
Rhenium and base co-catalyzed [3 + 2] annulations of N–H ketimines and alkynes to access unprotected tertiary indenamines through C–H bond activation†
Xiqing
Jin
,
Xiaoxu
Yang
,
Yunhui
Yang
and
Congyang
Wang
*
Beijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Molecular Recognition and Function, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: wangcy@iccas.ac.cn
First published on 24th December 2015
Abstract
Herein, a rhenium-catalyzed [3 + 2] carbocyclization of N–H ketimines and alkynes through C–H bond activation to afford unprotected tertiary indenamines is described. Notably, the catalytic use of a cheap base, sodium carbonate, was demonstrated to be crucial to the success of this reaction. The protocol is also highlighted by a high atom-economy, a broad substrate scope, and excellent regioselectivity for unsymmetrical alkynes.
 Congyang Wang | Congyang Wang was born in Jiangsu province, China in 1977. He obtained his BS degree from Nanjing University in 2000 and a Ph.D. degree from Peking University under the guidance of Professor Zhenfeng Xi in 2005. After a postdoctoral stay of two more years in the same group, he moved to the University of Münster, Germany working with Professor Frank Glorius as an Alexander von Humboldt Research Fellow. In 2010, he started his independent research at the Institute of Chemistry, Chinese Academy of Sciences (ICCAS) as a professor. His research interests include organometallic chemistry, transition metal catalysis, and synthesis of functional molecules. Currently, his group is focusing on manganese-group-metal catalysis. |
Due to the ubiquitous nature of C–H bonds in organic compounds and the inherent atom- and step-economies of direct C–H bond transformations, C–H bond activation has recently attracted immense efforts from chemical communities. As a result, it has evolved as an important tool for new chemical bond formation and the efficient construction of complex molecules. Transition metal catalysis is among the most powerful tool kits for C–H bond activation because of the versatile modes of C–H bond breakage promoted by transition metal catalysts, such as oxidative addition, concerted metalation/deprotonation, σ-bond metathesis, and so on.1 Late transition metal (e.g. Pd, Rh, Ru, Ir) catalysts are the predominant players in this field and have witnessed an explosive progress in a large variety of C–H activation reactions during the past three decades.1 In sharp contrast, early transition metal compounds are far less explored as efficient catalysts for C–H bond transformations. As a prominent example of early transition metals, rhenium has demonstrated intriguing reactivity toward inert C–H bond activation in particular for cyclization reactions.2–4 In 2005, Takai and Kuninobu et al. reported rhenium-catalyzed annulations of aromatic aldimines and alkynes leading to conjugated indenamines via a C–H addition/cyclization/isomerization sequence (Scheme 1a).4 An oxidative addition pathway was proposed for the C–H bond activation step in this reaction. Very recently, we have reported manganese-catalyzed [4 + 2] annulations of N–H ketimines and alkynes to approach diverse isoquinolines with the evolution of hydrogen gas (Scheme 1b).5a,6 Of note, only MnBr(CO)5 was used as the catalyst for this reaction without the need for any oxidant, external ligand, or additive. It remains a question whether the reaction of N–H ketimines and alkynes employing a congeneric rhenium catalyst would form the same isoquinoline products via [4 + 2] annulation or, alternatively, indenamine products through [3 + 2] cyclization. To address this query and based on our continuous interest in rhenium-catalysis,3,7 herein we describe rhenium-catalyzed [3 + 2] annulations of N–H ketimines and alkynes to access unprotected indenamines bearing a quaternary carbon center (Scheme 1c).8,9 Importantly, the catalytic use of a cheap base, sodium carbonate, is essential to the success of this transformation. Mechanistically, a base-assisted deprotonation pathway operates in the key C–H activation step, which is in sharp contrast to the previous work in Scheme 1a.4 Also, the reaction features a high atom-economy, excellent regioselectivity for unsymmetrical alkynes, and a broad substrate scope for both ketimines and alkynes.
