
Magnetic relaxations in four-coordinate Dy(III) complexes: effects of anionic surroundings and short Dy–O bonds†
Ke-Xin
Yu‡
a,
You-Song
Ding‡
a,
Tian
Han
a,
Ji-Dong
Leng
ab and
Yan-Zhen
Zheng
*a
aFrontier Institute of Science and Technology (FIST), State Key Laboratory of Mechanical Behavior of Materials and MOE Key Laboratory for Nonequilibrium Synthesis and Modulation of Condensed Matter, Xi'an Jiaotong University, Xi'an 710054, P. R. China. E-mail: zhengyanzhen@xjtu.edu.cn
bSchool of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 3rd June 2016
Abstract
Two four-coordinate Dy(III) complexes, [LiTHF4][Dy(NPh2)4] (1) and DyLi(Ph3CO)3(NPh2)THF (2), with similar distorted tetrahedral geometries and a pure anionic coordination environment were synthesized under anaerobic conditions. Magnetic studies reveal that only 2 having a short Dy–O bond (2.07 Å) shows slow magnetic relaxation behaviour under zero dc field. Quantitative analyses of energy-levels and eigenstates using the program PHI reveal that the ground state doublets for both the complexes are mixed with low-lying states, though complex 2 shows much higher purity of ground state doublets. This study presumably indicates that the spherically coordinated anions with evenly distributed charges are adverse to the enhancement of the axial magnetic anisotropy of the Dy(III) ions, while the strong Dy–O bond may break such a high symmetry and hence promote the axial magnetic anisotropy of the Dy(III) ions.
Introduction
In recent years the magnetic properties of mononuclear lanthanide complexes have been intensively investigated for single-molecule information storage materials, the so-called single-ion magnets (SIMs), because the large magnetic anisotropic energy barrier for spin-reversal (ΔE) originating from the strong spin–orbit coupling effect of unpaired 4f electrons may significantly enhance the magnetic blocking temperature.1–4 Since the spin–orbit coupling effect of the lanthanide ions is considered to be directly related to the crystal-field terms,5 the coordination environments, including the coordination number (CN), coordination symmetry and coordination ligands, are highly important in the design of high-performance lanthanide SIMs.Because the ionic radii of lanthanide ions are large, the CN of lanthanide complexes is often high, ranging from 7 to 12.4 From a literature review we can see that many low-coordinate (CN < 6) lanthanide complexes were synthesized by organometallic chemists,6 but their low temperature magnetic dynamics were seldom studied. Indeed, the geometric control of low-coordinate lanthanide complexes is relatively easier since the topological variants are much reduced as the coordination number decreases. Hence, the magnetic investigation of such complexes may be beneficial to understand the magneto-structural correlation of lanthanide SIMs and has already drawn some attention in recent studies.7–12 Surprisingly, most of the reported low-coordinate lanthanide SIMs that exhibit slow magnetic relaxation without the assistance of an external magnetic field are based on Er(III) ions.7,9 Low-coordinate, especially for CN = 3 or 4, Dy(III) complexes were rarely reported as SIMs.12
Recently we reported the magnetic properties of a series of Dy(III) complexes with crown ethers.13 We found that when the Dy(III) ions are coordinated by some counter-anions such as NO3− and OH−, the magnetic performance will be better. We then reasoned that if more anions were coordinated to the Dy(III) ion, the magnetic anisotropy may be further enhanced. Low-coordinate lanthanide complexes thus provide us a good testing bed.
Herein we show two four-coordinate Dy(III) complexes, [LiTHF4][Dy(NPh2)4] (1) and DyLi(Ph3CO)3(NPh2)THF (2), with a pure anionic coordination environment and similar distorted tetrahedral geometries. However, magnetic studies reveal that only complex 2 exhibits slow magnetic relaxation under zero dc field presumably due to the very short Dy–O (2.068(3) Å) bond when compared with other Dy–O/N bonds in both 1 and 2.
Results and discussion
Syntheses and crystal structures
Complexes 1 and 2 were prepared in similar reactions of deprotonated amine/alcohol ligands and anhydrous DyCl3 in THF overnight (Scheme 1). The solvent was then removed under vacuum and the residue was extracted with hot toluene for 1 and THF for 2, respectively. Single crystals suitable for X-ray analysis were obtained by cooling the toluene extracts to −35 °C overnight for 1, and layering pentane above the THF extracts for 2 (for details, see the Experimental section). | ||
| Scheme 1 Syntheses and structures of complexes 1 (a) and 2 (b). | ||
Complex 1 crystallizes in the orthorhombic space group Pbcn and comprises a [Dy(Ph2N)4]− anion balanced by a counterion [Li(C4H8O)4]+ (Fig. 1a). The Dy(III) ion in the [Dy(Ph2N)4]− anion is bound to four NPh2− groups, generating a distorted DyN4 tetrahedron. The Dy–N bonds are in distances ranging from 2.28 Å to 2.30 Å (Table 1), whereas the N–Dy–N angles are in a much wide range (from 93.84° to 134.09°, Table S1†). This reveals a significant deviation from the ideal tetrahedral angle of 109.28°. These results are similar to its Y(III) analogue as reported previously.14 The Li+ ion in the [Li(C4H8O)4]+ cation is four-coordinated in a tetrahedral geometry with four oxygen atoms from four THF molecules. The Li–O distances lie in the range of 1.93 Å–1.94 Å. The O–Li–O angles range from 101.53° to 112.86°. For the crystal packing of complex 1, the shortest intermolecular Dy⋯Dy separation is about 11.33 Å (Fig. S1†).
