
Rh(III)-catalyzed redox-neutral annulation of azo and diazo compounds: one-step access to cinnolines†
Peng
Sun
,
Youzhi
Wu
,
Yue
Huang
,
Xiaoming
Wu
,
Jinyi
Xu
,
Hequan
Yao
* and
Aijun
Lin
*
State Key Laboratory of Natural Medicines (SKLNM) and Department of Medicinal Chemistry, School of Pharmacy, China Pharmaceutical University, Nanjing, 210009, P. R. China. E-mail: ajlin@cpu.edu.cn; hyao@cpu.edu.cn
First published on 26th November 2015
Abstract
Reported herein is a Rh-catalyzed redox-neutral annulation reaction between azo and diazo compounds, thus leading to the direct synthesis of cinnolines under mild conditions. The procedure exhibited a broad substrate scope, scalability and step economy, obviating the pre-functionalization of substrates and the use of oxidants. Further utilization of the resulting cinnolines provided efficient routes to some bioactive structures, such as 4-amino-cinnoline and 1-aminoindole.
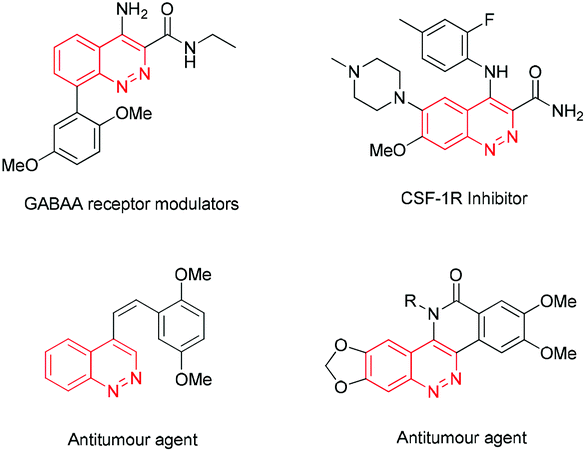
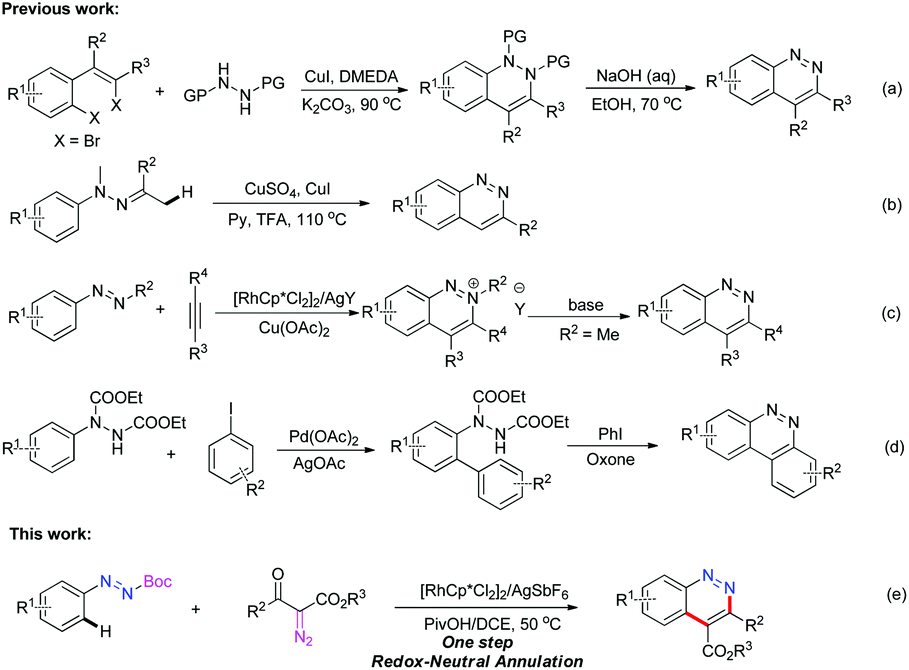
Cinnolines are privileged structural motifs widely found in materials science, natural products and pharmaceutical agents with bactericidal, anti-hypertensive, anti-inflammatory, anti-cancer activities, as well as luminescence and optical properties (Fig. 1).1 The classical synthetic approaches to cinnolines normally start from ortho-alkyl-anilines or ortho-vinyl-anilines, employing hydrochloric acid and sodium nitrite via intra-molecular ring-closing reactions.2 However, the restricted substrate scope and strongly acidic conditions limited their wide applications. Recently, transition-metal-catalyzed direct C–C or C–N bond formation reactions have emerged as versatile tools to cinnolines.3 In 2012, Willis4 reported a two-step route to cinnolines via Cu-mediated C–N bond-formation and base-promoted deprotecting processes (Scheme 1a). Ge
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 prepared C3 substituted cinnolines via Cu-catalyzed intramolecular cyclization and aromatization of hydrazones (Scheme 1b). In 2013, Cheng and You reported Rh-catalyzed annulation to cinnolinium salts with internal alkynes and azo compounds (Scheme 1c).6 After an extra base-mediated demethylation, the generated 2-methyl cinnolinium salts could be transformed to cinnolines. Additionally, in You's work, tert-butyl substituted azo compounds could react with di-alkyl alkynes to offer cinnolines by employing stoichiometric Cu(OAc)2 as an oxidant at 100–120 °C in moderate yields. Very recently, Reddy7 reported a two-step method to benzo[c]cinnolines from aryl-hydrazine dicarboxylates via the substrate-directed Pd-catalyzed C–H arylation and PhI-mediated C–N bond formation and deprotecting procedures (Scheme 1d). To improve the reaction efficiency and simplify the operational step, the exploitation of a practical method with readily available materials under mild conditions is of significance for further applications of cinnolines in organic synthesis and pharmaceutical chemistry.
