DOI:
10.1039/C6QI00096G
(Research Article)
Inorg. Chem. Front., 2016,
3, 1048-1057
Facile synthesis of diverse transition metal oxide nanoparticles and electrochemical properties†
Received
16th April 2016
, Accepted 29th May 2016
First published on 31st May 2016
Abstract
We herein report a facile method for synthesizing a series of metal oxides from the mixture of oleylamine and metal nitrates with heating at 180 °C. Several diverse nanoparticles with various morphologies are gained expectedly: 16.5 nm Mn3O4 nano-capsules, 22 nm CoO nano-cubes, 35 nm NiO nano-flowers and 4 nm Fe3O4 quantum dots. This method is convenient for preparing the dispersed nanoparticles at mild reaction temperature with low-cost metal sources. As-prepared CoO nano-cubes are employed as the electrode material of a pseudocapacitor in terms of implementation. The specific capacitance reaches a stable level of 750 F g−1. This material also exhibits a superior performance in high rate capability and maintains an excellent stability during 9000 recycles. The asymmetric supercapacitor device with a maximum voltage of 1.7 V is fabricated using CoO nano-cubes as the positive and activated carbon as the negative electrode.
1. Introduction
Transition metal oxides of MOx (M = Mn, Fe, Co and Ni) have been applied broadly in the areas of energy storage,1–5 catalyst preparation6–8 and nano-material synthesis in the last few years.9–15 Various shaped nanoparticles (NPs) synthesized by different methods have been reported such as cubes,15,16 spheres,17 rods,18 triangles,19 hollow,20 and other shapes.21,22 The materials could present specific physical and chemical properties when their morphologies are controlled on the nano scale.23,24 For example, hexagonal and cubic CoO nanocrystals were prepared by various processes and applied in research as magnetic semiconductors,25 along with Fe2O3,26,27 Fe3O4,28 ZnO,29 and Mn3O4.30,31 With different organic and inorganic dissolvents and even reducing agents, diverse materials, such as Au,8,32 Ag33,34 and Ni,6,35,36 are successfully obtained by kinetically tuned thermal deposition.37,38 The proper choice of reactants is important in wet-chemical routes for the synthesis of nanoparticles. Among numerous reagents as the reaction medium, researchers are focusing on alkylamine solvents, especially oleylamine (OAm).39–41 OAm is liquid at room temperature and can effectively provide high temperature because of its high boiling point (≈350 °C).41 Moreover, OAm can act as a versatile surfactant, mild reducing reagent and electron donor at enhanced temperature with amino and long chain alkenes. OAm has a much lower cost than the usually used pure alkylamines. These superior characteristics allow OAm to fabricate nanoparticles with special textures and target structures. As another important part of the reaction, however, many metal sources are organo-metallic precursors, such as acetylacetonate (acac) and carbonyl (CO) metal compounds, which are often expensive and hypertoxic. In addition, very high of a reaction temperature may also decrease the manipulation convenience for operation, in principle.42 Herein, we introduce a facile method for preparing nanoparticles through decomposition of metal nitrates with OAm. Metal nitrates are soluble, inexpensive and unstable. In addition, decomposition products of metal sources are explicit and simple. Therefore, metal nitrates are selected as the desired candidates. Diverse dispersed nanoparticles of transition metal oxides (MnOx, CoO, Fe3O4 and NiO) with various morphologies are gained successfully. More characteristics are investigated in detail, including the nanostructure, reaction mechanism and suitability for application in energy storage. Moreover, cobalt-based materials, such as CoO and Co3O4, are fascinating electrode materials because of their active electrochemical performance and easy synthesis. Therefore, as-prepared CoO nanoparticles are selected as a pseudocapacitor and aqueous asymmetric supercapacitor for potential energy applications.
2. Experimental section
All metal nitrates, including [Mn(NO3)2·6H2O] (98%, Alfa Aesar), [Fe(NO3)3·9H2O] (98+%, Alfa Aesar), [Co(NO3)2·6H2O] (98%, Alfa Aesar), [Ni(NO3)2·6H2O] (98%, Alfa Aesar), [AgNO3] (99%, Alfa Aesar), oleylamine (80%–90%, Aladdin) and other reagents were used without further purification.
