
Microwave-assisted simultaneous O,N-sulfonation in the synthesis of heparin-like oligosaccharides†
Peng
Xu
a,
Stephane
Laval
a,
Zheng
Guo
b and
Biao
Yu
*ab
aState Key Laboratory of Bio-organic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: byu@mail.sioc.ac.cn
bSchool of Physical Science and Technology, ShanghaiTech University, 100 Haike Road, Shanghai 201210, China
First published on 27th November 2015
Abstract
The synthesis of well-defined fragments of heparin (HP) and heparan sulfate (HS) is often hampered by difficult post-assembly manipulations on the fully elaborated oligosaccharides. In particular, the O- and N-sulfonation steps, usually performed separately, require long reaction times and often result in low yield due to problems associated with the incompletion of the reaction and the purification of highly sulfonated products, thus representing a bottle-neck in the synthesis. We report herein an effective microwave-assisted protocol for the simultaneous O,N-sulfonation of HP/HS-like saccharides. Complete O- and N-sulfonation were attained when using a SO3·NEt3 complex in a solvent mixture of NEt3/pyridine at 100 °C (MW heating) for 15 min, thus facilitating the purification process. Easy to implement, per-O,N-sulfonation of mono-, di-, tri- and tetra-saccharides with two to six reactive sites was performed effectively in short reaction times and excellent yields (>90%). Under smooth deprotection conditions, the resulting per-O,N-sulfonated saccharides were fully deprotected in high yields (>88%), providing saccharides pertinent to the synthesis of HP/HS-like fragments, including three tetrasaccharides relevant to the substrate of heparanase. Moreover, we developed a microwave-assisted protocol for the one-pot selective O-sulfonation/N-acetylation on disaccharide 7, which could be applied to synthesize the members of the GAG family bearing N-acetyl groups.
Introduction
Heparin (HP) and heparan sulfate (HS), the most complex members of the glycosaminoglycan (GAG) family, are linear, highly sulfated polysaccharides.1 These biopolymers modulate various cell–cell and cell–matrix communications. Structurally, HP/HS share a common backbone which consists of a repeating disaccharide unit of α-D-glucosamine (GlcN) 1→4-linked to either α-L-iduronic acid (IdoA) or β-D-glucuronic acid (GlcA). The position 2 of the uronic acid units and the positions 3 and 6 of the GlcN units can be free hydroxyl or sulfate groups. The nitrogen of the GlcN units can be sulfonated, acetylated or remains unchanged. It is well-known that the chain length, the position of the iduronic acid units and the sulfonation pattern of a specific HP/HS fragment are responsible for its selective binding to proteins.1,2 For instance, the N-sulfonate and 3-O-sulfonate groups of the fully synthetic pentasaccharide Arixtra (Fondaparinux) are essential for its anticoagulant activity.2,3This molecular variability is behind the lack of knowledge on their structure–activity relationships. Indeed, further study is hampered by the fact that HP/HS fragments are naturally produced in heterogeneous complex mixtures and in low yield. This difficulty drove scientists to develop chemical approaches to the syntheses of homogeneous fragments with well-defined structures.4–9 Despite the different strategies adopted, all share a common base: the assembly of the oligosaccharide fully protected with a judicious set of orthogonal protecting groups which is then subjected to selective deprotection, structural modification (oxidation, N-acetylation) and O,N-sulfonation.5–9 In addition to the difficulties in constructing stereoselectively the glycosidic linkages, the post-assembly manipulations on an elaborated oligosaccharide are difficult to monitor and require a long reaction time. In particular, the O- and N-sulfonation steps often result in low yield due to problems associated with the incompletion of the reaction and the purification of highly sulfonated products.
Usually, O-sulfonation of HP/HS-like oligosaccharides is achieved with a sulfur trioxide complex, such as SO3·NMe3,2b,5 SO3·NEt3,6 or SO3·Py (Py = pyridine),7–9 in dimethylformamide (DMF) or pyridine at room temperature or 50–60 °C. N-Sulfonation is performed with SO3·Py in pyridine, methanol or water (in the presence of a buffer) at 0 °C or room temperature, and often requires the addition of NEt3.5–9O- and N-sulfonation of oligosaccharide with multiple reaction sites usually necessitates a large excess of the sulfonating agent and long reaction times (from several hours to days). Even under harsh reaction conditions, only moderate yield can be attained.7a As the number of reactive sites increases, sulfonation becomes progressively difficult because of anion–anion repulsions, resulting in incomplete reaction and numerous partially sulfonated products.10 The major challenge is thus to drive the reaction to completion in order to sulfonate all the reactive sites available on the substrate. Current approaches involve adding a fresh sulfonating reagent during the reaction or after previous purification of the partially sulfonated crude mixture, albeit without guaranteeing high yield.6d–f,7b,d It is worth mentioning that the yield and reaction time are dependent upon the protecting groups’ pattern and must be optimized for each substrate. Moreover, the instability of highly sulfated HP/HS oligosaccharides limits the use of high temperature under prolonged reaction times.