5 prepared C3 substituted cinnolines via Cu-catalyzed intramolecular cyclization and aromatization of hydrazones (Scheme 1b). In 2013, Cheng and You reported Rh-catalyzed annulation to cinnolinium salts with internal alkynes and azo compounds (Scheme 1c).6 After an extra base-mediated demethylation, the generated 2-methyl cinnolinium salts could be transformed to cinnolines. Additionally, in You's work, tert-butyl substituted azo compounds could react with di-alkyl alkynes to offer cinnolines by employing stoichiometric Cu(OAc)2 as an oxidant at 100–120 °C in moderate yields. Very recently, Reddy7 reported a two-step method to benzo[c]cinnolines from aryl-hydrazine dicarboxylates via the substrate-directed Pd-catalyzed C–H arylation and PhI-mediated C–N bond formation and deprotecting procedures (Scheme 1d). To improve the reaction efficiency and simplify the operational step, the exploitation of a practical method with readily available materials under mild conditions is of significance for further applications of cinnolines in organic synthesis and pharmaceutical chemistry.
 | ||
| Fig. 1 Bioactive cinnoline derivatives. | ||
 | ||
| Scheme 1 Approach to cinnolines. | ||
Transition-metal-mediated carbene insertion into C–H bonds has become one of the preferred methods for direct C–C bond formation.8 Recently, the readily available diazo compounds or related derivatives have been well established as versatile carbene precursors in C–H activation processes,9 in particular in the synthesis of heterocycles.10 Inspired by the previous studies, and also in conjunction with our ongoing interest in the synthesis and applications of heterocycles via transition-metal-mediated C–H functionalization,11 herein we describe our results on the Rh(III)-catalyzed redox-neutral annulation of azo and diazo compounds through a tandem C–H activation and C–N bond formation strategy, thus representing an efficient and economical protocol to cinnolines under mild conditions. Moreover, the generated cinnolines could be readily transformed to substituted 1-aminoindole and other bioactive structures.
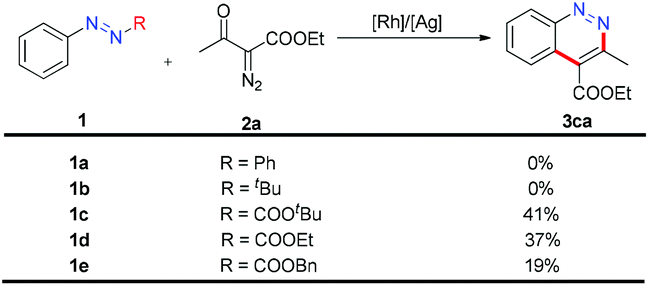
To obtain neutral cinnolines in a single step, a multitasking directing group is necessary to accomplish three targets in one process: site selectivity, cleavability and redox versatility. At first, several azo substrates were prepared and tested as shown in Scheme 2. Compounds 1a and 1b failed to yield the desired adduct 3ca with the starting material recovered using 2.5 mol% [Cp*RhCl2]2, 20 mol% AgSbF6 and 2 mL AcOH/DCE (v:v = 1:4) at 50 °C. Fortunately, 1c, 1d and 1e delivered 3ca in 41%, 37% and 19% yields respectively, which indicated that the carboxylate groups were the appropriate leaving groups and the tert-butyl carboxylate group was the best choice for this annulation reaction.