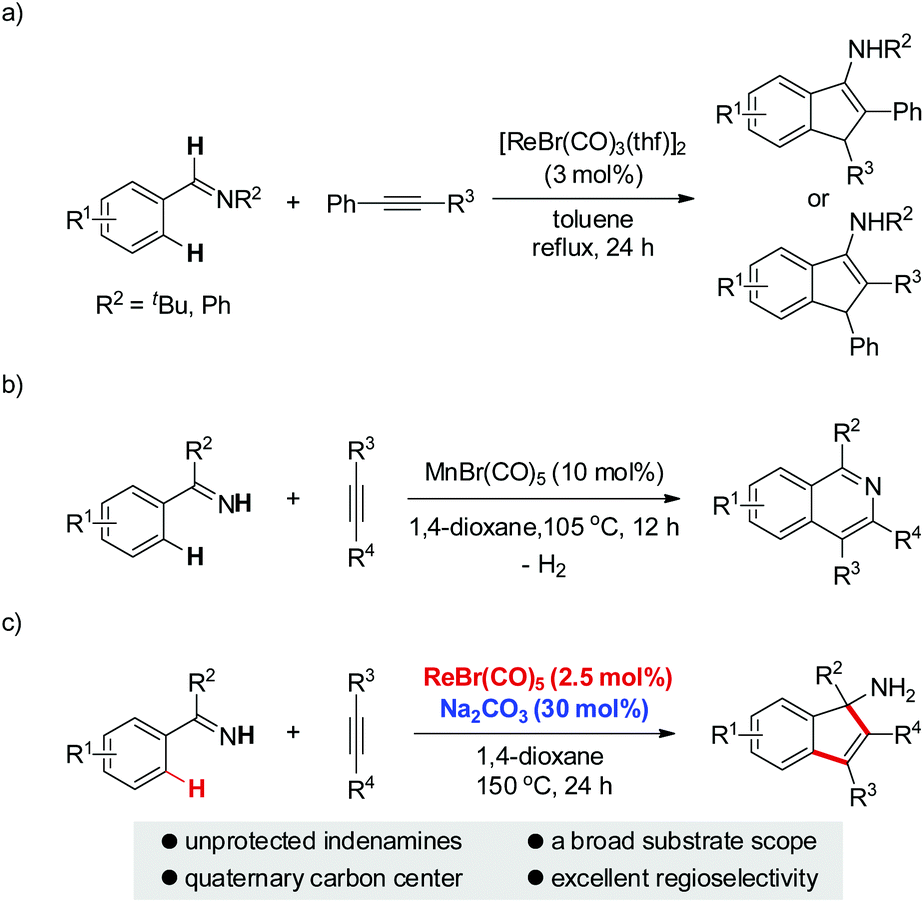 | ||
| Scheme 1 Manganese-group-metal catalyzed annulations of imines and alkynes via C–H bond activation. (a) Re-catalyzed annulations of aldimines and alkynes: Kuninobu and Takai. (b) Mn-catalyzed annulations of N–H ketimines and alkynes: our previous work. (c) Re-base co-catalyzed annulations of N–H ketimines and alkynes: this work. | ||
At the outset, we selected the model reaction of ketimine 1c and diphenylacetylene 2a to screen the reaction parameters (Table 1).10 No products were observed with the mere use of ReBr(CO)5 as a catalyst (entry 1). Inspired by our recent findings on acetate-accelerated Re-catalyzed C–H activation reactions,3c,e we tested the addition of a series of different bases in the current reaction. To our delight, with a 30 mol% amount of NaOAc the [3 + 2] annulation product 3ca was obtained in 20% 1H NMR yield without the formation of the [4 + 2] cyclization product 4ca (entry 2), which is in contrast to the reaction catalyzed by MnBr(CO)5.5 It was further shown that strong bases were detrimental to the reaction (entries 3–6), while relatively weak bases gave better results with Na2CO3 being the optimal (entries 7–10). Variations on the solvents showed that the use of 1,4-dioxane gave a nearly quantitative yield of 3ca comparable to that in THF (entries 11–13).10 As a result, 1,4-dioxane was selected as the optimal solvent because of its suitable boiling point for the reaction. Other rhenium and manganese carbonyl catalysts gave no better results in the reaction (entries 14–16). Further tunings on the reaction conditions demonstrated that the catalyst loading could be lowered to 2.5 mol% while maintaining equal reactivity (entry 17). Finally, 3ca was isolated as a pure compound in 95% yield. Of note, no excessive amount of either ketimine 1c or alkyne 2a was required in this reaction.
|
|
||||
|---|---|---|---|---|
| Entry | Cat. (5 mol%) | Additive (30 mol%) | Solvent | Yield of 3ca![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (%) b (%) |
| a Reaction conditions unless otherwise noted: 1c (0.2 mmol), 2a (0.2 mmol), catalyst (0.01 mmol), base (0.06 mmol), solvent (2 mL), 150 °C, 24 h. b Yields determined by 1H NMR analysis with an internal standard. c 140 °C. d No additive. e Isoquinoline 4ca was formed in 21% 1H NMR yield. f ReBr(CO)5 (2.5 mol%). g Isolated yield on the 1.0 mmol scale. PMP = p-methoxyphenyl. | ||||