Recently, Huang and coworkers reported the introduction of O-sulfonates pre-installed as sulfate esters in the disaccharide building blocks prior to the assembly of the full length oligosaccharide backbone.11 Deprotection of the sulfate esters with zinc/ammonium formate provides the corresponding O-sulfonated oligosaccharides. However, this approach limits the O-sulfonation pattern at the 6-O-position only whereas the amino groups are still sulfonated using a sulfonating reagent, i.e. the SO3·Py complex. Generally, O- and N-sulfonation are performed separately. Thus, O-sulfonation/azide reduction/N-sulfonation or sequential O-/N-sulfonation with an intermediate purification are the current methods. To the best of our knowledge, only one protocol described the simultaneous O,N-sulfonation of HP/HS-like oligosaccharides which required the use of the SO3·Py complex in pyridine at room temperature for 24 h firstly (N-sulfonation) and then at 50–55 °C for 16–24 h (O-sulfonation).8 However, the yield is still dependent upon the length of the oligosaccharide and the protecting groups’ pattern,6a and the long reaction time might represent a limitation.
de Paz et al. described a microwave-assisted O-sulfonation of heparin oligosaccharide intermediates with a SO3·NMe3 complex in DMF at 100 °C.12,13 Taking advantage of the ionic charged molecule and the polar solvent,14 per-O-sulfonation could be performed in a short reaction time, allowing a higher reaction temperature, and in high yield.7e,f They also reported microwave conditions for the N-sulfonation of one substrate only (SO3·NMe3, DMF, NEt3, 60 °C), performed after O-sulfonation and azide reduction. Their results highlighted the effectiveness of microwaves for the sulfonation of HP like-oligosaccharides. The significant short reaction time allowed the use of a temperature above 100 °C, essential to attain complete O-sulfonation. However, microwave-assisted simultaneous O,N-sulfonation has not been addressed so far.
Motivated by the rapid and efficient post-modification of HP/HS-like oligosaccharides,6d,9 we report herein an effective protocol for simultaneous O,N-sulfonation under microwave irradiation. This method is expected to reduce reaction times, facilitate the purification process and thus accelerate the preparation of highly-sulfated HP/HS-like fragments.
Results and discussion
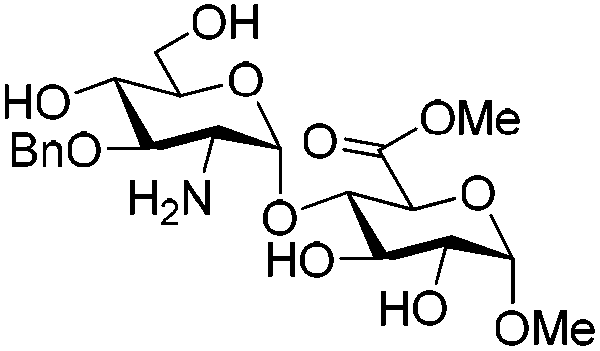
In the course of synthesizing a library of heparin tri- and tetra-saccharides relevant to ΔHexA(2S)–GlcN(NS,6S)–GlcA–GlcN(NS,6S), a reported substrate of heparanase,15 we faced difficulties to sulfonate the hydroxyl and amino groups of tetrasaccharide 2, simultaneously (Scheme 1).9 Indeed, under the standard reported conditions, that is the SO3·Py complex in pyridine at room temperature for 24 h followed by heating at 55 °C for an additional 24 h,8 only the N-sulfonated compound 4 was isolated. This result was confirmed by the mass analysis which revealed the presence of only two sulfonate groups (ESI-MS: [M − 2SO3 − 2H+]2− = 755.1) and by the 1H NMR spectrum in which the H-2 of the GlcN units was shifted over 3.00 ppm (2.62 ppm for 2 and 2.71 and 2.91 ppm for 5) (ESI†). This result, also encountered by Seeberger et al. on a relevant tetrasaccharide,6a prompted us to examine the simultaneous O- and N-sulfonation under microwave irradiation. | ||