 | ||
| Scheme 2 Exploration for the directing groups. | ||
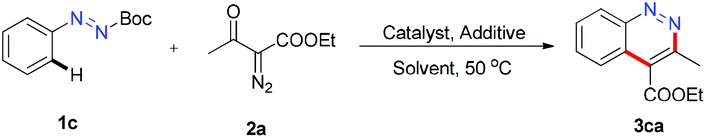
Next, our investigations were focused on the combination of (E)-tert-butyl 2-phenyldiazenecarboxylate (1c) and ethyl 2-diazo-3-oxobutanoate (2a) to optimize the reaction conditions (Table 1). Several silver salts were examined, and AgSbF6 offered 3ca in 41% yield (entries 1–3). The yield was raised to 77% with PivOH as a co-solvent (entry 6). Replacing PivOH with methylsulphonic acid (MSA), resulted in a trace product. When the reaction was performed in other solvent systems such as PivOH/DMF, PivOH/DMSO and PivOH/MeOH, no better outcomes were obtained (see the ESI† for details). Temperature screening suggested that neither a rise nor a drop in the temperature would promote the product generation (see the ESI† for details). An excellent yield (92%) was reached when the ratio of PivOH/DCE was changed to 1:20 (entry 9). The reaction outcome was slightly decreased to 83% when 10 mol% AgSbF6 was loaded (entry 10). Only trace 3ca was detected in the absence of PivOH (entry 11). Finally, the optimized conditions were determined to be 2.5 mol% [Cp*RhCl2]2, 20 mol% AgSbF6, and 2 mL PivOH/DCE (v:v = 1:20) at 50 °C for 24 hours.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Solvent (v:v) |
Yieldb (%) |
| a Reaction conditions: 1c (0.75 mmol, 1.5 equiv.), 2a (0.5 mmol, 1.0 equiv.), [Cp*RhCl2]2 (0.0125 mmol, 2.5 mol%), Ag salts (0.1 mmol, 20 mol%), solvent (2 mL), 50 °C, 24 hours under an air atmosphere. b Yields were determined by 1H NMR using dibromomethane (δ = 4.80) as an internal standard. c Isolated yield. d 10 mol% Ag salt was used. | |||
| 1 | [Cp*RhCl2]2/AgSbF6 | AcOH/DCE (1:4) |
41 |
| 2 | [Cp*RhCl2]2/AgOAc | AcOH/DCE (1:4) |
21 |
| 3 | [Cp*RhCl2]2/Ag2CO3 | AcOH/DCE (1:4) |
23 |
| 4 | [Cp*RhCl2]2/AgSbF6 | HCOOH/DCE (1:4) |
47 |
| 5 | [Cp*RhCl2]2/AgSbF6 | EtCOOH/DCE (1:4) |
40 |
| 6 | [Cp*RhCl2]2/AgSbF6 | PivOH/DCE (1:4) |
77 |
| 7 | [Cp*RhCl2]2/AgSbF6 | MSA/DCE (1:4) |
Trace |
| 8 | [Cp*RhCl2]2/AgSbF6 | PivOH/DCE (1:10) |
85 |
| 9 | [Cp*RhCl2]2/AgSbF6 | PivOH/DCE (1:20) |
92 (86c) |
| 10 | [Cp*RhCl2]2/AgSbF6d |
PivOH/DCE (1:20) |
83 |