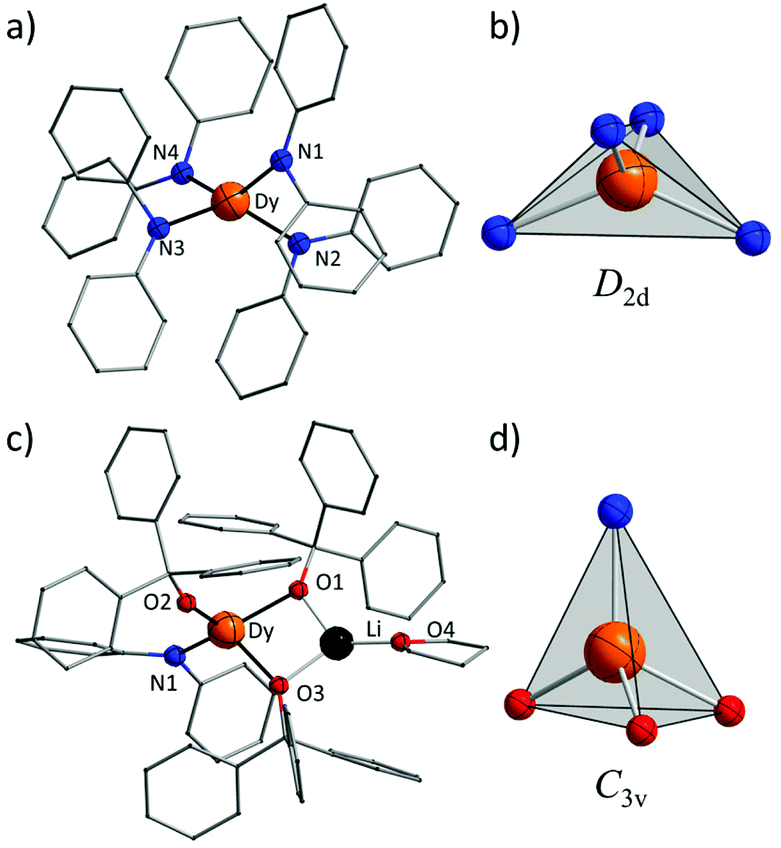 | ||
| Fig. 1 (a) Structure of [Dy(Ph2N)4]− anion in 1. The [Li(C4H8O)4]+ cation and H atoms are omitted for clarity. (b) The coordination polyhedron of 1. (c) Molecular structure of 2. H atoms have been omitted for clarity. (d) The coordination polyhedron for 2. | ||
| 1 | 2 | ||
|---|---|---|---|
| Dy–N(1) | 2.2948(15) | Dy–N(1) | 2.273(4) |
| Dy–N(2) | 2.2825(16) | Dy–O(1) | 2.195(3) |
| Dy–N(3) | 2.2947(15) | Dy–O(2) | 2.068(3) |
| Dy–N(4) | 2.2825(16) | Dy–O(3) | 2.152(3) |
Complex 2 crystallizes in the monoclinic space group P21/n. Different from 1, the Dy(III) ion is coordinated to three Ph3CO− and one NPh2− groups in a distorted DyO3N tetrahedron (Fig. 1c). Remarkably, the terminal triphenylmethanol provides a very strong Dy–O bond (2.068(3) Å) (Table 1), which is one of the shortest coordination bonds among all the reported Ln-SIMs.13 The Dy–O bond lengths for the two other triphenylmethanol ligands that bridge the Li+ ion and the Dy(III) ion are 2.15 Å and 2.20 Å, respectively (Table 1). The length of the Dy–N bond is 2.273(4) Å, which is a bit shorter than those in 1. The angles in the DyO3N tetrahedron range from 78.04° to 127.30° (Table S2†). The Li+ ion lies in a planar triangle geometry additionally coordinated to a THF molecule. In the crystal packing of 2, the shortest intermolecular Dy⋯Dy separation is 11.90 Å (Fig. S2†).