| Scheme 1 O,N-Sulfonation of tetrasaccharide 2 under standard conditions (oil bath heating). Bn = benzyl; Bz = benzoyl. | ||
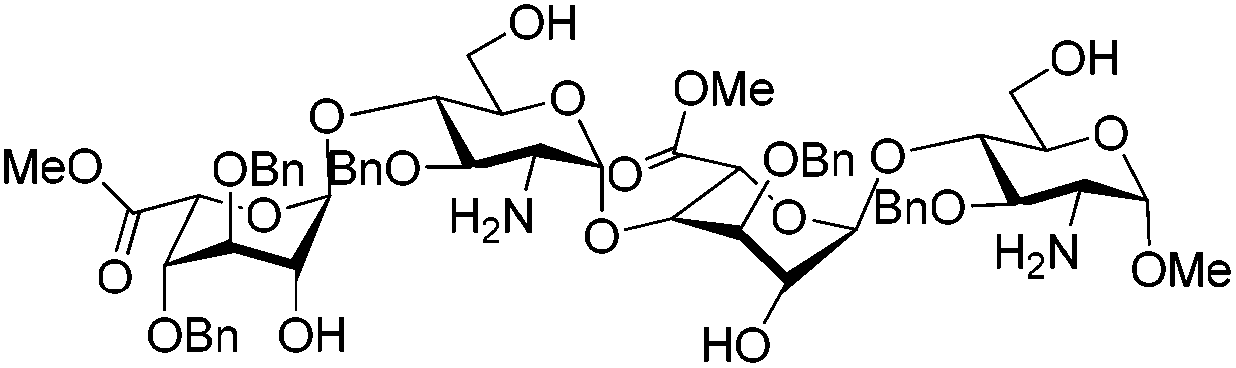
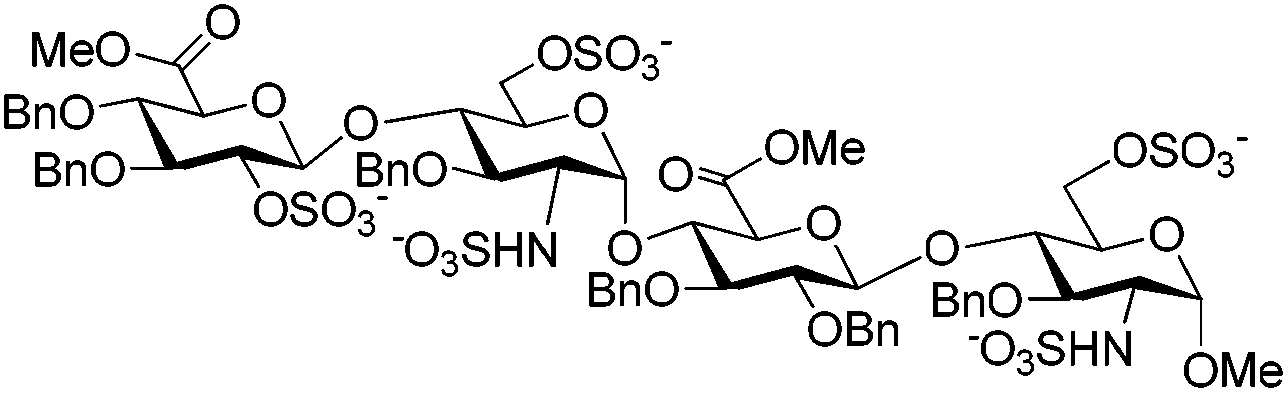
Owing to the structural complexity of tetrasaccharides, disaccharide 7 bearing sterically hindered 2,4′-OH and 2′-NH2 was selected as a model substrate (Scheme 2). In order to facilitate analysis of the reaction products, the targeted tri-O,N-sulfonated disaccharide 9 was synthesized from intermediate 6 under standard conditions (O-sulfonation/azide reduction/N-sulfonation).9
 | ||
| Scheme 2 Preparation of disaccharides 7 and 9. Mp = para-methoxyphenyl. | ||
As shown in Table 1, the nature of the sulfonating reagent, base, solvent, as well as the reaction temperature was assessed. Depending on the reaction conditions, per-O,N-sulfonated (9) and di-O-sulfonated disaccharides (10) were isolated as the main products, whereas mono-N-sulfonated compound 11 was not obtained. Close monitoring by TLC analysis revealed that the conditions using the SO3·Py complex in pyridine at 55 °C provided only di-O-sulfonated disaccharide 10, albeit with complete consumption of 7. Prolonging the reaction time to 45 min did not convert 10 to the tri-O,N-sulfonated disaccharide 9 and compound 10 was isolated in 66% yield after reverse-phase chromatographic purification (entry 1). Its structure was confirmed by its 1H NMR spectrum which matched one of the disaccharides 10 synthesized under standard conditions. Indeed, compound 10 is an intermediate in the synthesis of per-O,N-sulfonated disaccharide 9 from 8 (Scheme 2). Increasing the temperature to 100 °C also furnished 10, albeit in a higher yield and shorter reaction time (entry 2). The presence of NEt3 appeared to be pivotal in order to obtain the tri-O,N-sulfonated target 9 (entries 3–7). Indeed, under the previous conditions with NEt3 as the additive, 9 was isolated in 92% yield after 15 min at 100 °C (entry 3). Substituting pyridine by DMF in the presence of NEt3 provided 9 in a lower 30% yield (entry 4). However, using pyridine as the solvent, the substitution of NEt3 by DMAP did not furnish the targeted disaccharide 9 and the di-O-sulfonated compound 10 was isolated in 74% yield (entry 5). For the last two cases, the lower yields are explained by the presence of partially sulfonated byproducts. The use of the SO3·NEt3 complex in pyridine at 100 °C also provided the desired tri-O,N-sulfonated disaccharide 9 in a good 82% yield after 15 min (entry 6).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Sulfonating agenta | Additive | Solvent | T (°C) | Time | Product, yieldb |
a 5.0 equiv. per reactive site.
b % isolated yield after reverse-phase column chromatography.
c Ratio additive/solvent = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10.
d DMAP (10 equiv.). Py = pyridine; DMAP = 4-dimethylaminopyridine; DMF = dimethylformamide. :10.
d DMAP (10 equiv.). Py = pyridine; DMAP = 4-dimethylaminopyridine; DMF = dimethylformamide.