| 11 | [Cp*RhCl2]2/AgSbF6 | DCE | Trace |
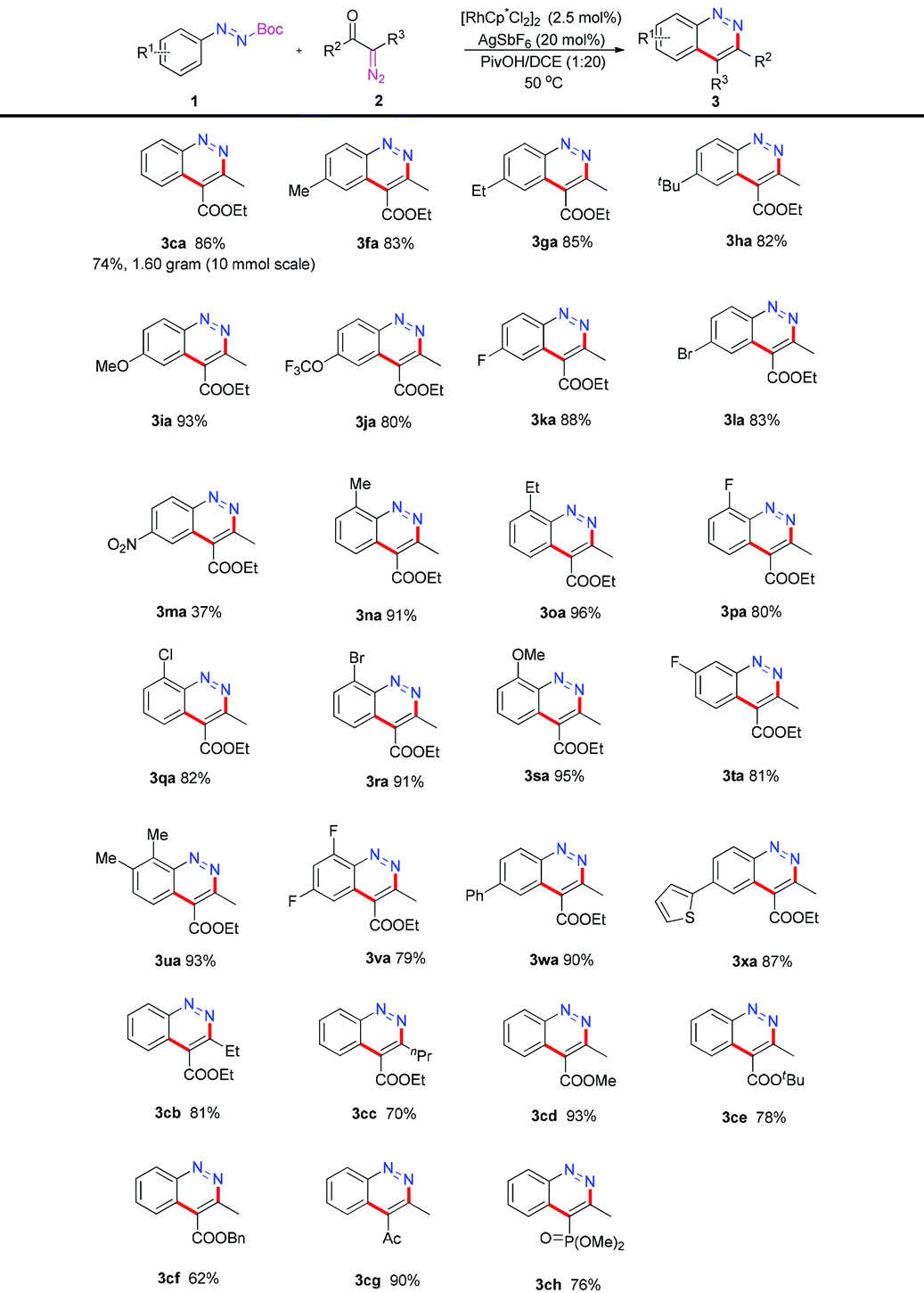
With the optimized conditions in hand, we then explore the substrate scope and limitation. At first, a variety of azo substrates were prepared and examined as shown in Table 2. Substrates with alkyl groups (1f–1h), methoxy group (1i) and trifluoromethoxy (1j) group at the para-position performed smoothly under the standard conditions and offered the corresponding cinnoline products 3fa–3ja in 80%–93% yields. para-Halogen-substituted azo compounds such as 1k and 1l yielded 3ka and 3la in 88% and 83% yields, which allowed further manipulation of the initial products. When electron-withdrawing group (NO2) substituted 1m was evaluated, the product 3ma was obtained in 37% yield. Substitutions with varying electronic properties in the ortho-position (1n–1s) also proved to be tolerated and the desired products 3na–3sa were generated in 80%–96% yields. meta-Substituted azo substrate 1t reacted with 2a giving 3ta in 81% yield. Furthermore, multi-substituted cinnoline products 3ua and 3va could also be obtained in 93% and 79% yields. It is worthy of mention that substrates carrying aryl substituents such as phenyl or thiophene groups offered 3wa and 3xa in 90% and 87% yields. When the reaction scale was amplified to 10 mmol, the model product 3ca (1.60 g) was obtained in 74% yield. Next, the substrate scope was extended to the diazo compounds. A range of substitutions were found to be tolerated under optimal conditions. R2 with the ethyl or n-propyl group afforded 3cb and 3cc in 81% and 70% yields. Methyl, tert-butyl and benzyl-substituted 2-diazo-3-oxobutanoic acid reacted with 1c smoothly to give 3cd, 3ce and 3cf in 93%, 78% and 62% yields. When the ester group of 2a was displaced with a ketone group, the desired cinnoline 3cg was generated in 90% yield. When dimethyl 1-diazo-2-oxopropylphosphonate (2h) was subjected to the optimized conditions, product 3ch was obtained in 76% yield.
|
a Reaction conditions: 1 (0.75 mmol, 1.5 equiv.), 2a (0.5 mmol, 1.0 equiv.), [Cp*RhCl2]2 (0.0125 mmol, 2.5 mol%), AgSbF6 (0.1 mmol, 20 mol%), 2 mL PivOH/DCE (v:v = 1:20), 50 °C, 24 hours under an air atmosphere.