| 1c | ReBr(CO)5 | —d | THF | 0 |
| 2c | ReBr(CO)5 | NaOAc | THF | 20 |
| 3c | ReBr(CO)5 | PhLi | THF | 0 |
| 4c | ReBr(CO)5 | PhMgBr | THF | 0 |
| 5c | ReBr(CO)5 | NaH | THF | Trace |
| 6c | ReBr(CO)5 | KOtBu | THF | Trace |
| 7c | ReBr(CO)5 | LiOtBu | THF | 85 |
| 8c | ReBr(CO)5 | Cs2CO3 | THF | 54 |
| 9c | ReBr(CO)5 | CsF | THF | 96 |
| 10c | ReBr(CO)5 | Na2CO3 | THF | 98 |
| 11 | ReBr(CO)5 | Na2CO3 | Toluene | 87 |
| 12 | ReBr(CO)5 | Na2CO3 | 1,4-Dioxane | 98 |
| 13 | ReBr(CO)5 | Na2CO3 | DMSO | 88 |
| 14 | ReCl(CO)5 | Na2CO3 | 1,4-Dioxane | 61 |
| 15 | Re2(CO)10 | Na2CO3 | 1,4-Dioxane | 74 |
| 16 | MnBr(CO)5 | Na2CO3 | 1,4-Dioxane | Tracee |
| 17f | ReBr(CO)5 | Na2CO3 | 1,4-Dioxane | 97 (95)g |
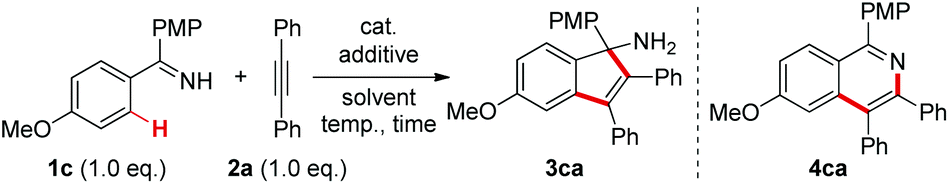
With the optimized conditions in hand, we first examined the reaction scope with regard to N–H ketimines as shown in Scheme 2. Both electron-donating and -withdrawing groups on the benzene moiety of N–H ketimines were well tolerated leading to products 3aa–ea in moderate to excellent yields. It was shown that electron-rich ketimines had better reactivity than the electron-deficient ones. Primary, secondary, and tertiary alkyl aryl N–H ketimines with increasing steric hindrance were all suitable substrates for this reaction giving the corresponding products 3fa–ha smoothly. The ortho-methyl substituent had no severe influence on the reaction yield of 3ia, which contrasted obviously with the reaction of aldimines and alkynes.4 When meta-substituted aromatic ketimine 1j was employed in the reaction, the sterically less congested C–H bond was preferably annulated affording indenamine 3ja as the major product. Of note, two regioisomeric products 3ka and 3ka′ were formed in the case of the unsymmetrical diaryl ketimine 1k. Finally, conjugated indenamine 3la was also obtained successfully from the annulation of aldimine 1l and diphenylacetylene 2a under slightly modified conditions.4
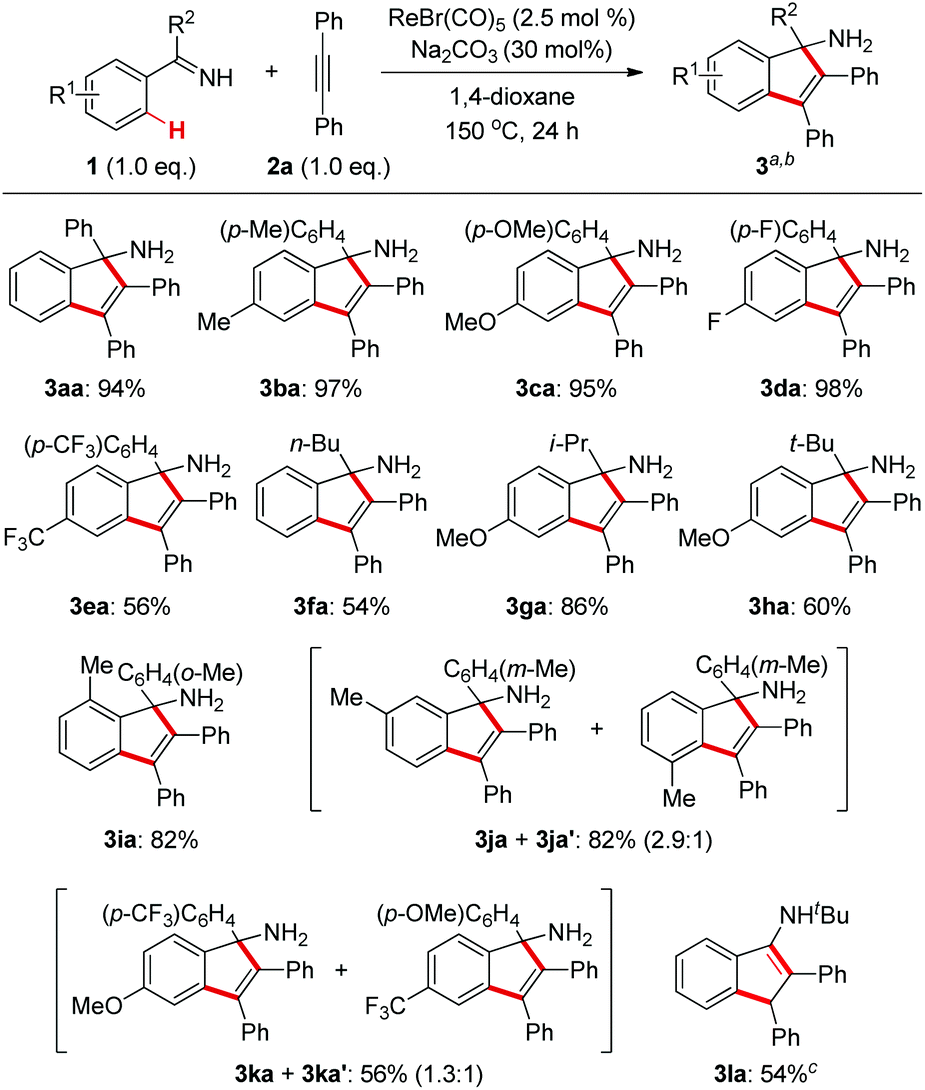 | ||
| Scheme 2 Substrate scope of N–H ketimines. aReaction conditions: 1 (1.0 mmol), 2a (1.0 mmol), ReBr(CO)5 (2.5 mol%), Na2CO3 (30 mol%), 1,4-dioxane (1.5 mL), 150 °C, 24 h. bIsolated yields of 3 are shown. cRe2(CO)10 (2.5 mol%) was used. | ||