|
||||||
| 1 | SO3·Py | — | Py | 55 | 15 min × 3 | 10, 66 |
| 2 | SO3·Py | — | Py | 100 | 15 min | 10, 96 |
| 3 | SO3·Py | NEt3c |
Py | 100 | 15 min | 9, 92 |
| 4 | SO3·Py | NEt3c |
DMF | 100 | 15 min | 9, 30 |
| 5 | SO3·Py | DMAPd | Py | 100 | 15 min | 10, 74 |
| 6 | SO3·NEt3 | — | Py | 100 | 15 min | 9, 82 |
| 7 | SO3·NEt3 | NEt3c |
Py | 100 | 15 min | 9, 96 |
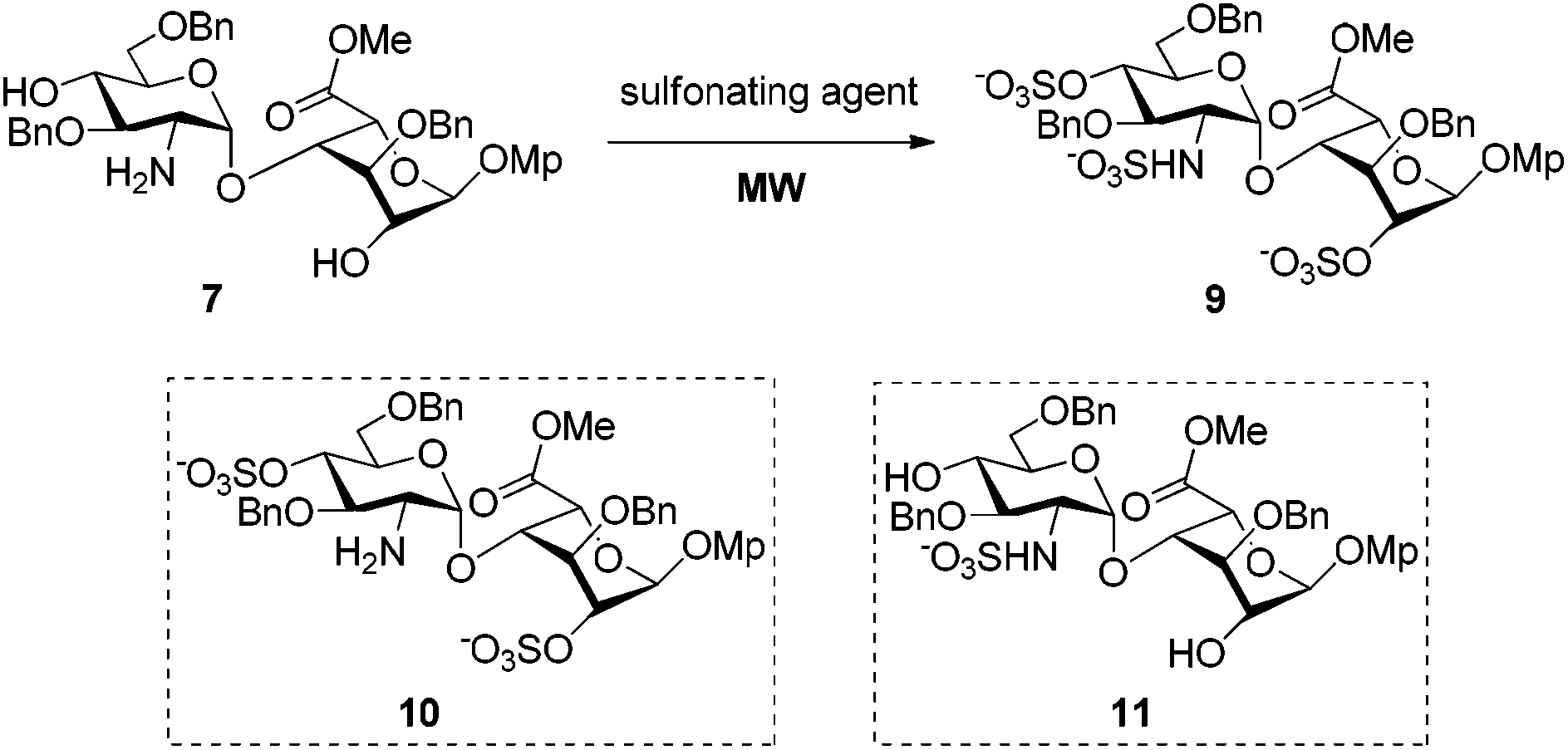
Moreover, the yield could be improved to 96% by the addition of NEt3, thus enhancing the pivotal role of NEt3 in this reaction (entry 7).
As shown in Scheme 3, the conditions using the SO3·Py complex in pyridine with NEt3 as a base were examined without microwave heating. Running the reaction at room temperature provided only the N-sulfonated disaccharide 11, isolated in 72% yield after 8 h. The N-sulfonated compound 11 was also detected as the main product by TLC monitoring when performing the reaction at 55 °C, even after a prolonged reaction time. Under these conditions the per-O,N-sulfonated target 9 was not detected. These results as well as the one obtained in Scheme 1 suggest that the amino group is more reactive than the hydroxyl groups under standard conditions, which is in contrast with our results obtained under microwave heating (Table 1, entries 1, 2, 5). To rationalize this observation, we assume that the hydroxyl groups of disaccharide 7 are involved in intra- and inter-molecular hydrogen bonding, which renders these OH unreactive at room temperature and 55 °C under oil bath heating.13a However, the activation energy provided by the fast microwave heating is able to break the hydrogen bonds, so that the O-sulfonation can occur. As soon as O-sulfonation proceeds, the system becomes acidic, resulting in the protonation of the amino group which is then not available to react with sulfur trioxide. Only a stronger base such as NEt3 is able to deprotonate the quaternary nitrogen and neutralize the system, thus allowing N-sulfonation.
 | ||
| Scheme 3
O,N-Sulfonation of disaccharide 7 under standard conditions (oil bath). Conditions: SO3·Py complex (5.0 equiv. per reactive site), ratio NEt3/Py = 1:10. RT = room temperature; n.d. = not determined. | ||
The microwave conditions giving the best results (Table 1, entries 3, 6 and 7) were applied to the more complex trisaccharide 12 bearing three hydroxyl and two amino groups (Table 2). As expected, the use of the SO3·NEt3 complex in pyridine at 100 °C provided the penta-O,N-sulfonated trisaccharide 13 after 15 min reaction in a good 74% yield, which can be improved to 95% by addition of NEt3 (entries 1 and 2). Surprisingly, per-sulfonated compound 13 was not detected when using the SO3·Py complex in pyridine at 100 °C in the presence of NEt3 (entry 3). For that reaction, we noticed that the addition of NEt3 after the SO3·Py complex resulted in a complex mixture of partially sulfonated byproducts whereas the reverse addition lead to no reaction with substrate 12 being totally recovered. Consequently, the microwave-assisted conditions employing the SO3·NEt3 complex in pyridine at 100 °C, in the presence of NEt3, appeared as the optimized reaction conditions.