To gain a further insight into the coordination geometry of these two complexes, the continuous shape measures (CSM) were used to determine the degree of the deviation from the ideal polyhedron (a larger value means a larger distortion).15 There are four kinds of ideal polyhedra for four-coordinate complexes: namely tetrahedron, seesaw, vacant trigonal-bipyramid and square. For 1, the lowest value of 4.73 was found for the tetrahedron geometry; for 2, two similar CSM values were obtained (3.36 for tetrahedron and 3.35 for vacant trigonal bipyramid) (Table 2), indicating two possibilities. Nevertheless, the coordination geometry for 1 (Fig. 1b) shows an oblate tetrahedron with a D2d local symmetry for the central Dy(III) ion and the umbrella-opening distortion from the ideal tetrahedron for 2 gives a C3v local symmetry (Fig. 1d).15
Magnetic properties
 | ||
| Fig. 2 The χT versus T plot of 1 (a) and 2 (b). Inset: The field-dependent magnetization plots at indicated temperatures. The solid lines are best fits. | ||
With the dc magnetic susceptibilities, quantitative information of energy-levels and eigenstates can be obtained by using the program PHI.16 For complex 1, its Dy(III) ion locates in the D2d local symmetry, and for 2, the local symmetry is C3v (see above), so their effective crystal-field (CF) Hamiltonians are proposed as:
| ĤCF = B20C20 + B40C40 + B44C44 + B60C60 + B64C64 |
| ĤCF = B20C20 + B40C40 + B43C43 + B60C60 + B63C63 + B66C66 |
The crystal-field parameters of the CF Hamiltonians were obtained by fitting their χT versus T plot and M–H at the same time (solid lines in Fig. 2), which were then used to simulate their energy-levels and eigenstates (Tables S5 and S6†). Both complexes 1 and 2 have impure ground state doublets. For 1, its ground state doublets are mixed with ±15/2 (69.8%), ±7/2 (30.1%) and ±1/2 (0.1%) mJ states, which are about 24.26 cm−1 separated from the excited Kramers doublets. For 2, ±15/2 (92.0%), ±9/2 (7.8%) and ±3/2 (0.2%) mJ states comprise its main components of ground state doublets. The barrier for the first excited state is about 59.9 cm−1. From these fitting results we can conclude: (1) the ground states for both 1 and 2 are all mixed; (2) the first excited states are not high in both 1 and 2. Obviously, the mixed mJ states are the key, because the mixture of different mJ states will cause strong quantum tunneling of magnetization (QTM).18 Relatively, complex 2 shows much higher purity of ground state doublets and ΔE compared to 1.