Next, we explored the substrate scope of alkynes with ketimine 1c as the reaction partner (Scheme 3). It turned out that the electronic variations on the diaryl alkynes showed no obvious effect on the reaction outcome (3cb and 3cc). Importantly, halogen functionalities such as F, Cl, and Br were all compatible with the reaction conditions (3cd–f). ortho- and meta-substituted diaryl alkynes gave the corresponding products successfully (3cg–h). As an example of heteroaryl alkynes, dithiophenylacetylene delivered the expected indenamine (3ci) in excellent yield. Furthermore, perfect regioselectivity was obtained when unsymmetrical aryl alkyl alkynes were employed in this reaction with the aryl group posed adjacent to the free amino functionality (3ak, 3cj, 3ck). The structure of 3ak was confirmed by comparison with known spectral data in the literature.10 Of particular note, 4-octyne bearing no aryl group was also amenable to this annulation protocol which again demonstrated its wider substrate scope than that of the reaction between aldimines and alkynes.4
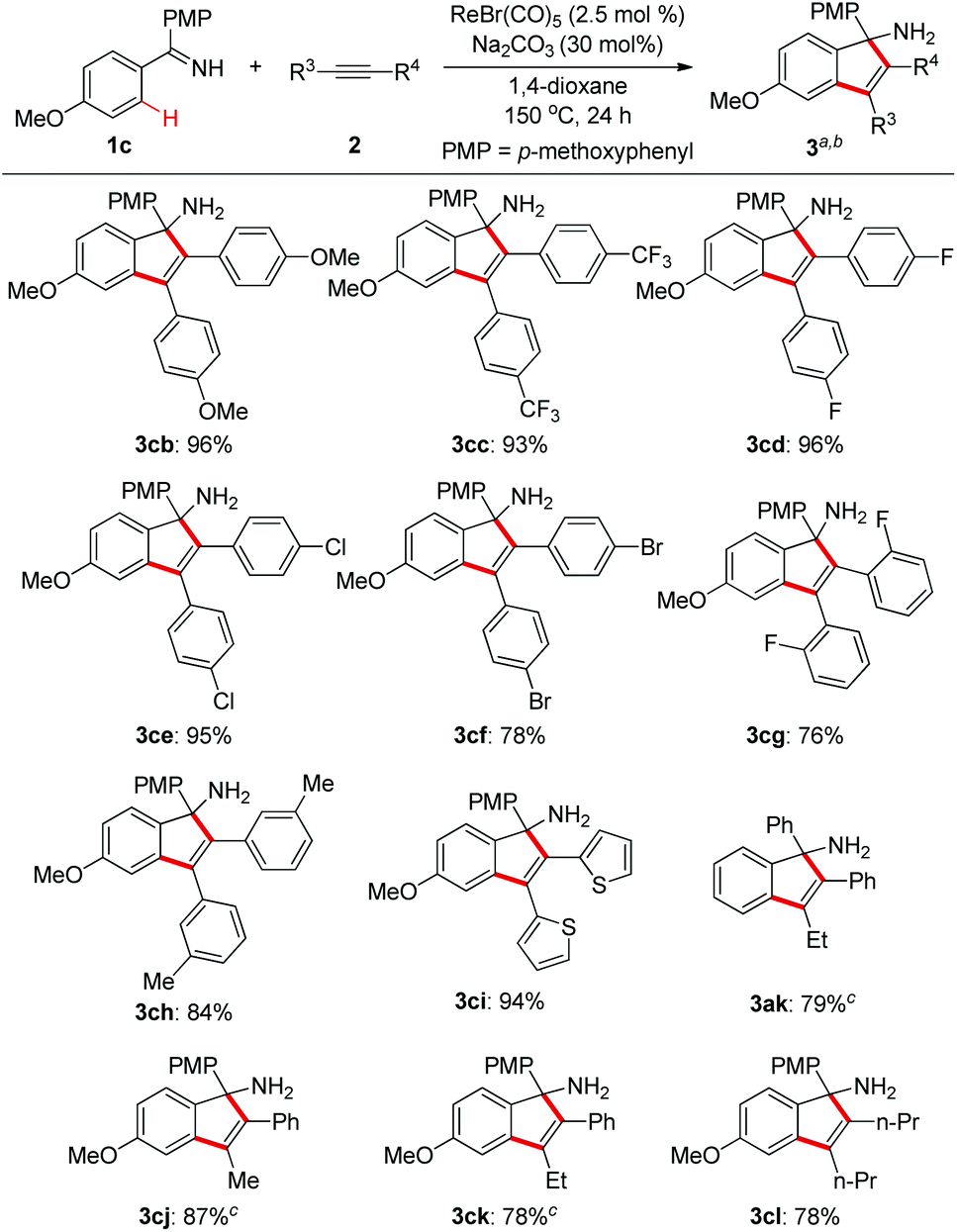 | ||
| Scheme 3 Substrate scope of alkynes. aReaction conditions: 1c (1.0 mmol), 2 (1.0 mmol), ReBr (CO)5 (2.5 mol%), Na2CO3 (30 mol%), 1,4-dioxane (1.5 mL), 150 °C, 24 h. bIsolated yields of 3 are shown. cSingle isomer was detected. | ||
To shed light on the possible reaction mechanism, competition experiments between two ketimines and alkynes bearing electronically varied groups were investigated. It turned out that ketimine 1c containing an electron-donating group (OMe) exhibited much better reactivity than that of ketimine 1e with an electron-withdrawing group (CF3) (Scheme 4a). This result might originate from the stronger coordination ability of the imino functionality of 1c with the rhenium center, which facilitated the following C–H activation step. On the other hand, alkyne 2c bearing an electron-withdrawing group (CF3) was slightly preferred to alkyne 2b having an electron-donating group (OMe) (Scheme 4b), which might arise from the higher propensity of 2c toward nucleophilic attack by the cyclic rhenium species formed after the C–H activation (vide infra).