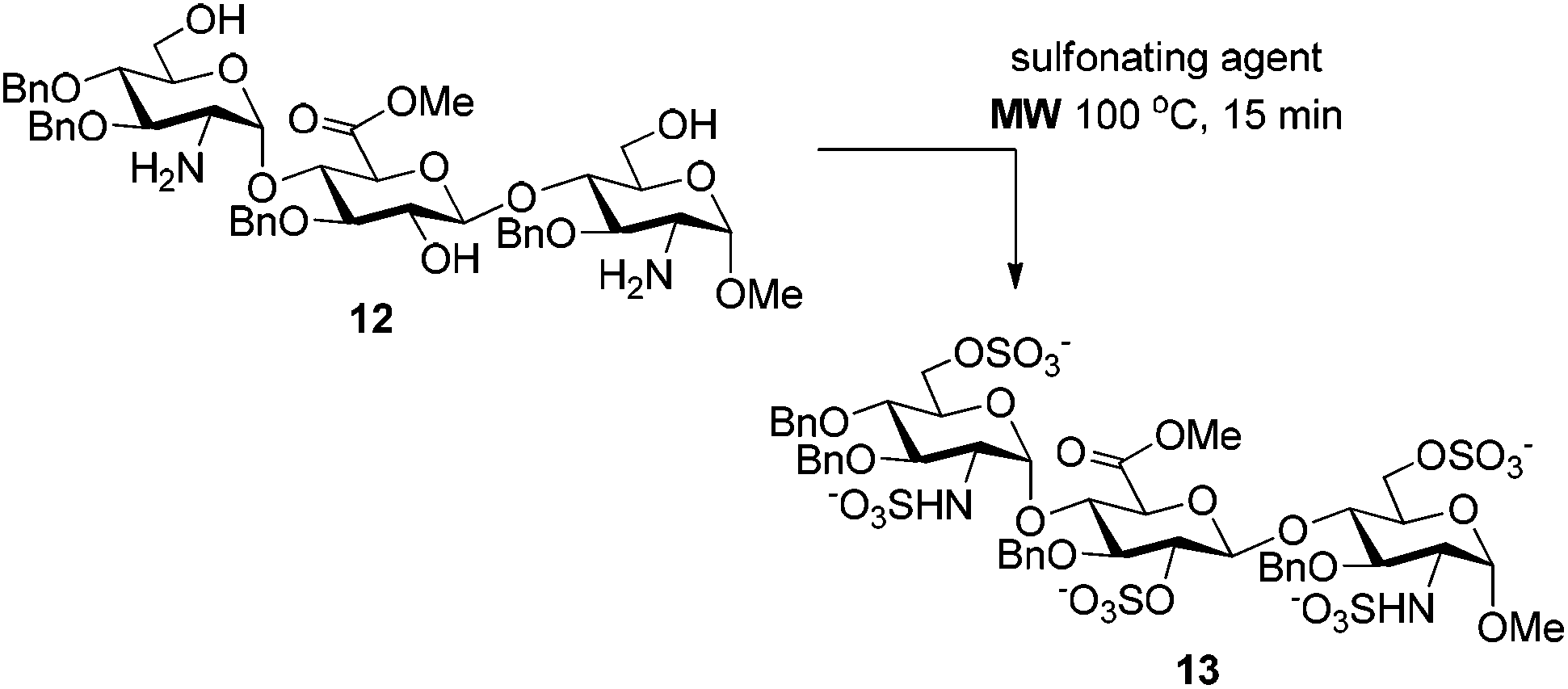
These conditions were then applied to various saccharides relevant for the synthesis of HP/HS-like fragments in order to test the generality of this protocol (Table 3). To our delight, this method provided excellent yields of per-O,N-sulfonated compounds for mono-, di- and tetra-saccharides with two to six reactive sites in short reaction times (15 min). Mono-saccharides 14, 16 and 18 possessing a 2-NH2 and a 6-OH were per-sulfonated in excellent yields (∼90%) (entries 1–3). As expected, the configuration of the anomeric carbon did not affect the yield of the O,N-sulfonation (entries 1 and 2). Disaccharides 20 and 22 bearing 5 reactive sites, that is 2-NH2, 6-OH and sterically hindered 2,3(3′),4′-OH were per-sulfonated without particular difficulties, providing 21 and 23 in 98% and 94% yields, respectively (entries 4 and 5). Tetrasaccharides 24 (6 reaction sites), 26 (5 reaction sites) and 2 (4 reaction sites) furnished the corresponding per-O,N-sulfonated compounds 25, 27 and 3 in excellent yields (>92%), thus evincing the powerfulness of this microwave-assisted O,N-sulfonation protocol (entries 6–8).


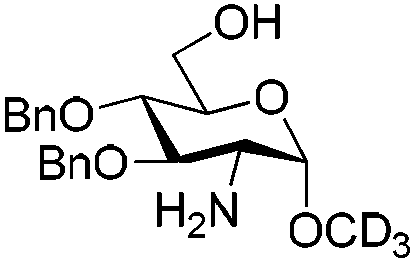


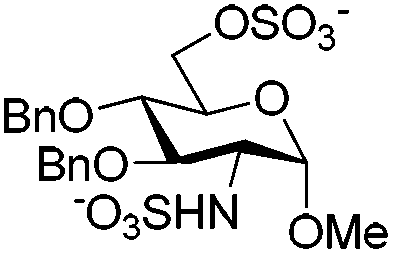







The per-O,N-sulfonated mono-, di-, tri- and tetra-saccharides were then subjected to full deprotection in one (Method A) or two steps (Methods B or C) depending on the protecting groups’ pattern (Scheme 4). The methyl esters were saponified with 1 M LiOH and 30% H2O2 in THF at 0 °C; the benzoyl groups were completely removed using 3 M KOH in methanol; and hydrogenolysis of the benzyl groups was achieved with H2 and Pd(OH)2/C in a mixture of methanol and water buffer. All these steps provided excellent yields (>88%) of the fully deprotected per-O,N-sulfonated saccharides (28–35). The number of sulfonates was well confirmed by ESI-MS analyses and the NMR spectra, especially the easily diagnosable chemical shifts of the anomeric protons and carbons were found to be identical to those reported in the literature for the analogous saccharides.9 This approach involving the microwave-assisted simultaneous O,N-sulfonation followed by full deprotection allowed us to synthesize effectively three potential substrates of heparanase (33–35),15 a mammalian endo-β-D-glucuronidase involved in tumor progression and metastasis by degrading HP/HS.