2.1 Synthesis of 21.8 ± 0.7 nm CoO nano-cubes
[Co(NO3)2·6H2O] (0.2 g, 0.69 mmol) and OAm (10 mL, 29 mmol) were mixed and stirred in a three-necked flask. The mixture was heated at 110 °C with vigorous stirring for 0.5 h, producing a red-orange homogeneous solution. Then, the mixture was flash-heated to 180 °C. While maintaining this temperature for 12 h, the color of the solution became deep-brown. The solution was cooled down to room temperature and then, the as-prepared nanoparticles were separated by ethanol (30 mL). After the suspension was centrifuged and washed with ethanol three times, the final product, CoO nano-cubes, was dispersed in hexane (10 mL).
2.2 Synthesis of other nanoparticles
Four types of nanoparticles were prepared under similar reaction conditions: 16.5 ± 2 nm Mn3O4 nano-capsules (0.5 mL, 3.2 mmol, 1 min), 35 ± 5 nm NiO nano-flowers (0.2 g, 0.69 mmol, 12 h), and 3.75 ± 1.5 nm Fe3O4 quantum dots (0.15 g, 0.37 mmol, 3 h).
2.3 Compositional and structural characterization
Powder X-ray diffraction (XRD) was performed on a Rigaku D/max-2500 diffractometer with Cu Kα radiation for characterizing the architecture of the synthesized NPs. The detailed morphology of the NPs was studied with a transmission electron microscope (TEM, Philips Tecnai G2 F20) equipped with an energy dispersive X-ray spectrometer (EDX) for elemental analysis. X-ray photoelectron spectroscopy (XPS, PHI 5000 Versaprobe, ULVAC PHI) was employed to investigate the valence of elements. The Fourier transform infrared (FTIR) spectrum was obtained for the KBr diluted sample using a FTIR-650 spectrometer (Tianjin Gangdong, resolution of 4 cm−1).
2.4 Electrochemical measurements
For the electrochemical measurements, the working electrodes were constructed by mixing the active materials, acetylene black and a polyvinylidene fluoride (PVDF) binder. CoO (80%), acetylene black (10%) and the PVDF binder (10%) were dispersed in an N-methyl-2-pyrrolidinone (NMP) solvent and ground thoroughly to form slurry. Then, the slurry was pressed at 20 MPa to a piece of nickel foam (10 mm × 10 mm) and dried under vacuum at 80 °C for 12 h. An Hg/HgO reference electrode and one piece of nickel foam were used as a reference electrode and counter electrode, respectively. The electrolyte was 2 M KOH aqueous solution. The electro-chemical measurements were conducted in a three compartment cell by a LAND battery test instrument (CT2001A). Cyclic voltammetry (CV) measurements were conducted using a Zahner IM6e electrochemical workstation with voltage scan rates of 1 mV s−1, 5 mV s−1, 10 mV s−1, 20 mV s−1, 50 mV s−1, and 100 mV s−1. The galvanostatic charge–discharge tests were conducted at the currents of 1 A g−1, 2 A g−1, 5 A g−1, 10 A g−1, and 20 A g−1. The electrochemical impedance spectroscopy (EIS) measurements were carried out using an AC voltage with 5 mV amplitude in a frequency range from 0.1 Hz to 0.1 MHz at open circuit potential.
3. Results and discussion
3.1 Synthesis process of nanoparticles
The growth and transformation of Mn3O4 nano-capsules are studied using a model by our method. The 16.5 ± 2 nm Mn3O4 nano-capsules are synthesized by heating [Mn(NO3)2·6H2O] with OAm (10 mL) for one minute at 180 °C. While extending reaction time for 10 min, we get 100 nm rods, which are the mixtures of MnO and Mn3O4 in Fig. 1. X-ray powder diffraction (XRD) experiment is employed to analyze the composition and structure of the nanoparticles. As shown in Fig. 1S,† all peaks of the MnOx compounds structures are clearly identified. Mn3O4 completely transforms into the MnO phase after nearly 60 min of heating for reaction. Moreover, the short rods elongate themselves through the Ostwald ripening process. Based on the HRTEM images and fast Fourier transform (FFT) patterns of tetragonal phase Mn3O4, the measured spacing of the lattice planes are 0.250 nm and 0.491 nm. The short rods transform to 100 nm smooth rods after 10 min of reaction. However, Fig. 1S† shows that the 100 nm nano-rods are a mixture of Mn3O4 and MnO, also shown in Fig. 1h. By extending the reaction time to 30 min, 80 nm Mn3O4 and MnO nano-rods are obviously obtained, but the 80 nm rods are less smooth than the 100 nm rods. The reason for this is the breaking of long rods; the atomic lattice fringes in Fig. 1k have an obvious distance of 0.118 nm. Fig. 1l is simpler than Fig. 1d and h, revealing that the MnO phase is favored in the mixture. After 60 min of reaction, all the nano-rods are broken and aggregated remarkably. The atomic lattice fringes in Fig. 1o have a distance of 0.157 nm, which implied that only the MnO phase exists after heating for 60 min in the OAm system, as shown in Fig. S1p.† The transformation also demonstrates the weak reducibility of OAm during the reaction to form the low valence state of the metal oxide.