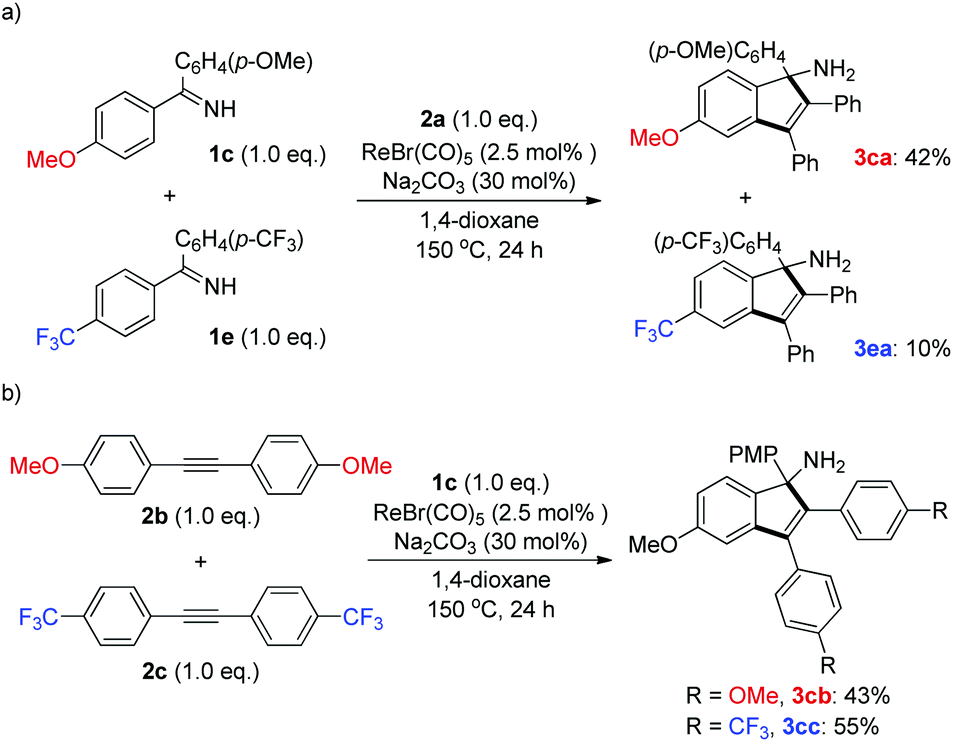 | ||
| Scheme 4 Competition experiments. (a) Competition experiments of ketimines; (b) competition experiments of alkynes. | ||
Furthermore, the deuterium-labeling experiments were carried out. First, decadeuterated ketimine 1a-d10 was synthesized and subjected to a reaction with alkyne 2a (Scheme 5a). After 6 h, the reaction was stopped and analyzed. It was shown that partial deuterium-loss occurred at the ortho-positions of the indenamine product 3aa-d9. A similar phenomenon was found in the deuterated ketone 5a-d10 isolated from the hydrolysis of unconverted ketimine. These results suggested a reversible deprotonative C–H activation step, which is in agreement with the previously reported mechanism in Re/base systems.3 In addition, a competition experiment as well as two parallel reactions for KIE measurements between 1a and 1a-d10 were conducted (Scheme 5b and c). The results indicated that the cleavage of the C–H bond might be involved in the rate-determining step of the reaction.