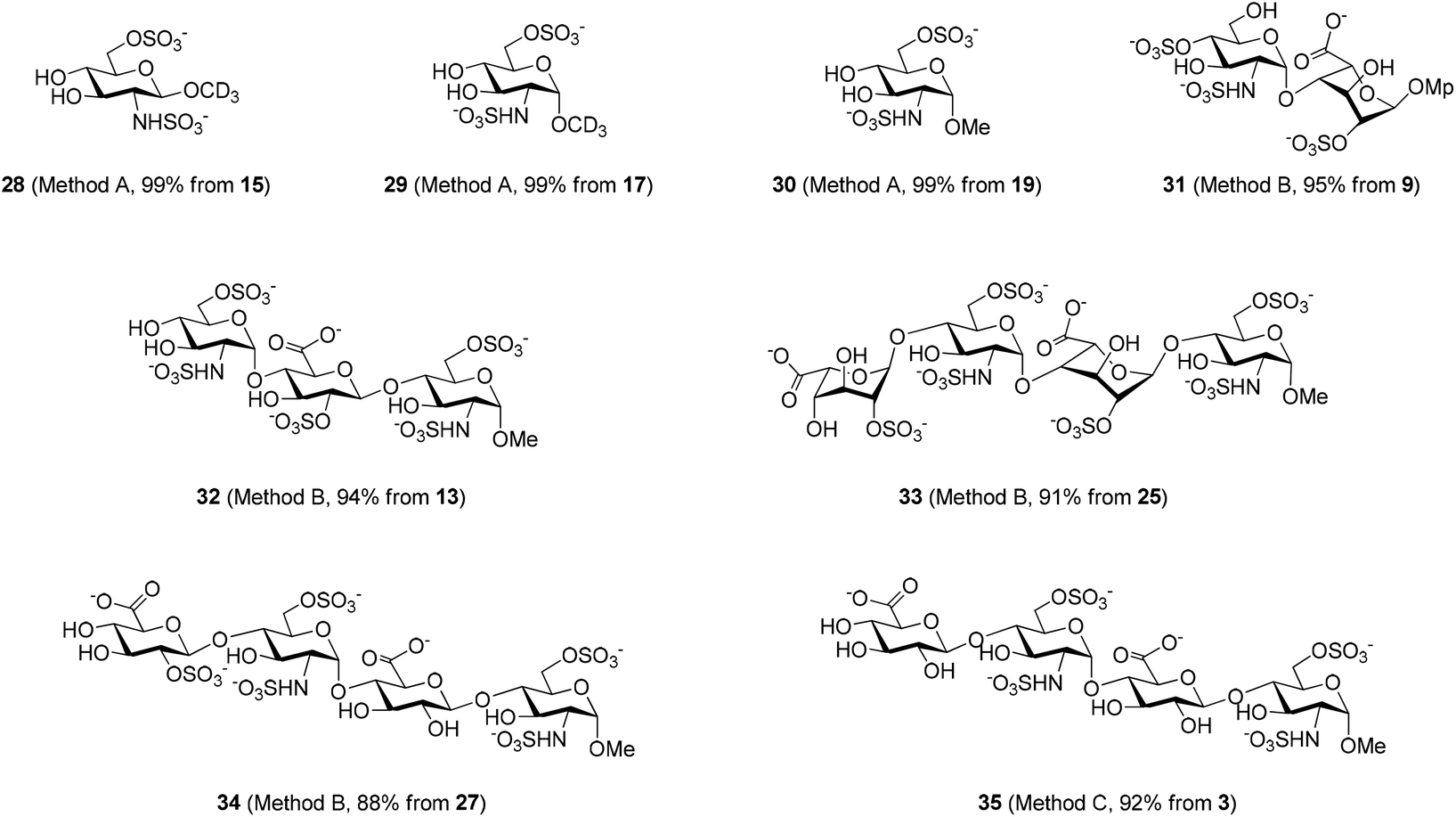 | ||
| Scheme 4 HP/HS-like saccharides (28–35) synthesized by full deprotection of the corresponding per-O,N-sulfonated substrates. Method A: 10 atm H2, Pd(OH)2/C, MeOH/H2O. Method B: (1) 1 M LiOH, 30% H2O2, THF, 0 °C; (2) H2, Pd(OH)2/C, MeOH/buffer H2O. Method C: (1) (i) 1 M LiOH, 30% H2O2, THF, 0 °C; (ii) 3 M KOH, methanol, 0 °C; (2) H2, Pd(OH)2/C, MeOH/buffer H2O. Isolated yield after size exclusion chromatography (Sephadex G-10) is shown. | ||
To further explore the present microwave-assisted conditions, we attempted the selective O-sulfonation/N-acetylation in one-pot under microwave irradiation (Scheme 5). Disaccharide 7 was selectively O-sulfonated under the microwave conditions using the SO3·Py complex in pyridine at 100 °C for 15 min (see Table 1, entry 2). Then, N-acetylation was performed by the addition of acetic anhydride to the crude reaction followed by microwave heating at 50 °C for 15 min. To our delight, the desired O-sulfonated/N-acetylated disaccharide 36 was isolated in 90% yield after reverse-phase column chromatography. Disaccharide 36 was then fully deprotected (Method B) to provide disaccharide 37 in an excellent 92% yield. To the best of our knowledge, this result represents the first example of a microwave-assisted selective O-sulfonation/N-acetylation in one-pot. This approach could be applied to synthesize other members of the GAG oligosaccharides bearing more N-acetyl groups such as dermatan, chondroitin and keratan sulfates.16
 | ||
| Scheme 5 One-pot selective O-sulfonation and N-acetylation of disaccharide 7 under microwave irradiation. | ||
Conclusions
We have developed a general and effective microwave-assisted protocol for the simultaneous O,N-sulfonation of HP/HS-like saccharides. Under the optimized conditions [SO3·NEt3 complex, NEt3/Py, MW 100 °C, 15 min], mono-, di-, tri- and tetra-saccharides with two to six reactive sites were effectively and efficiently per-O,N-sulfonated in short reaction times and excellent yields (>90%). The resulting saccharides could be fully deprotected under smooth deprotection conditions in high yields (>88%). By this approach, three tetrasaccharides relevant to the substrate of heparanase were synthesized. In addition, a one-pot selective O-sulfonation/N-acetylation was developed under microwave heating. This method provided excellent yield of O-sulfonated/N-acetylated disaccharide 36/37 and could be applied to synthesize other members of the GAG oligosaccharides. The generality of these conditions, the short reaction time, and the easy purification process resulting from the complete sulfonation of all the reactive sites under microwave irradiation is expected to accelerate the preparation of HP/HS-like oligosaccharides, thus overcoming one of the difficult bottle-necks in the synthesis of GAG.Acknowledgements
Financial support from the Ministry of Science and Technology of China (2012CB822102) and the National Natural Science Foundation of China (21432012) is gratefully acknowledged.Notes and references
- (a) I. Capila and R. J. Linhardt, Angew. Chem., Int. Ed., 2002, 41, 390–412 CrossRef CAS; (b) N. S. Gandhi and R. L. Mancera, Chem. Biol. Drug Des., 2008, 72, 455–482 CrossRef CAS PubMed.