Under an optimized dc field, the QTM is effectively reduced so that obvious signals were observed in both χ′(T) and χ′′(T) plots for complexes 1 and 2 (Fig. 3). For 1, under a 1600 Oe dc field, ac signals appear below 3.5 K (Fig. 3a) at high frequencies. Its frequency-dependent ac susceptibility data were recorded from 2.0 K to 3.5 K to evaluate the relaxation times (Fig. S3†), which were fitted by the generalized Debye function using the CC-FIT program.19 The α parameters range from 0.17 to 0.35 between 2.0 and 3.5 K (Table S3†). For 2, the optimized dc field is 700 Oe. The peaks of χ′′ signals became sharp and move to a slightly higher temperature than that of 1. For a frequency of 1302 Hz, a maximum signal appears at about 4.0 K (Fig. 3b). Frequency-dependent ac susceptibility data were measured from 2.0 K to 4.9 K (Fig. S6†). The distribution coefficient value (α) is 0.18–0.26 (Table S4†).
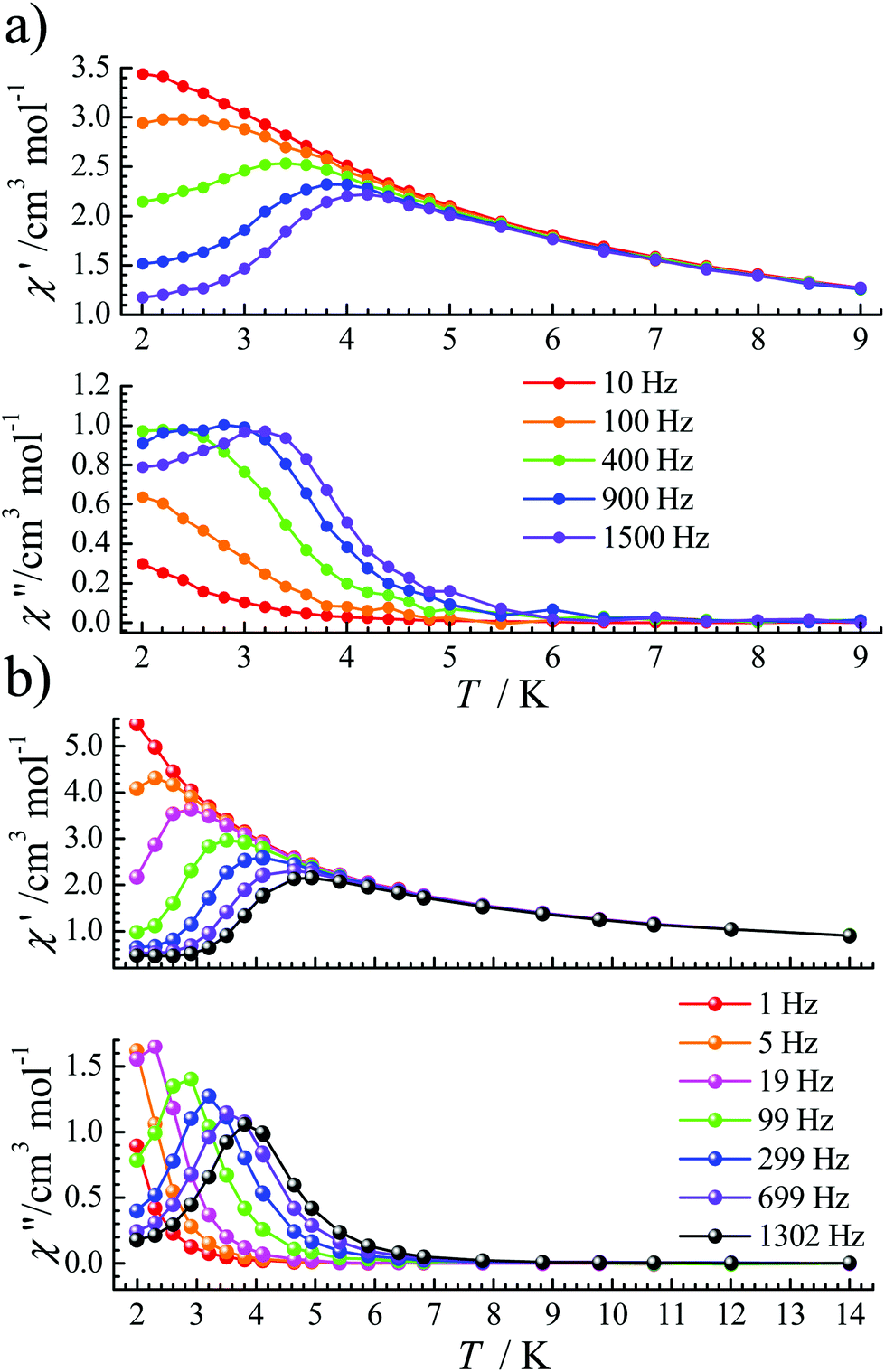 | ||
| Fig. 3 Temperature-dependence of the χ′ and χ′′ susceptibility signals under the optimized dc field (1600 Oe for 1 and 700 Oe for 2) at the indicated frequencies for 1 (a) and 2 (b). The solid lines are guides for the eye. | ||
The relaxation time τ is one of the main parameters to evaluate the success of SMMs. Only when the relaxation time is long enough to observe the magnetic hysteresis, it can be called a magnet.1a In our cases, the relaxation time for 2 is more than 100 times longer than that for 1 at the same temperature (Fig. 4). For complex 1, a large curvature appears at a low temperature, which is probably caused by QTM. If an Orbach process and the QTM are assumed to be dominant relaxation processes, fitting those data with the equation τ−1 = τQTM−1 + τ0−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−ΔE/T) gives ΔE = 24.8 K with τQTM = 2.89 × 10−4 s and τ0 = 6.41 × 10−8 s for 1. The ln(τ) values increase linearly with 1/T at a higher temperature, and then a slight curvature appears for 2. Fitting of the high temperature regime with the Arrhenius law τ = τ0exp(ΔE/T) gives ΔE = 35.9 K and τ0 = 1.22 × 10−8 s. The curvature at a low temperature is usually caused by other relaxation processes, such as direct and Raman.20 The latter process may be dominant at low temperatures for 2 as we can well simulate the data below 3 K using τ−1 = cTn with c = 0.59 K−n s−1 and n = 6.53 (Fig. S8†).
exp(−ΔE/T) gives ΔE = 24.8 K with τQTM = 2.89 × 10−4 s and τ0 = 6.41 × 10−8 s for 1. The ln(τ) values increase linearly with 1/T at a higher temperature, and then a slight curvature appears for 2. Fitting of the high temperature regime with the Arrhenius law τ = τ0exp(ΔE/T) gives ΔE = 35.9 K and τ0 = 1.22 × 10−8 s. The curvature at a low temperature is usually caused by other relaxation processes, such as direct and Raman.20 The latter process may be dominant at low temperatures for 2 as we can well simulate the data below 3 K using τ−1 = cTn with c = 0.59 K−n s−1 and n = 6.53 (Fig. S8†).