|
|---|
|
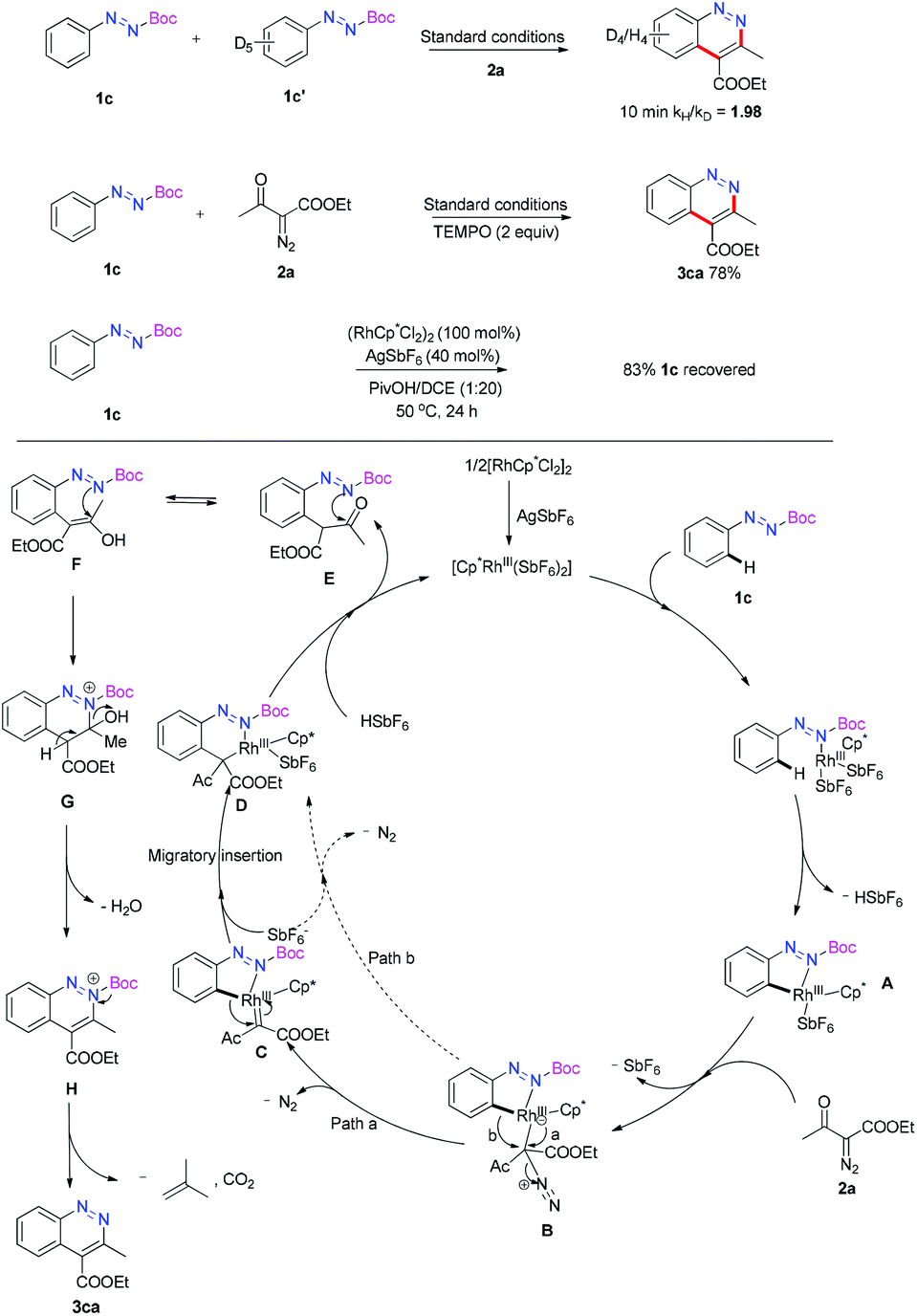
After the reaction scope test, the competitive reaction was conducted between model substrate 1c and electron-donating substrate 1i (see the ESI† for details). The mixture of 1c and 1i (1:1) with 2a was performed in the standard reaction system and 3ca and 3ia were obtained in 26% and 47% yield, which indicated that the high electron-density of the phenyl ring was beneficial for the reaction. Furthermore, kinetic isotope effect studies were conducted to gain mechanical insight into this reaction. The kH/kD value was determined to be 1.98 in 10 minutes (see the ESI† for details). This result suggested that the C–H activation process was the rate determining step rather than carbene insertion. When 2 equivalents of the radical scavenger TEMPO was added, the reaction outcome was not influenced. This result ruled out the possibility of a radical mechanism.12 When 1c was treated with a stoichiometric [Rh]-catalyst without the addition of 2a, 83% of 1c was recovered after 24 hours, and no de-Boc product was detected, which revealed that the Boc group was not removed before the C–H activation step.
Based on the experiments above, the reaction pathway was proposed as shown in Scheme 3. At first, 1c was coordinated with the active catalyst [Cp*Rh(SbF6)2], which was generated from anion exchange with AgSbF6, and a facile ortho C–H activation occurred to deliver the intermediate A. Then the insertion of diazo substrate 2a produced the species B. The intermediate D could be formed through an Rh-carbene C migratory insertion process.13 Meanwhile, an intramolecular 1,2-aryl shift pathway might also be possible to offer the same intermediate D.14 The detailed mechanism about the formation of intermediate D is still unclear. The following Rh-proton exchange step produced tautomer E or F. Subsequent nuclear addition of the N atom to the carbonyl group gave the intermediate G. Then the sequential elimination of a water, a carbon dioxide and a 2-methylprop-1-ene led to the target molecule 3ca.
