In addition, compound 10 was completely transformed to the final product 11 in the presence of the gold(I) catalyst (Scheme 6c). These results provided direct evidence of a stepwise mechanism of our catalytic Povarov-type reaction. Thus, the enamine intermediate C approaches the corresponding iminium ion species D tautomerized from Cvia a Mannich reaction to afford intermediate E. A final intramolecular aza-Friedel–Crafts reaction activated with excellent stereoselectivity control via a favorable chairlike six-membered transition state F, in which the methyl group is placed in a pseudoequatorial position and the situation of the nitrogen atom of the iminium ion in a pseudoaxial position favored by the anomeric effect,6 followed by rearomatization and protodemetallation leads to final product 2. As a net result, two new C–C bonds and two C–N bonds are stereoselectively generated from this tandem annulation, as well as three new rings containing one spiro ring. On the other hand, the formation of dipyrroloquinoline ring framework 4 from 1,3-aminoalkyne 3 is also proposed in Scheme 6b.
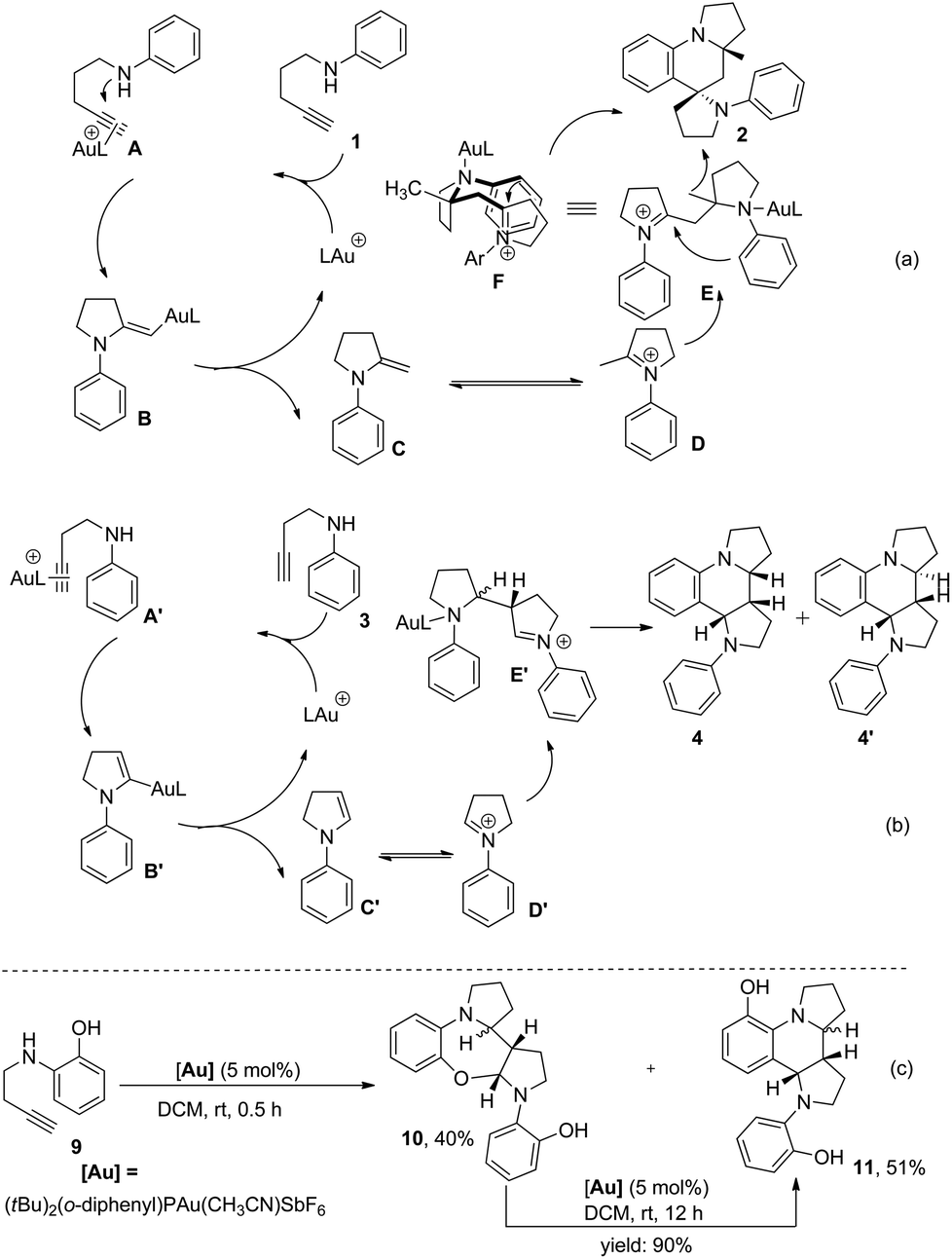 | ||
| Scheme 6 Proposed mechanism and control experiment. | ||
Conclusion
In summary, a catalytic transformation has been achieved on the basis of an intramolecular hydroamination and an aza-Diels–Alder tandem process of aminoalkynes with high regio- and diastereoselectivity and up to almost complete chemoselectivity in a broad spectrum of substrates. The developed gold-catalyzed tandem catalytic methodology showed great efficiency in multiple-bond formation and establishing spiro-dipyrroloquinolines with densely multiple stereogenic centers including quaternary carbons in a stereocontrolled fashion, and therefore provides a convenient one-step access to the complex azaheterocylic molecules of medicinal interest. This methodology allowed the first one-step synthesis of incargranine B aglycone and (±)-seneciobipyrrolidine (I) in good yield which represents the first application of the gold-catalyzed tandem methodology toward the highly efficient one-step synthesis of natural products. Further studies, including an asymmetric variant of this transformation and its synthetic application to chiral incargranine B and seneciobipyrrolidine (I), are currently underway in our laboratory.Acknowledgements
Financial support from the National Natural Science Foundation of China (no. 215722096, 21302088), the Shenzhen overseas high level talents innovation plan of technical innovation project (KQCX20150331101823702), the Shenzhen special funds for the development of biomedicine, Internet, new energy, and new material industries (JCYJ20150430160022517) and the South University of Science and Technology of China (FRG-SUSTC1501A-16) is greatly appreciated.Notes and references
- For selected recent examples, see: (a) D. L. Boger, C. W. Boyce, R. M. Garbaccio and J. A. Goldberg, Chem. Rev., 1997, 97, 787 CrossRef CAS PubMed; (b) S. M. Weinreb, Chem. Rev., 2006, 106, 2531 CrossRef CAS PubMed.