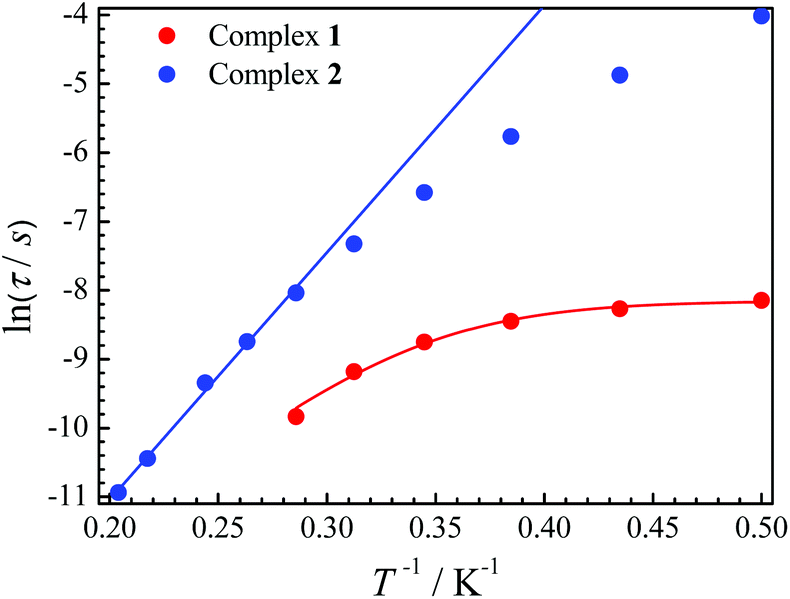 | ||
| Fig. 4 Plots of ln(τ) versus 1/T for 1 and 2. The solid lines are best fits. | ||
Moreover, the correlation between the ground state magnetic anisotropy and the coordination environment for Dy(III) complexes was performed using the program Magellan (Fig. 5).21 This program clearly shows that the principal magnetic axis of 1 is not aligned in any of the Dy–N bonds, whereas the principal magnetic axis for 2 lies exactly along the shortest Dy–O bond direction. The result for 1 may suggest competition of the anionic donors in such a cubic symmetry environment, which is fairly even and there is no obvious winner in each direction. However, for 2 there exists a Dy–O bond with a obvious shorter distance that may affect the charge distribution of the central metal ion more profoundly than in other directions, which in other words, may enhance the axial magnetic anisotropy of Dy(III) ions. So complex 2 has a higher energy barrier for magnetization reversal and hence displays magnetic relaxation under zero dc field even in such a cubic environment.
 | ||
| Fig. 5 Principal magnetic axis (green arrow) for 1 (a) and 2 (b). | ||
Discussions
As far as we know, only a limited number of low-coordinate mononuclear lanthanide complexes have been magnetically studied in the literature, and some Er(III) analogues exhibit SIM behaviour.7,9,12 For instance, the three-coordinated Er{N(SiMe3)2}3 displays magnetic relaxation behaviours under zero dc field with an ΔE of 122 K,7 while its Dy(III) analogue did not show any SIM behaviour above 2 K; the four-coordinated [Li(THF)4[Er{N(SiMe3)2}3Cl]·2THF shows an ΔE of 66.4 K,9 but its Dy(III) counterpart was reported to have a ΔE of only 17 K under the assistance of a 600 Oe dc field.12 The reason behind these phenomena is that both the equatorial and the spherical coordination environments often generate small or mixed ground mJ state doublets,7,12 while the latter situation is rather similar to 1 and 2.However, a short coordination bond may weaken such disadvantages as we can find high performance Dy(III) single-molecule magnets to usually associate it.22–27 For example, the lanthanide alkoxides often contain short coordination bonds, so that some of their Dy(III) complexes are reported as high performance SIMs.22 In particular the polymetallic complex Dy@[Y4K2O(OtBu)12], which shows an ΔE of 842 K, contains a very short Dy–O bond of 2.07 Å.22b Other high performance Dy(III) SIMs, such as [Zn2DyL′2(MeOH)]NO3·3MeOH·H2O with a ΔE of 439 K,23 [Dy(BIPMTMS)2][K(18C6)(THF)2] with a ΔE of 813 K,24 Dy(Cy3PO)2(H2O)5]Br3·2(Cy3PO)·2H2O·2EtOH with the blocking temperature 20 K,25 Dy(bbpen)Br with a ΔE of 1025 K (ref. 26) and [L′′2Dy(H2O)5][I]3·L′′2·(H2O) with a ΔE of 735 K,27 the axial coordination bonds are also found to be much shorter than the equatorial bonds. Hence, we suspect that the short Dy–O bond in 2 is vital to fight against the adverse spherical symmetry, which results in a slightly higher ΔE compared to 1. Nevertheless, short Dy–O bonds cannot ensure a high ΔE for Dy-SIMs. Though the Dy–O bond in 2 is shorter than any reported Dy-SIMs, its magnetic property is not better than others. Thus, the combination of highly axial local symmetry is also very important to strengthen the magnetic anisotropy of the Dy(III) ions.