 | ||
| Scheme 3 Plausible reaction mechanism. | ||
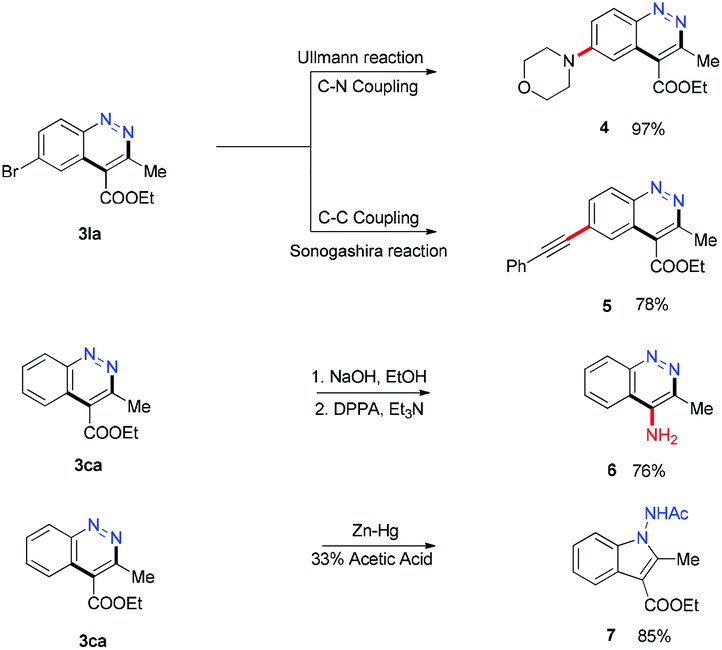
To expand the applications of our method, the generated cinnolines were transformed as shown in Scheme 4. The C–Br bond of 3la was functionalized through the Ullmann reaction15 and the Sonogashira reaction16 to prepare 4 and 5 in 97% and 78% yields. The ester group at the 4-position of 3ca could be converted to an amino group via sequential hydrolysis and Curtis rearrangement reactions.17 When 3ca was treated with Zn–Hg and 33% acetic acid,18 a ring contraction product 1-aminoindole 7 was obtained in 85% yield, which was an important structural motif in many bioactive agents.19 Numerous efforts have been devoted to the synthesis of the materials in recent years.20
 | ||
| Scheme 4 Transformation of generated cinnolines. | ||
Conclusions
In conclusion, we have developed a Rh-catalyzed redox-neutral annulation reaction between azo and diazo compounds, representing an efficient and economical protocol to cinnolines in one step under mild conditions. The procedure involved C–H activation and C–N bond formation, exhibited good functional group tolerability, scalability and step economy. Meanwhile, our method provided an efficient route to some bioactive structures, such as 6-morpholinocinnoline, 4-aminocinnoline, 6-(phenylethynyl)cinnoline and 1-aminoindole.Acknowledgements
Generous financial support from the National Natural Science Foundation of China (NSFC21572272 and NSFC21502232), the Natural Science Foundation of Jiangsu Province (BK20140655 and BK20130645) and the Foundation of State Key Laboratory of Natural Medicines (ZZJQ201306) is gratefully acknowledged.Notes and references
- For selected examples see: (a) C. Alhambra, C. Becker, T. Blake, A. Chang, J. R. Damewood, T. Daniels, B. T. Dembofsky, D. A. Gurley, J. E. Hall, K. J. Herzog, C. L. Horchler, C. J. Ohnmacht, R. J. Schmiesing, A. Dudley, M. D. Ribadeneira, K. S. Knappenberger, C. Maciagc, M. M. Stein, M. Chopra, X. F. Liu, E. P. Christian, J. L. Arriza and M. J. Chapdelaine, Bioorg. Med. Chem., 2011, 19, 2927–2938 CrossRef CAS PubMed; (b) D. A. Scott, L. A. Dakin, D. J. Del Valle, R. B. Diebold, L. Drew, T. W. Gero, C. A. Ogoe, C. A. Omer, G. Repik, K. Thakur, Q. Ye and X. Zheng, Bioorg. Med. Chem. Lett., 2011, 21, 1382–1384 CrossRef CAS PubMed; (c) Y. Yu, S. K. Singh, A. Liu, T.-K. Li, L. F. Liu and E. J. LaVoie, Bioorg. Med. Chem., 2003, 11, 1475–1491 CrossRef CAS PubMed; (d) E. Hu, R. K. Kunz, S. Rumfelt, N. Chen, R. Bürli, C. Li, K. L. Andrews, J. Zhang, S. Chmait, J. Kogan, M. Lindstrom, S. A. Hitchcock and J. Treanor, Bioorg. Med. Chem. Lett., 2012, 22, 2262–2265 CrossRef CAS PubMed; (e) T. Mitsumori, M. Bendikov, J. Sedo and F. Wudl, Chem. Mater., 2003, 15, 3759–3768 CrossRef CAS; (f) H. Tsuji, Y. Yokoi, Y. Sato, H. Tanaka and E. Nakamura, Chem. – Asian J., 2011, 6, 2005–2008 CrossRef CAS PubMed.