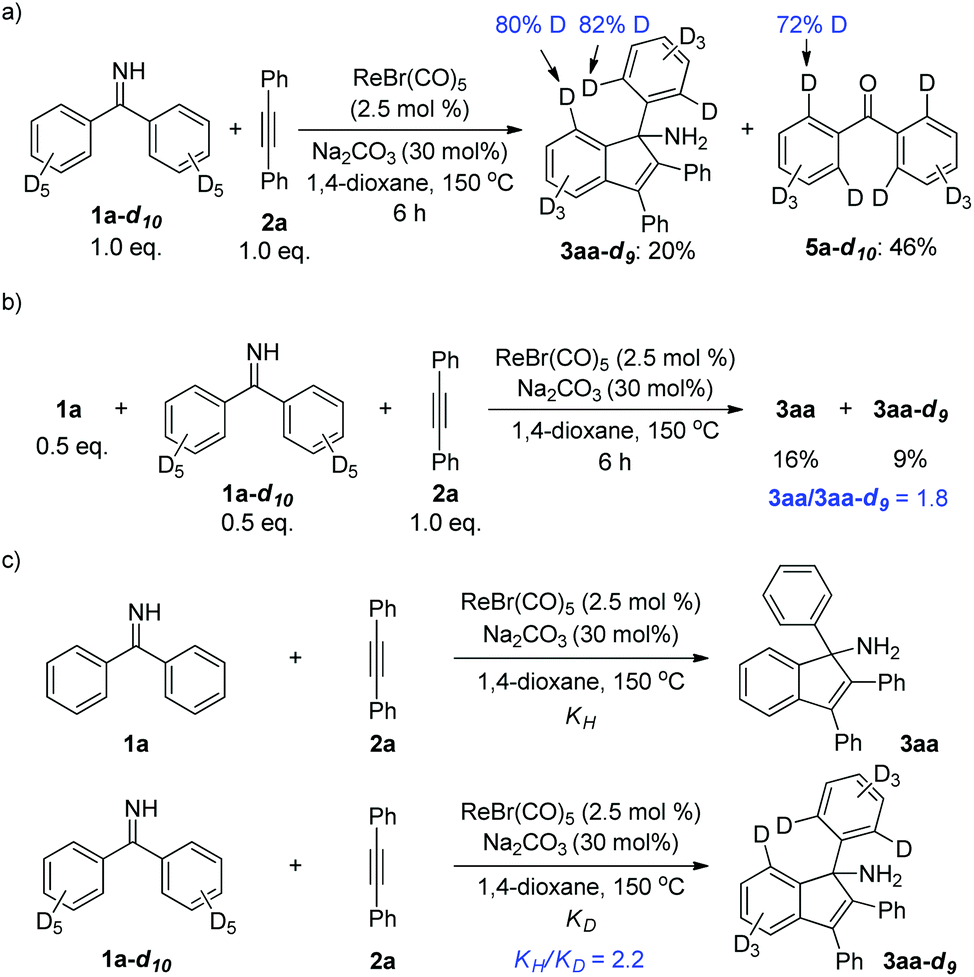 | ||
| Scheme 5 Deuterium-labeling experiments. (a) H/D scrambling experiment; (b) KIE determined from intermolecular competition; (c) KIE determined by two parallel reactions. | ||
Based on these results and the literature clues,3,4 a tentative reaction mechanism was proposed in Scheme 6. First, ReBr(CO)5 reacted with ketimine 1a in the presence of Na2CO3 to generate rhenacycle A. The ligand exchange of CO with alkyne 2a gave the rhenium-alkyne complex B, which led to the seven-membered rhenacycle C through a migratory insertion of the alkyne. The intramolecular nucleophilic attack of the C–Re bond on the imino group in C resulted in the formation of intermediate D. Protonation of D gave the target [3 + 2] annulation product 3aa and formed the rhenium species E, which promoted the C–H bond cleavage of 1a with the assistance of the base regenerating rhenacycle A. The base, Na2CO3, acting as a proton shuttle in the reaction, facilitated the deprotonative C–H activation of 1a and the protonation of species D.
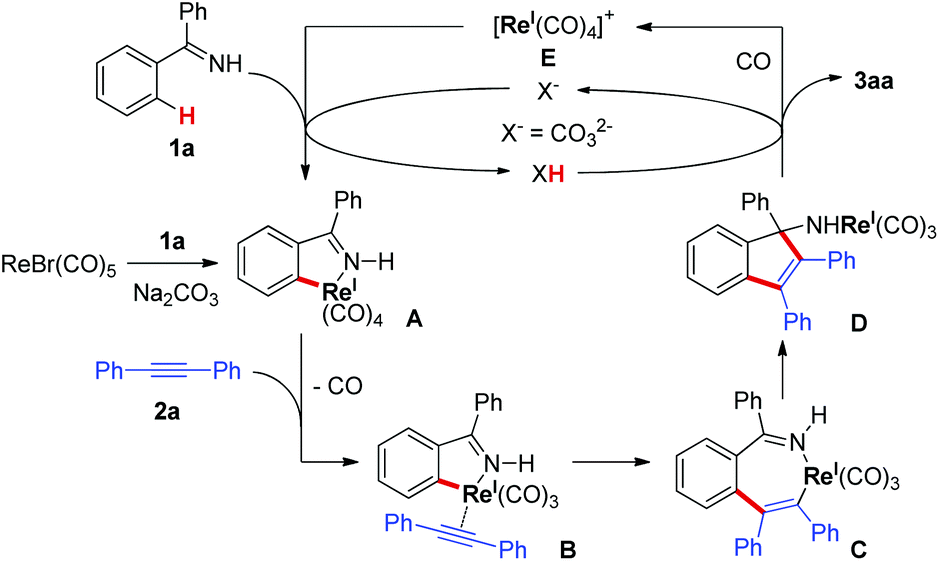 | ||
| Scheme 6 A tentative reaction mechanism. | ||
In conclusion, we have developed rhenium and base co-catalyzed [3 + 2] annulations of N–H ketimines and alkynes to approach the unprotected tertiary indenamines through C–H bond activation. The reaction demonstrates a high atom-economy, a broad substrate scope for both ketimines and alkynes, and excellent regioselectivity for unsymmetrical alkyne insertion. Mechanistic studies suggested a base-assisted deprotonation pathway occurring in the C–H bond activation step. Further implementations of rhenium/base co-catalysis in novel C–H transformations are underway in our laboratory.