Conclusions
In conclusion, the magnetic properties of two four-coordinate Dy(III) complexes with a distorted cubic Td symmetry have been investigated. All the coordinated atoms are negatively charged, providing an overall spherical anionic coordination environment, which we believe is disadvantageous to enhance the axial magnetic anisotropy for Dy(III) ions. Only complex 2 with a shorter Dy–O bond can differentiate its direction from the others and hence displays magnetic relaxation under zero dc field. For higher performance Dy-SIMs the combination of the local axial symmetry of the Dy(III) ions is obviously necessary.Experimental section
Materials and methods
All the reactions were carried out under a dry and oxygen-free argon atmosphere by using Schlenk techniques or in a glovebox. All the solvents used were dried and degassed by standard techniques. Anhydrous LnCl3 salts were prepared according to the literature procedure.28 The ligands were purchased from TCI and used without further treatment. n-Butyllithium (2.5 mol L−1 in n-hexane) was purchased from Chemetal.Synthesis of [Li(C4H8O)4][Dy(Ph2N)4] (1)
Diphenylamine (0.85 g, 5 mmol) was dissolved in 5 ml THF, and 2.0 ml of n-butyllithium (2.5 mol L−1) in n-hexane solution was added through a syringe at 0 °C, which produced a yellow solution. The solution was stirred for further 2 h at 0 °C. Then anhydrous DyCl3 (0.27 g, 1 mmol) was added, forming a cloudy solution immediately. The reaction mixture was heated to reflux overnight. After evaporation of the solvent followed by extraction of the residue with hot toluene, the crystals of [Li(C4H8O)4][Dy(Ph2N)4] were obtained by cooling the extracts. The products were filtered and washed with n-pentane (2 × 5 ml), then dried under vacuum. Yield 0.85 g (75.2% based on metal Dy).Synthesis of DyLi(Ph3CO)3(NPh2)THF (2)
Diphenylamine (0.085 g, 0.5 mmol) and triphenylmethanol (0.39 g, 1.5 mmol) were dissolved in 5 ml THF, and then 0.8 ml of n-butyllithium (2.5 mol L−1) in n-hexane solution was added using a syringe at 0 °C, producing a yellow cloudy solution, which was stirred for further 2 h at 0 °C. Then anhydrous DyCl3 (0.14 g, 0.5 mmol) was added. The reaction mixture was heated to reflux overnight. After evaporation of the solvent followed by extraction of the residue with THF, the crystals of DyLi(Ph3CO)3(NPh2)THF were obtained by layering n-pentane above the extracts. The products were filtered and washed with n-pentane (2 × 5 ml), and then dried under vacuum. Yield 0.48 g (80.8% based on metal Dy).X-ray crystallography data
Single crystals suitable for X-ray analysis were coated with deoxygenated Paratone-N oil. All the data were collected on a Bruker Apex II DUO area-detector diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). Cell refinement and data reduction were accomplished with the SAINT processing program. The structures were solved by direct methods and refined on F2 using SHELXTL (Table 3). CCDC 1469847 (1) and 1469848 (2) contain the supplementary crystallographic data for this paper.| 1 | 2 | |
|---|---|---|
| Empirical formula | C64H72DyLiN4O4 | C73H63DyLiNO4 |
| Formula weight (g mol−1) | 1130.70 | 1187.68 |
| Crystal system | Orthogonal | Monoclinic |
| Space group | Pbcn | P21/n |
| T (K) | 150 | 150 |
| a (Å) | 17.2073(11) | 17.1728(13) |
| b (Å) | 15.8244(10) | 18.0592(13) |
| c (Å) | 20.0712(13) | 19.3313(14) |
| β (°) | 90 | 95.0640(10) |
| V (Å3) | 5465.3(6) | 5971.8(8) |
| Z | 4 | 4 |
| T (K) | 150(2) | 150(2) |
| ρ calcd (g cm−3) | 1.374 | 1.321 |
| F (000) | 2340 | 2436 |
| Reflns collected | 31463 |
67577 |
| Unique reflns | 6288 | 13802 |
| R int | 0.02 | 0.0326 |
| GOF | 1.043 | 1.184 |
| R 1 [I > 2σ(I)] | 0.0223 | 0.0552 |
| wR2 [all data] | 0.0545 | 0.1105 |
| Largest diff. peak/hole (e Å−3) | 0.899/−0.473 | 2.018/−1.888 |
Magnetic properties
Magnetic susceptibility measurements have been carried out with a Quantum Design MPMS-XL7 SQUID magnetometer upon cooling from 300 to 2 K in variable applied fields. Ac susceptibility measurements have been performed at frequencies between 1 and 1500 Hz with an ac field of 3.5 Oe and with a variable dc applied field. Powder samples were embedded in eicosane to avoid any field induced crystal reorientation. A diamagnetic correction has been calculated from Pascal constants and embedding eicosane has been applied to the observed magnetic susceptibility.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the “973 projects” (2012CB619401 and 2012CB619402), the NSFC (21473129, 21503155 and IRT13034), the “National Young 1000-Plan” program, the Fundamental Research Funds for the Central Universities and the Wuhan National High Magnetic Field Center (2015KF06).Notes and references
- (a) D. Gatteschi, R. Sessoli and J. Villain, Molecular nanomagnets, Oxford University Press, 2006 Search PubMed; (b) C. Benelli and D. Gatteschi, Introduction to Molecular Magnetism: From Transition Metals to Lanthanides, John Wiley & Sons, 2015 Search PubMed; (c) S. Gao, Molecular Nanomagnets and Related Phenomena, Springer, 2015 Search PubMed.