- For review on the synthesis of cinnolines see: O. V. Vinogradova and I. A. Balova, Chem. Heterocycl. Compd., 2008, 44, 501–522 CrossRef CAS.
- (a) K. Hasegawa, N. Kimura, S. Arai and A. Nishida, J. Org. Chem., 2008, 73, 6363–6368 CrossRef CAS PubMed; (b) D. B. Kimball, T. J. R. Weakley and M. M. Haley, J. Org. Chem., 2002, 67, 6395–6405 CrossRef CAS PubMed.
- C. J. Ball, J. Gilmore and M. C. Willis, Angew. Chem., Int. Ed., 2012, 51, 5718–5722 CrossRef CAS PubMed.
- G. Zhang, J. Miao, Y. Zhao and H. Ge, Angew. Chem., Int. Ed., 2012, 51, 8318–8321 CrossRef CAS PubMed.
- (a) K. Muralirajan and C.-H. Cheng, Chem. – Eur. J., 2013, 19, 6198–6202 CrossRef CAS PubMed; (b) D. Zhao, Q. Wu, X. Huang, F. Song, T. Lv and J. You, Chem. – Eur. J., 2013, 19, 6239–6244 CrossRef CAS PubMed.
- B. V. S. Reddy, C. R. Reddy, M. R. Reddy, S. Yarlagadda and B. Sridhar, Org. Lett., 2015, 17, 3730–3733 CrossRef CAS PubMed.
- For reviews on metal-catalyzed carbene insertions to C–H bonds see: (a) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS PubMed; (b) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS PubMed.
- For representative examples of diazo compounds involved in C–H activation reactions see: (a) B. Xu, M.-L. Li, X.-D. Zuo, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2015, 137, 8700–8703 CrossRef CAS PubMed; (b) Z. Yu, B. Ma, M. Chen, H.-H. Wu, L. Liu and J. Zhang, J. Am. Chem. Soc., 2014, 136, 6904–6907 CrossRef CAS PubMed; (c) F. Ye, X. Ma, Q. Xiao, H. Li, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2012, 134, 5742–5745 CrossRef CAS PubMed; (d) X. Zhao, G. Wu, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2011, 133, 3296–3299 CrossRef CAS PubMed; (e) A. DeAngelis, V. W. Shurtleff, O. Dmitrenko and J. M. Fox, J. Am. Chem. Soc., 2011, 133, 1650–1653 CrossRef CAS PubMed.
- For representative examples see: (a) W.-W. Chan, S.-F. Lo, Z. Zhou and W.-Y. Yu, J. Am. Chem. Soc., 2012, 134, 13565–13568 CrossRef CAS PubMed; (b) Z. Shi, D. C. Koester, M. Boultadakis-Arapinis and F. Glorius, J. Am. Chem. Soc., 2013, 135, 12204–12207 CrossRef CAS PubMed; (c) T. K. Hyster, K. E. Ruhl and T. Rovis, J. Am. Chem. Soc., 2013, 135, 5364–5367 CrossRef CAS PubMed; (d) S. Cui, Y. Zhang, D. Wang and Q. Wu, Chem. Sci., 2013, 4, 3912–3916 RSC; (e) B. Ye and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 7896–7899 CrossRef CAS PubMed; (f) Y. Liang, K. Yu, B. Li, S. Xu, H. Song and B. Wang, Chem. Commun., 2014, 50, 6130–6133 RSC; (g) S. Yu, S. Liu, Y. Lan, B. Wan and X. Li, J. Am. Chem. Soc., 2015, 137, 1623–1631 CrossRef CAS PubMed; (h) X. G. Li, M. Sun, K. Liu, Q. Jin and P. N. Liu, Chem. Commun., 2015, 51, 2380–2383 RSC; (i) F. Hu, Y. Xia, C. Ma, Y. Zhang and J. Wang, Chem. Commun., 2015, 51, 7986–7995 RSC; (j) J. Shi, J. Zhou, Y. Yan, J. Jia, X. Liu, H. Song, H. E. Xu and W. Yi, Chem. Commun., 2015, 51, 668–671 RSC; (k) L. Qiu, D. Huang, G. Xu, Z. Dai and J. Sun, Org. Lett., 2015, 17, 1810–1813 CrossRef CAS PubMed; (l) J.-Y. Son, S. Kim, W. H. Jeon and P. H. Lee, Org. Lett., 2015, 17, 2518–2521 CrossRef CAS PubMed; (m) S. Sharma, S. H. Han, S. Han, W. Ji, J. Oh, S.-Y. Lee, J. S. Oh, Y. H. Jung and I. S. Kim, Org. Lett., 2015, 17, 2852–2855 CrossRef CAS PubMed; (n) G. Xu, C. Zhu, W. Gu, J. Li and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 883–887 CrossRef CAS PubMed; (o) D. Zhang, G. Xu, D. Ding, C. Zhu, J. Li and J. Sun, Angew. Chem., Int. Ed., 2014, 53, 11070–11074 CrossRef CAS PubMed; (p) K. Liu, C. Zhu, J. Min, S. Peng, G. Xu and J. Sun, Angew. Chem., Int. Ed., 2015, 54, 12962–12967 CrossRef CAS PubMed.