- (a) C. A. A. van Boeckel and M. Petitou, Angew. Chem., Int. Ed. Engl., 1993, 32, 1671–1690 CrossRef; (b) M. Petitou and C. A. van Boeckel, Angew. Chem., Int. Ed., 2004, 43, 3118–3133 CrossRef CAS PubMed.
- (a) L. H. Lam, J. E. Silbert and R. D. Rosenberg, Biochem. Biophys. Res. Commun., 1976, 69, 570–577 CrossRef CAS PubMed; (b) R. D. Rosenberg and L. Lam, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 1218–1222 CrossRef CAS PubMed; (c) U. Lindahl, G. Backstrom, M. Hook, L. Thunberg, L. A. Fransson and A. Linker, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 3198–3202 CrossRef CAS PubMed; (d) A. G. G. Turpie, B. I. Eriksson, M. R. Larssen and K. A. Bauer, Curr. Opin. Hematol., 2003, 10, 327–332 CrossRef PubMed.
- (a) G. J. S. Lohman and P. H. Seeberger, J. Org. Chem., 2004, 69, 4081–4093 CrossRef CAS PubMed; (b) R. Lucas, D. Hamza, A. Lubineau and D. Bonnaffé, Eur. J. Org. Chem., 2004, 2107–2117 CrossRef CAS; (c) S. U. Hansen, G. J. Miller, M. Baráth, K. R. Broberg, E. Avizienyte, M. Helliwell, J. Raftery, G. C. Jayson and J. M. Gardiner, J. Org. Chem., 2012, 77, 7823–7843 CrossRef CAS PubMed; (d) N. Guedes, P. Czechura, B. Echeverria, A. Ruiz, O. Michelena, M. Martin-Lomas and N.-C. Reichardt, J. Org. Chem., 2013, 78, 6911–6934 CrossRef CAS PubMed; (e) J. Li, Y. Dai, W. Li, S. Laval, P. Xu and B. Yu, Asian J. Org. Chem., 2015, 4, 756–762 CrossRef CAS.
- (a) P. Sinaÿ, J.-C. Jacquinet, M. Petitou, P. Duchaussoy, I. Lederman, J. Choay and G. Torri, Carbohydr. Res., 1984, 132, C5–C9 CrossRef; (b) M. Petitou, P. Duchaussoy, I. Lederman, J. Choay, J.-C. Jacquinet, P. Sinaÿ and G. Torri, Carbohydr. Res., 1987, 167, 67–75 CrossRef CAS PubMed.
- (a) C. Noti, J. L. de Paz, L. Polito and P. H. Seeberger, Chem. – Eur. J., 2006, 12, 8664–8686 CrossRef CAS PubMed; (b) A. Adibekian, P. Bindschädler, M. S. M. Timmer, C. Noti, N. Schützenmeister and P. H. Seeberger, Chem. – Eur. J., 2007, 13, 4510–4522 CrossRef CAS PubMed; (c) T. Polat and C.-H. Wong, J. Am. Chem. Soc., 2007, 129, 12795–12800 CrossRef CAS PubMed; (d) J. Chen, Y. Zhou, C. Chen, W. Xu and B. Yu, Carbohydr. Res., 2008, 343, 2853–2862 CrossRef CAS PubMed; (e) Z. Wang, Y. Xu, B. Yang, R. Liu, G. Tiruchinapally, B. Sun, S. Dulaney, J. Liu and X. Huang, Chem. – Eur. J., 2010, 16, 8365–8375 CrossRef CAS PubMed; (f) Y.-P. Hu, S.-Y. Lin, C.-Y. Huang, M. M. L. Zulueta, J.-Y. Liu, W. Chang and S.-C. Hung, Nat. Chem., 2011, 3, 557–563 CrossRef CAS PubMed; (g) M. M. L. Zulueta, S.-Y. Lin, Y.-T. Lin, C.-J. Huang, C.-C. Wang, C.-C. Ku, Z. Shi, C.-L. Chyan, D. Irene, L.-H. Lim, T.-I. Tsai, Y.-P. Hu, S. D. Arco, C.-H. Wong and S.-C. Hung, J. Am. Chem. Soc., 2012, 134, 8988–8995 CrossRef CAS PubMed; (h) Y.-P. Hu, Y.-Q. Zhong, Z.-G. Chen, C.-Y. Chen, Z. Shi, M. M. L. Zulueta, C.-C. Ku, P.-Y. Lee, C.-C. Wang and S.-C. Hung, J. Am. Chem. Soc., 2012, 134, 20722–20727 CrossRef CAS PubMed; (i) S.-C. Hung, X.-A. Lu, J.-C. Lee, M. D.-T. Chang, S.-L. Fang, T.-C. Fan, M. M. L. Zulueta and Y.-Q. Zhong, Org. Biomol. Chem., 2012, 10, 760–772 RSC; (j) C.-H. Chang, L. S. Lico, T.-Y. Huang, S.-Y. Lin, C.-L. Chang, S. D. Arco and S.-C. Hung, Angew. Chem., Int. Ed., 2014, 53, 9876–9879 CrossRef CAS PubMed.