- (a) R. Sessoli and A. K. Powell, Coord. Chem. Rev., 2009, 253, 2328–2341 CrossRef CAS; (b) D. N. Woodruff, R. E. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed; (c) H. L. Feltham and S. Brooker, Coord. Chem. Rev., 2014, 276, 1–33 CrossRef CAS; (d) P. Zhang, L. Zhang and J. Tang, Dalton Trans., 2015, 44, 3923–3929 RSC; (e) M. Ren and L.-M. Zheng, Acta Chim. Sin., 2015, 73, 1091–1113 CrossRef CAS; (f) K. Liu, X. Zhang, X. Meng, W. Shi, P. Cheng and A. K. Powell, Chem. Soc. Rev., 2016, 45, 2423–2439 RSC.
- (a) M. Urdampilleta, S. Klyatskaya, J.-P. Cleuziou, M. Ruben and W. Wernsdorfer, Nat. Mater., 2011, 10, 502–506 CrossRef CAS PubMed; (b) S. Thiele, F. Balestro, R. Ballou, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Science, 2014, 344, 1135–1138 CrossRef CAS PubMed.
- (a) N. Ishikawa, M. Sugita, T. Ishikawa, S.-Y. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed; (b) J. Tang, I. Hewitt, N. T. Madhu, G. Chastanet, W. Wernsdorfer, C. E. Anson, C. Benelli, R. Sessoli and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 1729–1733 CrossRef CAS PubMed; (c) M. A. AlDamen, J. M. Clemente-Juan, E. Coronado, C. Martí-Gastaldo and A. Gaita-Ariño, J. Am. Chem. Soc., 2008, 130, 8874–8875 CrossRef CAS PubMed; (d) S. D. Jiang, B. W. Wang, G. Su, Z. M. Wang and S. Gao, Angew. Chem., Int. Ed., 2010, 122, 7610–7613 CrossRef; (e) S.-D. Jiang, B.-W. Wang, H.-L. Sun, Z.-M. Wang and S. Gao, J. Am. Chem. Soc., 2011, 133, 4730–4733 CrossRef CAS PubMed; (f) J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, J. Am. Chem. Soc., 2011, 133, 14236–14239 CrossRef CAS PubMed; (g) J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, Nat. Chem., 2011, 3, 538–542 CrossRef CAS PubMed; (h) J. W. Sharples, Y. Z. Zheng, F. Tuna, E. J. L. McInnes and D. Collison, Chem. Commun., 2011, 47, 7650–7652 RSC; (i) F. Tuna, C. A. Smith, M. Bodensteiner, L. Ungur, L. F. Chibotaru, E. J. L. McInnes, R. E. P. Winpenny, D. Collison and R. A. Layfield, Angew. Chem., Int. Ed., 2012, 51, 6976–6980 CrossRef CAS PubMed; (j) L. Zou, L. Zhao, P. Chen, Y.-N. Guo, Y. Guo, Y.-H. Li and J. Tang, Dalton Trans., 2012, 41, 2966–2971 RSC; (k) J. J. Le Roy, M. Jeletic, S. I. Gorelsky, I. Korobkov, L. Ungur, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2013, 135, 3502–3510 CrossRef CAS PubMed; (l) K. R. Meihaus and J. R. Long, J. Am. Chem. Soc., 2013, 135, 17952–17957 CrossRef CAS PubMed; (m) S. Demir, M. Nippe, M. I. Gonzalez and J. R. Long, Chem. Sci., 2014, 5, 4701–4711 RSC; (n) S. Demir, M. Nippe, M. I. Gonzalez and J. R. Long, Chem. Sci., 2014, 5, 4701–4711 RSC; (o) Y. N. Guo, L. Ungur, G. E. Granroth, A. K. Powell, C. Wu, S. E. Nagler, J. Tang, L. F. Chibotaru and D. Cui, Sci. Rep., 2014, 4, 5471 CAS; (p) W. Cao, C. Gao, Y.-Q. Zhang, D. Qi, T. Liu, K. Wang, C. Duan, S. Gao and J. Jiang, Chem. Sci., 2015, 6, 5947–5954 RSC; (q) W. Cao, C. Gao, Y.-Q. Zhang, D. Qi, T. Liu, K. Wang, C. Duan, S. Gao and J. Jiang, Chem. Sci., 2015, 6, 5947–5954 RSC.
- (a) J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC; (b) S.-D. Jiang and S.-X. Qin, Inorg. Chem. Front., 2015, 2, 613–619 RSC.