- (a) L. Zhao, Z. Li, L. Chang, J. Xu, H. Yao and Xi. Wu, Org. Lett., 2012, 14, 2066–2069 CrossRef CAS PubMed; (b) Z. Li, L. Ma, J. Xu, L. Kong, X. Wu and H. Yao, Chem. Commun., 2012, 48, 3763–3765 RSC; (c) Y. Su, H. Zhou, J. Chen, J. Xu, X. Wu, A. Lin and H. Yao, Org. Lett., 2014, 16, 4884–4887 CrossRef CAS PubMed; (d) P. Sun, Y. Wu, T. Yang, X. Wu, J. Xu, A. Lin and H. Yao, Adv. Synth. Catal., 2015, 357, 2469–2473 CrossRef CAS; (e) H. Jiang, S. Gao, J. Xu, X. Wu, A. Lin and H. Yao, Adv. Synth. Catal., 2015 DOI:10.1002/adsc.201500769.
- S. K. Fehler, G. Pratsch and M. R. Heinrich, Angew. Chem., Int. Ed., 2014, 53, 11361–11365 CrossRef CAS PubMed.
- Carbene migratory insertion process was reported in many C–H activation reactions. See: (a) Z. Shi, D. C. Koester, M. Boultadakis-Arapinis and F. Glorius, J. Am. Chem. Soc., 2013, 135, 12204–12207 CrossRef CAS PubMed; (b) S. Yu, S. Liu, Y. Lan, B. Wan and X. Li, J. Am. Chem. Soc., 2015, 137, 1623–1631 CrossRef CAS PubMed.
- For report on the 1, 2-aryl shift mechanism, see: W.-W. Chan, S.-F. Lo, Z. Zhou and W.-Y. Yu, J. Am. Chem. Soc., 2012, 134, 13565–13568 CrossRef CAS PubMed.
- D. Cheng, D. Han, W. Gao, Q. Jing, J. Jiang, Y. Wan, N. P. Englund, T. Tuntland, X. Wu and S. Pan, Bioorg. Med. Chem. Lett., 2012, 22, 6573–6576 CrossRef CAS PubMed.
- L. W. Hardy, M. L. R. Heffernan, F. X. Wu, K. L. Spear and L. D. Saraswat, WO2011075699, 2011 Search PubMed.
- S. Sunami, T. Sagara, M. Ohkubo and H. Morishima, Tetrahedron Lett., 1999, 40, 1721–1724 CrossRef CAS.
- L. S. Besford and J. M. Bruce, J. Chem. Soc., 1964, 4037–4044 RSC.
- For bioactive application of 1-aminoindoles see: (a) T. Itoh, M. Miyazaki, H. Maeta, Y. Matsuya, K. Nagata and A. Ohsawa, Bioorg. Med. Chem., 2000, 8, 1983–1989 CrossRef CAS PubMed; (b) G. Saxty, D. Norton, K. Affleck, D. Clapham, A. Cleasby, J. Coyle, P. Day, M. Frederickson, A. Hancock, H. Hobbs, J. Hutchinson, J. Le, M. Leveridge, R. McMenamin, P. Mortenson, L. Page, C. Richardson, L. Russell, E. Sherriff, S. Teague, S. Uddin and S. Hodgson, Med. Chem. Commun., 2014, 5, 134–141 RSC.
- For the synthesis of 1-aminoindoles see: (a) M. Watanabe, T. Yamamoto and M. Nishiyama, Angew. Chem., Int. Ed., 2000, 39, 2501–2504 CrossRef CAS; (b) T. T. H. Luong, J.-D. Brion, M. Alami and S. Messaoudi, J. Org. Chem., 2015, 80, 751–761 CrossRef CAS PubMed; (c) D. Y. Li, H. J. Chen and P. N. Liu, Org. Lett., 2014, 16, 6176–6179 CrossRef CAS PubMed; (d) Y. Liang, K. Yu, B. Li, S. Xu, H. Song and B. Wang, Chem. Commun., 2014, 50, 6130–6133 RSC; (e) E. Brachet, S. Messaoudi, J.-F. Peyrat, J.-D. Brion and M. Alami, Adv. Synth. Catal., 2012, 354, 2829–2839 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00331h |
| This journal is © the Partner Organisations 2016 |