Financial support from the National Basic Research Program of China (973 Program) (No. 2012CB821600) and the National Natural Science Foundation of China (21322203, 21272238, 21472194) is gratefully acknowledged.
Notes and references
- For selected reviews, see: (a) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (b) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS PubMed; (c) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (d) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed; (e) E. Nakamura and N. Yoshikai, J. Org. Chem., 2010, 75, 6061 CrossRef CAS PubMed; (f) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (g) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (h) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (i) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed; (j) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (k) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2011, 44, 814 Search PubMed; (l) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (m) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (n) C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464 RSC; (o) A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177 CrossRef CAS PubMed; (p) M. C. White, Science, 2012, 335, 807 CrossRef CAS PubMed; (q) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (r) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed; (s) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed; (t) C. Wang, Synlett, 2013, 1606 CrossRef CAS; (u) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208 CrossRef CAS PubMed; (v) R. He, Z.-T. Huang, Q.-Y. Zheng and C. Wang, Tetrahedron Lett., 2014, 55, 5705 CrossRef CAS; (w) F. Jia and Z. Li, Org. Chem. Front., 2014, 1, 194 RSC; (x) Y. Yang and C. Wang, Sci. China: Chem., 2015, 58, 1266 CrossRef CAS; (y) B. Liu and B.-F. Shi, Tetrahedron Lett., 2015, 56, 15 CrossRef CAS; (z) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC.
- For a review, see: (a) Y. Kuninobu and K. Takai, Chem. Rev., 2011, 111, 1938 CrossRef CAS PubMed. For selected examples, see: (b) H. Chen and J. F. Hartwig, Angew. Chem., Int. Ed., 1999, 38, 3391 CrossRef CAS; (c) Y. Kuninobu, Y. Nishina, C. Nakagawa and K. Takai, J. Am. Chem. Soc., 2006, 128, 12376 CrossRef CAS PubMed; (d) Y. Kuninobu, Y. Nishina, T. Matsuki and K. Takai, J. Am. Chem. Soc., 2008, 130, 14062 CrossRef CAS PubMed; (e) Q. Liu, Y.-N. Li, H.-H. Zhang, B. Chen, C.-H. Tung and L.-Z. Wu, J. Org. Chem., 2011, 76, 1444 CrossRef CAS PubMed; (f) Y. Fukumoto, M. Daijo and N. Chatani, J. Am. Chem. Soc., 2012, 134, 8762 CrossRef CAS PubMed; (g) H. Peng, A. Lin, Y. Zhang, H. Jiang, J. Zhou, Y. Cheng, C. Zhu and H. Hu, ACS Catal., 2012, 2, 163 CrossRef CAS; (h) H. Jin, J. Xie, C. Pan, Z. Zhu, Y. Cheng and C. Zhu, ACS Catal., 2013, 3, 2195 CrossRef CAS; (i) S. Sueki, Y. Guo, M. Kanai and Y. Kuninobu, Angew. Chem., Int. Ed., 2013, 52, 11879 CrossRef CAS PubMed; (j) H. Jin, Z. Zhu, N. Jin, J. Xie, Y. Cheng and C. Zhu, Org. Chem. Front., 2015, 2, 378 RSC.
- (a) D. Xia, Y. Wang, Z. Du, Q.-Y. Zheng and C. Wang, Org. Lett., 2012, 14, 588 CrossRef CAS PubMed; (b) Q. Tang, D. Xia, X. Jin, Q. Zhang, X.-Q. Sun and C. Wang, J. Am. Chem. Soc., 2013, 135, 4628 CrossRef CAS PubMed; (c) X. Geng and C. Wang, Org. Lett., 2015, 17, 2434 CrossRef CAS PubMed; (d) H. Gu and C. Wang, Org. Biomol. Chem., 2015, 13, 5880 RSC; (e) X. Geng and C. Wang, Org. Biomol. Chem., 2015, 13, 7619 RSC.
- (a) Y. Kuninobu, A. Kawata and K. Takai, J. Am. Chem. Soc., 2005, 127, 13498 CrossRef CAS PubMed; (b) Y. Kuninobu, Y. Tokunaga, A. Kawata and K. Takai, J. Am. Chem. Soc., 2006, 128, 202 CrossRef CAS PubMed; (c) Y. Kuninobu, H. Ueda, A. Kawata and K. Takai, J. Org. Chem., 2007, 72, 6749 CrossRef CAS PubMed; (d) Y. Kuninobu, Y. Nishina, A. Kawata, M. Shouho and K. Takai, Pure Appl. Chem., 2008, 80, 1149 CrossRef CAS.
- (a) R. He, Z.-T. Huang, Q.-Y. Zheng and C. Wang, Angew. Chem., Int. Ed., 2014, 53, 4950 CrossRef CAS PubMed. For related annulations: (b) W. Liu, D. Zell, M. John and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 4092 CrossRef CAS PubMed.