 |
| | Fig. 1 TEM, High Resolution Transmission Electron Microscopy (HRTEM) and fast Fourier transform (FFT) images of series of MnOx synthesized at different reaction times: 1 min (a–d), 10 min (e–h), 30 min (i–l), and 60 min (m–p). | |
Based on the analysis of TEM and XRD results, we know that the formation of MnOx nanoparticles can be affected by the reaction time and reducibility of OAm. After the Mn3O4 nuclei are formed, nano-capsules are capped by OAm immediately, and the classical Ostwald ripening process will occur and lead to the formation of Mn3O4 nano-capsules. However, because of the reducibility of OAm, Mn3O4 is gradually reduced to MnO with increasing reaction time. When the reaction time is 60 min, Mn3O4 turns into MnO overall. Due to formation of nanocrystal structures, long MnO nano-rods fall apart and become aggregated.
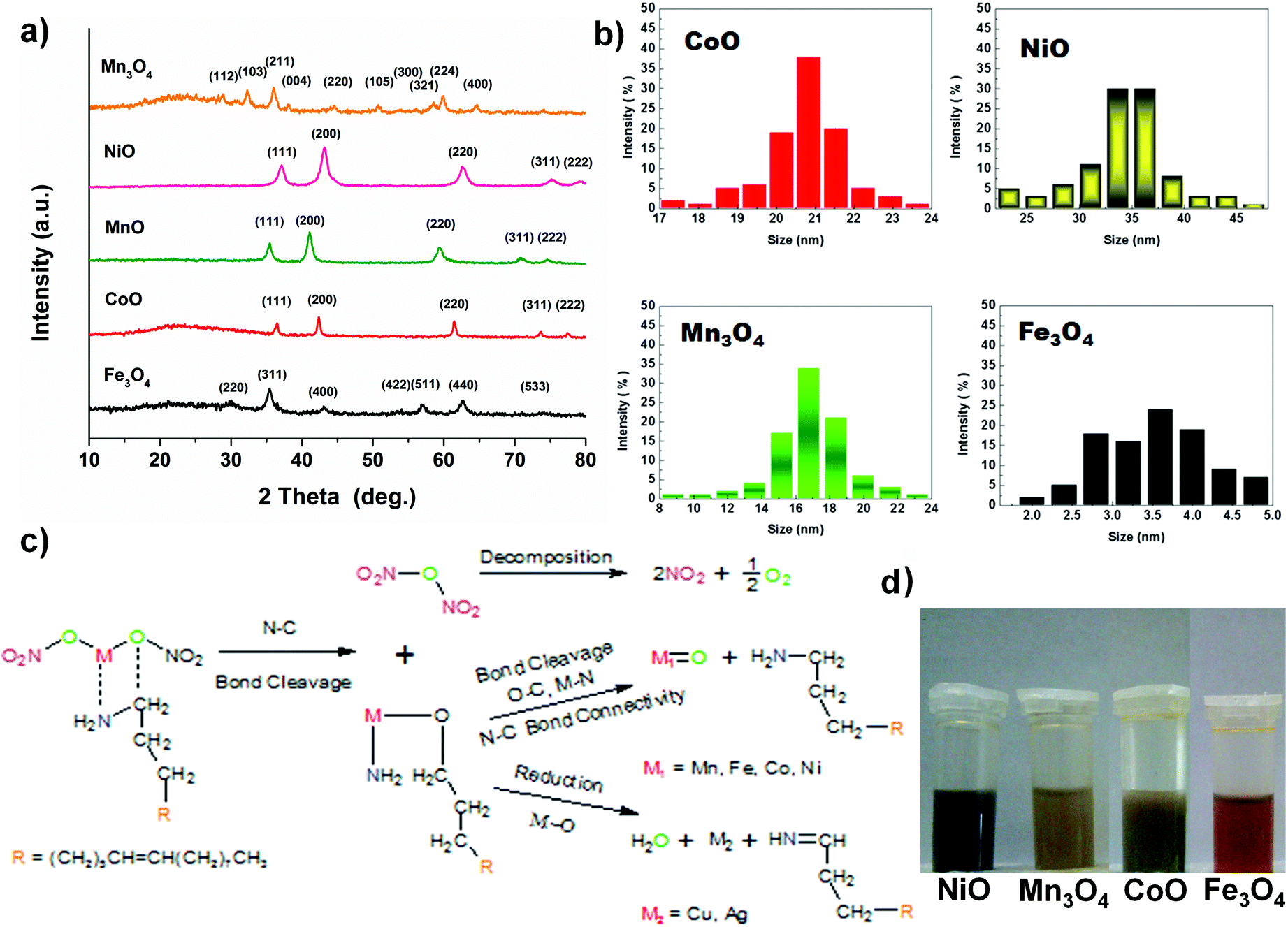
Other monodisperse nanoparticles with different morphologies are obtained via this method, i.e. 21.8 ± 0.7 nm CoO nano-cubes, 35 ± 5 nm NiO nano-flowers and 3.75 ± 1.5 nm Fe3O4 quantum dots, by different reaction