- (a) M. Lappert, A. Protchenko, P. Power and A. Seeber, Metal amide chemistry, John Wiley & Sons, 2008 Search PubMed; (b) C.-H. Huang, Rare earth coordination chemistry: fundamentals and applications, John Wiley & Sons, 2011 Search PubMed.
- P. Zhang, L. Zhang, C. Wang, S. Xue, S.-Y. Lin and J. Tang, J. Am. Chem. Soc., 2014, 136, 4484–4487 CrossRef CAS PubMed.
- S. K. Singh, T. Gupta, M. Shanmugam and G. Rajaraman, Chem. Commun., 2014, 50, 15513–15516 RSC.
- A. J. Brown, D. Pinkowicz, M. R. Saber and K. R. Dunbar, Angew. Chem., Int. Ed., 2015, 127, 5962–5966 CrossRef.
- N. F. Chilton, C. A. Goodwin, D. P. Mills and R. E. Winpenny, Chem. Commun., 2015, 51, 101–103 RSC.
- N. F. Chilton, Inorg. Chem., 2015, 54, 2097–2099 CrossRef CAS PubMed.
- P. Zhang, J. Jung, L. Zhang, J. Tang and B. Le Guennic, Inorg. Chem., 2016, 55, 1905–1911 CrossRef CAS PubMed.
- Y.-S. Ding, T. Han, Y.-Q. Hu, M. Xu, S. Yang and Y.-Z. Zheng, Inorg. Chem. Front., 2016, 3, 798–807 RSC.
- Y. Yao, X. Lu, Q. Shen and K. Yu, J. Chem. Crystallogr., 2004, 34, 275–279 CrossRef CAS.
- J. Cirera, P. Alemany and S. Alvarez, Chem. – Eur. J., 2004, 10, 190–207 CrossRef CAS PubMed.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- C. Görller-Walrand, K. Binnemans, K. A. Gschneidner and L. Eyring, Handbook on the Physics and Chemistry of Rare Earths, 1996, vol. 23 Search PubMed.
- Z.-X. Jiang, J.-L. Liu, Y.-C. Chen, J. Liu, J.-H. Jia and M.-L. Tong, Chem. Commun., 2016, 52, 6261–6264 RSC.
- Y.-N. Guo, G.-F. Xu, Y. Guo and J. Tang, Dalton. Trans., 2011, 40, 9953–9963 RSC.
- K. S. Pedersen, L. Ungur, M. Sigrist, A. Sundt, M. Schau-Magnussen, V. Vieru, H. Mutka, S. Rols, H. Weihe and O. Waldmann, Chem. Sci., 2014, 5, 1650–1660 RSC.
- N. F. Chilton, D. Collison, E. J. McInnes, R. E. Winpenny and A. Soncini, Nat. Commun., 2013, 4, 2551 RSC.
- (a) R. J. Blagg, C. A. Muryn, E. J. L. McInnes, F. Tuna and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2011, 50, 6530–6533 CrossRef CAS PubMed; (b) R. J. Blagg, L. Ungur, F. Tuna, J. Speak, P. Comar, D. Collison, W. Wernsdorfer, E. J. L. McInnes, L. F. Chibotaru and R. E. P. Winpenny, Nat. Chem., 2013, 5, 673–678 CrossRef CAS PubMed.
- J.-L. Liu, Y.-C. Chen, Y.-Z. Zheng, W.-Q. Lin, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M.-L. Tong, Chem. Sci., 2013, 4, 3310–3316 RSC . L′ = 2,2′,2′′-(((nitrilotris(ethane-2,1-diyl))tris(azanediyl)).
- M. Gregson, N. F. Chilton, A.-M. Ariciu, F. Tuna, I. F. Crowe, W. Lewis, A. J. Blake, D. Collison, E. J. McInnes and R. E. Winpenny, Chem. Sci., 2016, 7, 155–165 RSC.
- Y. C. Chen, J. L. Liu, L. Ungur, J. Liu, Q. W. Li, L. F. Wang, Z. P. Ni, L. F. Chibotaru, X. M. Chen and M. L. Tong, J. Am. Chem. Soc., 2016, 138, 2829–2837 CrossRef CAS PubMed.
- J. Liu, Y.-C. Chen, J.-L. Liu, V. Vieru, L. Ungur, J.-H. Jia, L. F. Chibotaru, Y. Lan, W. Wernsdorfer, S. Gao, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2016, 138, 5441–5450 CrossRef CAS PubMed . bbpen = N,N′-Bis(2-hydroxybenzyl)-N,N′-bis(2-methylpyridyl) ethylenediamine.
- S. K. Gupta, T. Rajeshkumar, G. Rajaraman and R. Murugavel, Chem. Sci., 2016 10.1039/C6SC00279J . L′′ = (tBuPO(NHiPr)2.
- J. B. Reed, B. Hopkins, L. Audrieth, P. Selwood, R. Ward and J. Dejong, Inorg. Synth., 1939, 1, 28–33 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1469847 and 1469848. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qi00117c |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2016 |