- For selected [4 + 2] annulations of imines and alkynes, see: (a) S.-G. Lim, J. H. Lee, C. W. Moon, C. J.-B. Hong and C.-H. Jun, Org. Lett., 2003, 5, 2759 CrossRef CAS PubMed; (b) N. Guimond and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 12050 CrossRef CAS PubMed; (c) T. Fukutani, N. Umeda, K. Hirano, T. Satoh and M. Miura, Chem. Commun., 2009, 5141 RSC; (d) X. Wei, M. Zhao, Z. Du and X. Li, Org. Lett., 2011, 13, 4636 CrossRef CAS PubMed; (e) K. Parthasarathy and C.-H. Cheng, J. Org. Chem., 2009, 74, 9359 CrossRef CAS PubMed; (f) T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 11846 RSC; (g) R. K. Chinnagolla, S. Pimparkar and M. Jeganmohan, Org. Lett., 2012, 14, 3032 CrossRef CAS PubMed; (h) C. Kornhaaß, J. Li and L. Ackermann, J. Org. Chem., 2012, 77, 9190 CrossRef PubMed; (i) H. Wang, C. Grohmann, C. Nimphius and F. Glorius, J. Am. Chem. Soc., 2012, 134, 19592 CrossRef CAS PubMed; (j) L. Zheng, J. Ju, Y. Bin and R. Hua, J. Org. Chem., 2012, 77, 5794 CrossRef CAS PubMed; (k) T. Gerfaud, L. Neuville and J. Zhu, Angew. Chem., Int. Ed., 2009, 48, 572 CrossRef CAS PubMed; (l) P. C. Too, Y.-F. Wang and S. Chiba, Org. Lett., 2010, 12, 5688 CrossRef CAS PubMed; (m) W. Liu, X. Hong and B. Xu, Synthesis, 2013, 2137 CAS; (n) X.-C. Huang, X.-H. Yang, R.-J. Song and J.-H. Li, J. Org. Chem., 2014, 79, 1025 CrossRef CAS PubMed; J. Jayakumar, K. Parthasarathy and C.-H. Cheng, Angew. Chem., Int. Ed., 2012, 51, 197 Search PubMed; (o) G. Zhang, L. Yang, Y. Wang, Y. Xie and H. Huang, J. Am. Chem. Soc., 2013, 135, 8850 CrossRef CAS PubMed; (p) S. Zhang, D. Huang, G. Xu, S. Cao, R. Wang, S. Peng and J. Sun, Org. Biomol. Chem., 2015, 13, 7920 RSC; (q) L. Qiu, D. Huang, G. Xu, Z. Dai and J. Sun, Org. Lett., 2015, 17, 1810 CrossRef CAS PubMed; (r) X. Peng, W. Wang, C. Jiang, D. Sun, Z. Xu and C.-H. Tung, Org. Lett., 2014, 16, 5354 CrossRef CAS PubMed; (s) W. Wang, X. Peng, X. Qin, X. Zhao, C. Ma, C.-H. Tung and Z. Xu, J. Org. Chem., 2015, 80, 2835 CrossRef CAS PubMed; (t) B. Liu, F. Hu and B.-F. Shi, Adv. Synth. Catal., 2014, 356, 2688 CrossRef CAS.
- (a) Y. Wang, L. Zhang, Y. Yang, P. Zhang, Z. Du and C. Wang, J. Am. Chem. Soc., 2013, 135, 18048 CrossRef CAS PubMed; (b) G. Mao, B. Jia and C. Wang, Chin. J. Org. Chem., 2015, 35, 284 CrossRef CAS.
- For Rh- and Ru-catalyzed [3 + 2] annulations of N–H ketimines and alkynes, see: (a) Z.-M. Sun, S.-P. Chen and P. Zhao, Chem. – Eur. J., 2010, 16, 2619 CrossRef CAS PubMed; (b) D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 11098 CrossRef CAS PubMed; (c) J. Zhang, A. Ugrinov and P. Zhao, Angew. Chem., Int. Ed., 2013, 52, 6681 CrossRef CAS PubMed.
- For [3 + 2] annulations of N-substituted imines and alkynes, see: (a) P. Zhao, F. Wang, K. Han and X. Li, Org. Lett., 2012, 14, 5506 CrossRef CAS PubMed; (b) Y. Chen, F. Wang, W. Zhen and X. Li, Adv. Synth. Catal., 2013, 355, 353 CAS; (c) L. Dong, C.-H. Qu, J.-R. Huang, W. Zhang, Q.-R. Zhang and J.-G. Deng, Chem. – Eur. J., 2013, 19, 16537 CrossRef CAS PubMed; (d) J.-R. Huang, Q. Song, Y.-Q. Zhu, L. Qin, Z.-Y. Qian and L. Dong, Chem. – Eur. J., 2014, 20, 16882 CrossRef CAS PubMed; (e) M. Nagamoto and T. Nishimura, Chem. Commun., 2014, 50, 6274 RSC; (f) X. Zhou, S. Yu, Z. Qi and X. Li, Sci. China: Chem., 2015, 58, 1297 CrossRef CAS; (g) J.-R. Huang, L. Qin, Y.-Q. Zhu, Q. Song and L. Dong, Chem. Commun., 2015, 51, 2844 RSC.
- For more details, see the ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data and NMR spectra of all new compounds. See DOI: 10.1039/c5qo00336a |
| This journal is © the Partner Organisations 2016 |