times, as shown in Fig. 2. The results of XRD measurements prove the success of synthesis of these metal oxides and metal and representative peaks of the materials are obviously identified (Fig. 3a).
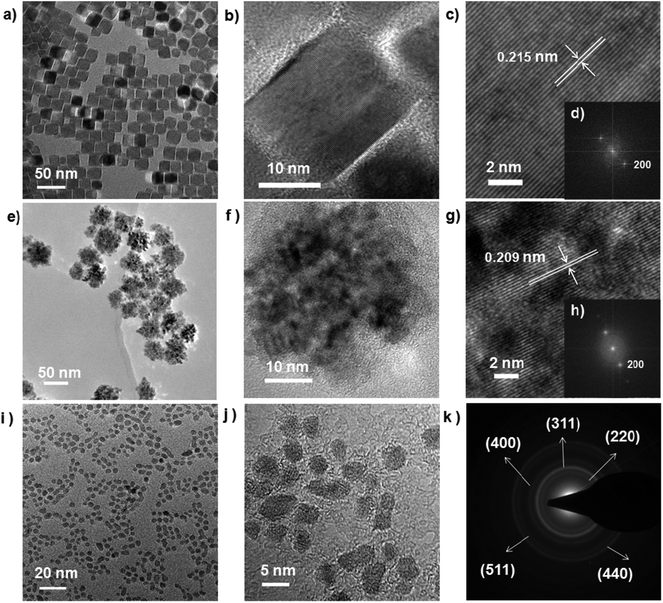 |
| | Fig. 2 TEM, HRTEM, SADE and FFT images of 21.8 ± 0.7 nm CoO nano-cubes (a–d), 35 ± 5 nm NiO nano-flowers (e–h), 3.75 ± 1.5 nm Fe3O4 quantum dots (i–k) nanoparticles. | |
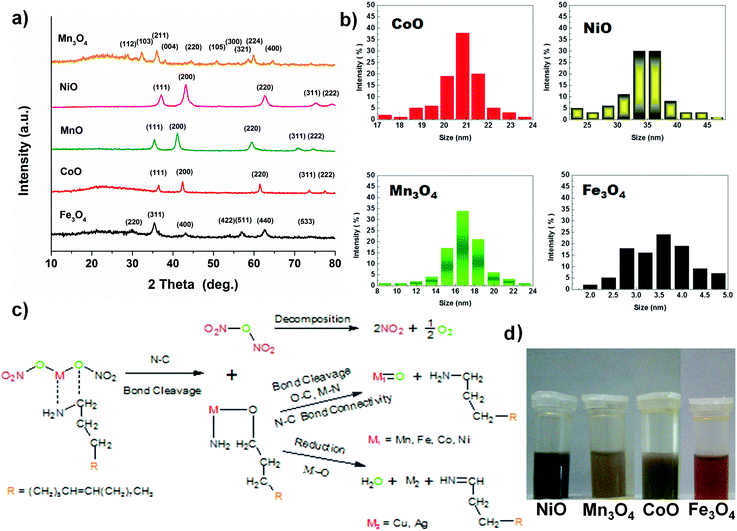 |
| | Fig. 3 (a) XRD patterns of metal oxides and metal particles. (b) The size distribution maps of dispersed nanoparticles. (c) Possible reaction pathways: the generation of as-prepared nanoparticles and (d) images of nanoparticles dispersed in hexane. | |
All the diverse metal oxides distinctly take regular and independent structures. The nanoparticles, protected by the long chain of OAm, can be well dispersed in non-polar solvent. All the nanoparticles have quite apparent crystallizations and thermally stable properties, though their morphologies are varying due to the variation of metal sources (Fig. 3 and Table S1 in the ESI†).