- (a) E. F. Rogers, H. R. Snyder and R. F. Fischer, J. Am. Chem. Soc., 1952, 74, 1987 CrossRef CAS; (b) P. Yates, F. N. MacLachlan, I. D. Rae, M. Rosenberger, A. G. Szabo, C. R. Willis, M. P. Cava, M. Behforouz, M. V. Lakshmikantham and W. Zeiger, J. Am. Chem. Soc., 1973, 95, 7842 CrossRef CAS; (c) A. J. Blackman and C. Li, Tetrahedron, 1993, 49, 8645 CrossRef CAS; (d) C. Boonlarppradab, C. A. Kauffman, P. R. Jensen and W. Fenical, Org. Lett., 2008, 10, 5505 CrossRef CAS PubMed; (e) M. C. Wilson, S.-J. Nam, T. A. M. Gulder, C. A. Kauffman, P. R. Jensen, W. Fenical and B. S. Moore, J. Am. Chem. Soc., 2011, 133, 1971 CrossRef CAS PubMed; (f) C. Dufour, J. Wink, M. Kurz, H. Kogler, H. Olivan, S. Sablé, W. Heyse, M. Gerlitz, L. Toti, A. Nuber, A. Rey, C. Couturier, A. Bauer and M. Brönstrup, Chem. – Eur. J., 2012, 18, 16123 CrossRef CAS PubMed.
- (a) X.-Y. Liu and C.-M. Che, Angew. Chem., Int. Ed., 2008, 47, 3805 CrossRef CAS PubMed; (b) X.-Y. Liu and C.-M. Che, Angew. Chem., Int. Ed., 2009, 48, 2367 CrossRef CAS PubMed.
- (a) J. Han, B. Xu and G. B. Hammond, J. Am. Chem. Soc., 2010, 132, 916 CrossRef CAS PubMed; (b) J. Han, B. Xu and G. B. Hammond, Org. Lett., 2011, 13, 3450 CrossRef CAS PubMed.
- For selected recent reviews on the aza Diels–Alder reaction, see: (a) P. Buonora, J.-C. Olsen and T. Oh, Tetrahedron, 2001, 57, 6099 CrossRef CAS; (b) V. V. Kouznetsov, Tetrahedron, 2009, 65, 2721 CrossRef CAS; (c) P. R. Girling, T. Kiyoi and A. Whiting, Org. Biomol. Chem., 2011, 9, 3105 RSC; (d) X. Jiang and R. Wang, Chem. Rev., 2013, 113, 5515 CrossRef CAS PubMed; (e) G. Masson, C. Lalli, M. Benohoud and G. Dagousset, Chem. Soc. Rev., 2013, 42, 902 RSC.
- For some selected elegant examples on the transition-metal-catalyzed tandem aza Diels–Alder reaction, see: (a) J. Barluenga, A. Mendoza, F. Rodrίguez and F. J. Fañanás, Angew. Chem., Int. Ed., 2008, 47, 7044 CrossRef CAS PubMed; (b) J. Barluenga, A. Mendoza, F. Rodrίguez and F. J. Fañanás, Chem. – Eur. J., 2008, 14, 10892 CrossRef CAS PubMed; (c) J. Barluenga, A. Braham, F. Rodrίguez and F. J. Fañanás, Angew. Chem., Int. Ed., 2009, 48, 1644 CrossRef CAS PubMed.
- Aza Diels–Alder reactions of preformed enamines as the dienophiles in the literature are restricted to the relative stable enamines, see: Y. Nomura, M. Kimura, Y. Takeuchi and S. Tomoda, Chem. Lett., 1978, 268 Search PubMed.