- (a) J. L. de Paz and M. Martín-Lomas, Eur. J. Org. Chem., 2005, 1849–1858 CrossRef CAS; (b) S. Arungundram, K. Al-Mafraji, J. Asong, F. E. Leach III, I. J. Amster, A. Venot, J. E. Turnbull and G.-J. Boons, J. Am. Chem. Soc., 2009, 131, 17394–17405 CrossRef CAS PubMed; (c) S. U. Hansen, G. J. Miller, G. C. Jayson and J. M. Gardiner, Org. Lett., 2013, 15, 88–91 CrossRef CAS PubMed; (d) S. Roy, A. El Hadri, S. Richard, F. Denis, K. Holte, J. Duffner, F. Yu, Z. Galcheva-Gargova, I. Capila, B. Schultes, M. Petitou and G. V. Kaundinya, J. Med. Chem., 2014, 57, 4511–4520 CrossRef CAS PubMed; (e) G. J. Miller, S. U. Hansen, E. Avizienyte, G. Rushton, C. Cole, G. C. Jayson and J. M. Gardiner, Chem. Sci., 2013, 4, 3218–3222 RSC; (f) S. U. Hansen, G. J. Miller, M. J. Cliff, G. C. Jayson and J. M. Gardiner, Chem. Sci., 2015, 6, 6158–6164 RSC.
- (a) A. Lubineau, H. Lortat-Jacob, O. Gavard, S. Sarrazin and D. Bonnaffé, Chem. – Eur. J., 2004, 10, 4265–4282 CrossRef CAS PubMed; (b) L.-D. Lu, C.-R. Shie, S. S. Kulkarni, G.-R. Pan, X.-A. Lu and S.-C. Hung, Org. Lett., 2006, 8, 5995–5998 CrossRef CAS PubMed; (c) A. Dilhas, R. Lucas, L. Loureiro-Morais, Y. Hersant and D. Bonnaffé, J. Comb. Chem., 2008, 10, 166–169 CrossRef CAS PubMed; (d) F. Baleux, L. Loureiro-Morais, Y. Hersant, P. Clayette, F. Arenzana-Seisdedos, D. Bonnaffé and H. Lortat-Jacob, Nat. Chem. Biol., 2009, 5, 743–748 CrossRef CAS PubMed.
- P. Xu, W. Xu, Y. Dai, Y. Yang and B. Yu, Org. Chem. Front., 2014, 1, 405–414 RSC.
- R. A. Al-Horani and U. R. Desai, Tetrahedron, 2010, 66, 2907–2918 CrossRef CAS PubMed.
- G. Tiruchinapally, Z. Yin, M. El-Dakdouki, Z. Wang and X. Huang, Chem. – Eur. J., 2011, 17, 10106–10112 CrossRef CAS PubMed.
- For microwave-assisted O-sulfonation of heparin-like oligosaccharides, see: (a) S. Maza, J. L. de Paz and P. M. Nieto, Tetrahedron Lett., 2011, 52, 441–443 CrossRef CAS; (b) S. Maza, G. Macchione, R. Ojeda, J. López-Prados, J. Angulo, J. L. de Paz and P. M. Nieto, Org. Biomol. Chem., 2012, 10, 2146–2163 RSC.
- For microwave-assisted O-sulfonation of other organic scaffolds, see: (a) H. Kiyota, D. J. Dixon, C. K. Luscombe, S. Hettstedt and S. V. Ley, Org. Lett., 2002, 4, 3223–3226 CrossRef CAS PubMed; (b) A. Raghuraman, M. Riaz, M. Hindle and U. R. Desai, Tetrahedron Lett., 2007, 48, 6754–6758 CrossRef CAS PubMed.
- C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250–6284 CrossRef CAS PubMed.
- (a) D. S. Pikas, J.-P. Li, I. Vlodavsky and U. Lindahl, J. Biol. Chem., 1998, 273, 18770–18777 CrossRef CAS PubMed; (b) Y. Okada, S. Yamada, M. Toyoshima, J. Dong, M. Nakajima and K. Sugahara, J. Biol. Chem., 2002, 277, 42488–42495 CrossRef CAS PubMed.
- (a) V. H. Pomin, Int. J. Biol. Macromol., 2015, 72, 282–289 CrossRef CAS PubMed; (b) J. Kandasamy, F. Schuhmacher, H. S. Hahm, J. C. Klein and P. H. Seeberger, Chem. Commun., 2014, 50, 1875–1877 RSC; (c) S. Maza, M. M. Kayser, G. Macchione, J. López-Prados, J. Angulo, J. L. de Paz and P. M. Nieto, Org. Biomol. Chem., 2013, 42, 3510–3525 RSC; (d) N. A. Karst and R. J. Linhardt, Curr. Med. Chem., 2003, 10, 1993–2031 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, and NMR spectra for new compounds. See DOI: 10.1039/c5qo00320b |
| This journal is © the Partner Organisations 2016 |