3.2 Characterization of CoO nano-cubes
CoO has excellent electrochemical performance and a less complex structure than other metal oxides we synthesized, so CoO nano-cube was investigated in detail. To obtain further detailed information about the CoO, the Fourier transform infrared (FT-IR) spectrum is used to characterize the structure of OAm molecules and growing process of the as-synthesized CoO nanoparticles. The spectra of CoO and Ag NPs in OAm are similar to that of the pure OAm in Fig. 4a. The principal part of OAm is not transformed to another structure, except for the –NH2 bending mode changing as reported.41,43 The peak at 3300–3400 cm−1 of Ag in OAm is much smaller than that of pure OAm and CoO, perhaps because the –NH2 oxidized.15,41,44 On the basis of the characteristic peaks and structures of the nanoparticles, the proposed reaction pathways of the metal oxides synthesized are displayed clearly in Fig. 3b.15,44 In Fig. 4b, five diffraction peaks could be well indexed to the standard card of cubic CoO (JCPDS no. 48-1719). Removing the OAm has no obvious effect on the characteristic peaks (annealing for 4 hours at 350 °C). The X-ray photoelectron spectroscopy (XPS) provides the binding energy of CoO in Fig. 4c and d. The peaks located at 781.2 eV and 797.1 eV in the high-resolution Co 2p spectrum correspond to Co 2p3/2 and Co 2p1/2. The shake-up peaks at 786.7 and 803.1 eV could also indicate the existence of CoO. The well-resolved O 1s peak at 529.8 eV (Fig. 4d) is well matched with the normal Co–O bonds in the CoO lattice, and the peak at 531.3 eV corresponds to O from the OH and oxides.
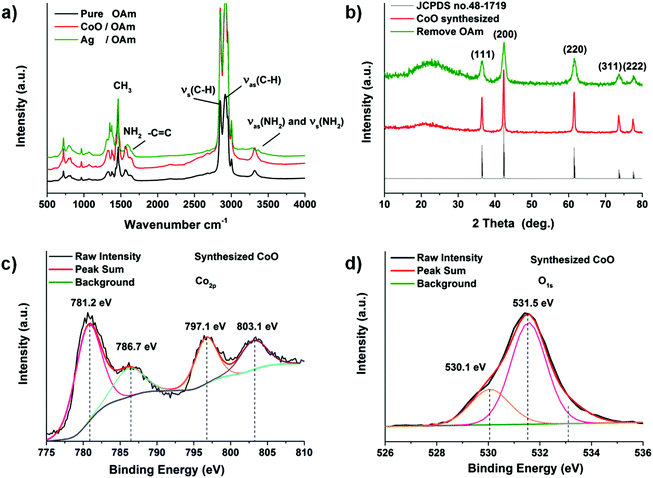 |
| | Fig. 4 (a) FT-IR patterns of metal particles and OAm. (b) XRD patterns of CoO. XPS analysis of CoO sample: (c) Co 3d and (d) O 1s. | |
3.3 Electrochemical properties of CoO nano-cubes
The as-prepared CoO nano-cubes are chosen as electrode materials, because of their high capacity and long cycle life, for pseudocapacitors and asymmetric supercapacitors.45–47 A series of electrochemical measurement results are shown and studied in detail in Fig. 5. The CV (cyclic voltammetry) measurements of the CoO electrode are performed at sweep rates of 1, 5, 10, 20, 50, and 100 mV s−1 (versus Hg/HgO). The loop curves expand as the sweep rates increase while keeping a pair of redox peaks with obviously the same shapes. The average specific capacitance of the CoO nano-cubes electrode could be calculated from the CV curves by the following equation:| | 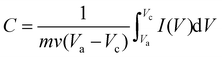 | (1) |
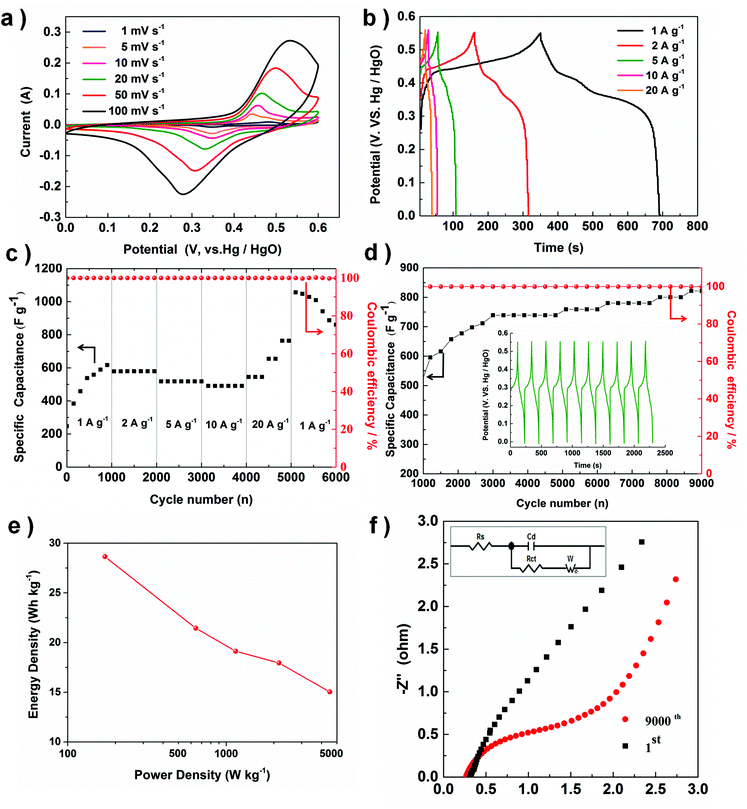 |