- For selected reviews on gold-catalyzed reactions, see: (a) D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395 CrossRef CAS PubMed; (b) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef CAS PubMed; (c) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239 CrossRef CAS PubMed; (d) E. Jiménez-Núñez and A. M. Echavarren, Chem. Rev., 2008, 108, 3326 CrossRef PubMed; (e) D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef PubMed; (f) S. M. Abu Sohel and R.-S. Liu, Chem. Soc. Rev., 2009, 38, 2269 RSC; (g) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2010, 49, 5232 CrossRef PubMed.
- For recent reviews on catalytic applications of NHC–Au complexes, see: (a) N. Marion and S. P. Nolan, Chem. Soc. Rev., 2008, 37, 1776 RSC; (b) S. P. Nolan, Acc. Chem. Res., 2011, 44, 91 CrossRef CAS PubMed.
- C. Nieto-Oberhuber, S. López and A. M. Echavarren, J. Am. Chem. Soc., 2005, 127, 6178 CrossRef PubMed.
- CCDC 1013665 (2g) and 1013666 (4′) contain the supplementary crystallographic data for this paper.
- For selected examples for the synthesis of a dipyrroloquinoline ring framework, see: (a) G. H. Kerr, O. Meth-Cohen, E. B. Mullock and H. Suschitzky, J. Chem. Soc., Perkin Trans. 1, 1974, 1614 RSC; (b) B. B. Snider, Y. Ahn and B. M. Foxman, Tetrahedron Lett., 1999, 40, 3339 CrossRef CAS; (c) D. W. Ma, C. F. Xia, J. Q. Jiang and J. H. Zhang, Org. Lett., 2001, 3, 2189 CrossRef CAS PubMed; (d) D. W. Ma, C. F. Xia, J. Q. Jiang, J. H. Zhang and W. J. Tang, J. Org. Chem., 2003, 68, 442 CrossRef CAS PubMed; (e) M. Buswell and I. Fleming, Chem. Commun., 2003, 202 RSC; (f) M. Buswell, I. Fleming, U. Ghosh, S. Mack, M. Russella and B. P. Clarkb, Org. Biomol. Chem., 2004, 2, 3006 RSC; (g) S. Fustero, P. Bello, J. Miro, M. Sanchez-Rosello, M. A. Maestro, J. Gonzalez and C. D. Pozo, Chem. Commun., 2013, 49, 1336 RSC; (h) M. Chang, S. Abbas and S. Daniel, Org. Lett., 2014, 16, 2756 CrossRef PubMed.
- (a) Y.-H. Shen, Y.-Q. Su, J.-M. Tian, S. Lin, H.-L. Li, J. Tang and W.-D. Zhang, Helv. Chim. Acta, 2010, 93, 2393 CrossRef CAS; (b) P. D. Brown, A. C. Willis, M. S. Sherburn and A. L. Lawrence, Angew. Chem., Int. Ed., 2013, 52, 13273 CrossRef CAS PubMed.
- D. Tan, G. Chou and Z. Wang, Chem. Nat. Compd., 2014, 50, 329 CrossRef CAS.
- (a) N. D. Shapiro and F. D. Toste, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2779 CrossRef CAS; (b) R. L. LaLonde, J. W. E. Brenzovich, D. Benitez, E. Tkatchouk, K. Kelley, W. A. Goddard III and F. D. Toste, Chem. Sci., 2010, 1, 226 RSC.
- For selected reviews, see: (a) C. Bruneau, Angew. Chem., Int. Ed., 2005, 44, 2328 CrossRef CAS PubMed; (b) L. Zhang, J. Sun and S. A. Kozmin, Adv. Synth. Catal., 2006, 348, 2271 CrossRef CAS; (c) V. Michelet, P. Y. Toullec and J.-P. Genêt, Angew. Chem., Int. Ed., 2008, 47, 4268 CrossRef CAS PubMed.
- For selected examples, see: (a) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Angew. Chem., Int. Ed., 2004, 43, 1566 CrossRef CAS PubMed; (b) D. Uraguchi and M. Terada, J. Am. Chem. Soc., 2004, 126, 5356 CrossRef CAS PubMed; (c) M. Terada, K. Machioka and K. Sorimachi, Angew. Chem., Int. Ed., 2006, 45, 2254 CrossRef CAS PubMed; (d) M. Terada, K. Machioka and K. Sorimachi, J. Am. Chem. Soc., 2007, 129, 10336 CrossRef CAS PubMed; (e) M. Terada, K. Soga and N. Momiyama, Angew. Chem., Int. Ed., 2008, 47, 4122 CrossRef CAS PubMed; (f) T. Yue, M.-X. Wang, D.-X. Wang, G. Masson and J. Zhu, Angew. Chem., Int. Ed., 2009, 48, 6717 CrossRef CAS PubMed; (g) C. Luo and Y. Huang, J. Am. Chem. Soc., 2013, 135, 8193 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, 1H and 13C NMR spectra, mechanism studies and biological assay. CCDC 1013665 and 1013666. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00354g |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2016 |