| | Fig. 5 The electrochemical characterizations of the as-synthesized CoO nano-cubes: (a) CV curves at various scan rates, (b) galvanostatic charge–discharge curves at various discharge current densities, (c) rate performances at different discharge current densities, (d) the dependences of the discharge specific capacitance and the coulombic efficiency on the charge–discharge cycle numbers, with galvanostatic charge and discharge curves at 5 A g−1 (inset), (e) Ragone plot (energy density vs. power density) of the CoO electrode. (f) Impedance Nyquist plots of the high frequency region of CoO nano-cube electrode (inset: Randles equivalent circuit). | |
The average specific capacitances of the CoO nano-cubes electrode are 572.9, 428.8, 382.4, 358.9, 300.6 and 247.6 F g−1 at the scan rates of 1, 5, 10, 20, 50 and 100 mV s−1, respectively. Galvanostatic charge and discharge measurements are employed to evaluate the electrochemical performance in detail, shown in Fig. 5b. The discharge plateau of the CoO electrode is located at 0.35–0.40 V, which is characteristic of pseudocapacitance. The specific capacitance is calculated by eqn (2):
| | 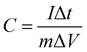 | (2) |
The results show that the CoO electrode exhibits great capacitance of 636.4, 585.5, 509.1, 472.7 and 727.3 F g−1 at the discharge current densities of 1, 2, 5, 10 and 20 A g−1, respectively. The measured results suggest that with the increasing current density, capacitance decreases slowly, because it is difficult to hold the oxidation–reduction transitions entirely, because this is beyond the ability of the active sites. Rate capability is also a significant parameter for supercapacitors, which possibly limits the practical application, shown in Fig. 5c.
We have calculated the specific capacitance according to formula (2), it is interesting that the capacitance at 20 A g−1 is higher than that at 10 A g−1 (727.7 vs. 472.7 F g−1), and the value calculated is different from other materials. We suggest two probable reasons. One reason is that we found that the discharge time for 20 A g−1 is not much lower than that for 10 A g−1 (20 vs. 26 s), but the current of 20 A g−1 is twice than 10 A g−1, which improves the value of the capacitance calculated. Another underlying reason might be that 20 A g−1 is too high for our CoO electrode materials; we have actually have CoO nanocubes, but there are still OAm molecules present, and the OAm would prevent OAm from aggregating and instead keep the special morphology. Very high current causes an organic substance such as OAm to decompose, and this process might cause the capacitance to be improved slightly.48 Focusing on this case, we still have a lot of study to do to investigate the mechanism in detail; therefore, we will make the contents and system more complete in the future. The outstanding capacitance performances mean that the CoO electrode has a good rate capability, especially in high current density.
Then, long-term cycle stability is evaluated by the charging-discharging measurement at a constant current density of 5 A g−1. After stimulating the process for 3000 cycles, the specific capacitance reaches a stable level of 750 F g−1, but after 9000 cycles, it slowly increases to 800 F g−1. Ragone plots derived from the discharge curve are shown in Fig. 5e; the energy density (E) and power density (P) could be calculated from the galvanostatic charge and discharge data using the eqn (3) and (4), where C (F g−1) is the discharge specific capacitance, V (V) is the potential window and Δt (h) is the discharge time.
The energy density could be estimated to be 28.6 Wh kg−1 at a power density of 171.9 W kg−1. The energy density of the CoO nano-cubes could also be estimated to be 15.0 Wh kg−1 at a power density of 2153.5 W kg−1 from Fig. 5e. The excellent cycling stability also reflects in the results of the electrochemical impedance spectra (EIS), after 9000 subsequent cycles, in which the impedance does not distinctly increase after the first cycle in Fig. 5f.
To affirm the phase and structural transformation of the CoO nano-cubes, TEM and XPS are employed, and the results are shown in Fig. 6. The CoO nano-cubes also maintain obvious cubic morphologies without visible smashing and aggregation in the foamed nickel mesh after 9000 cycles, as shown in Fig. 6a–d. From XPS, the shake-up peaks at 789.9 eV, 796.2 eV and 805.3 eV of Co 2p become weaker than those in Fig. 4c, because the content of CoO in the surface decreases after 9000 cycles. In addition, the peak at 529.8 eV increases obviously, indicating the existence of remaining Co(OH)2 or CoOOH, in Fig. 6e.49,50 Moreover, a new broad peak at 533.0 eV of O 1s in Fig. 6f shows the existence of Co3O4. The phase and structural transformation is identified after the cycling process through the above analysis. Possible eqn (5) and (6) of the redox reactions during the charge and discharge process are:51–55
| | | CoO + OH− = CoOOH + e− | (5) |
| | | 3CoOOH + e− = Co3O4 + OH− + H2O | (6) |
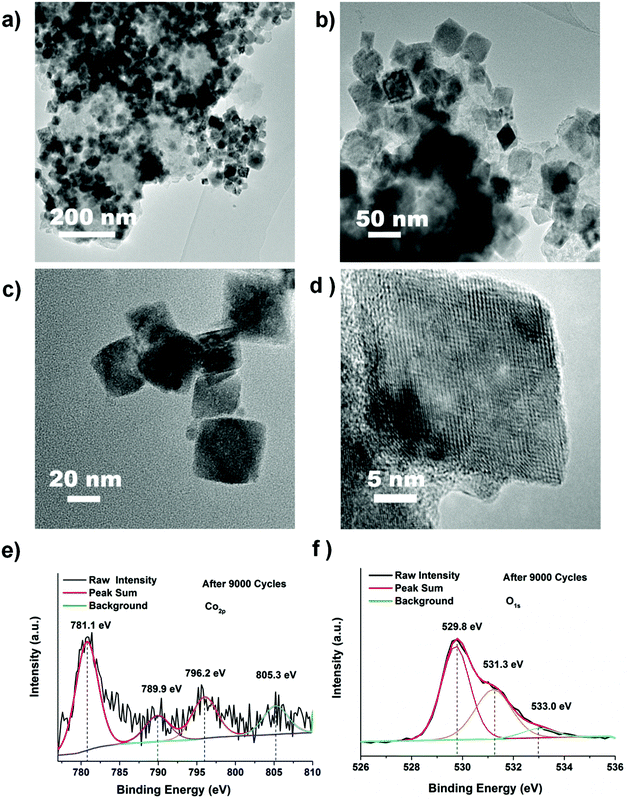 |
| | Fig. 6 (a–d) TEM, HRTEM and (e, f) XPS measurements of CoO nanoparticles after 9000 cycles. | |
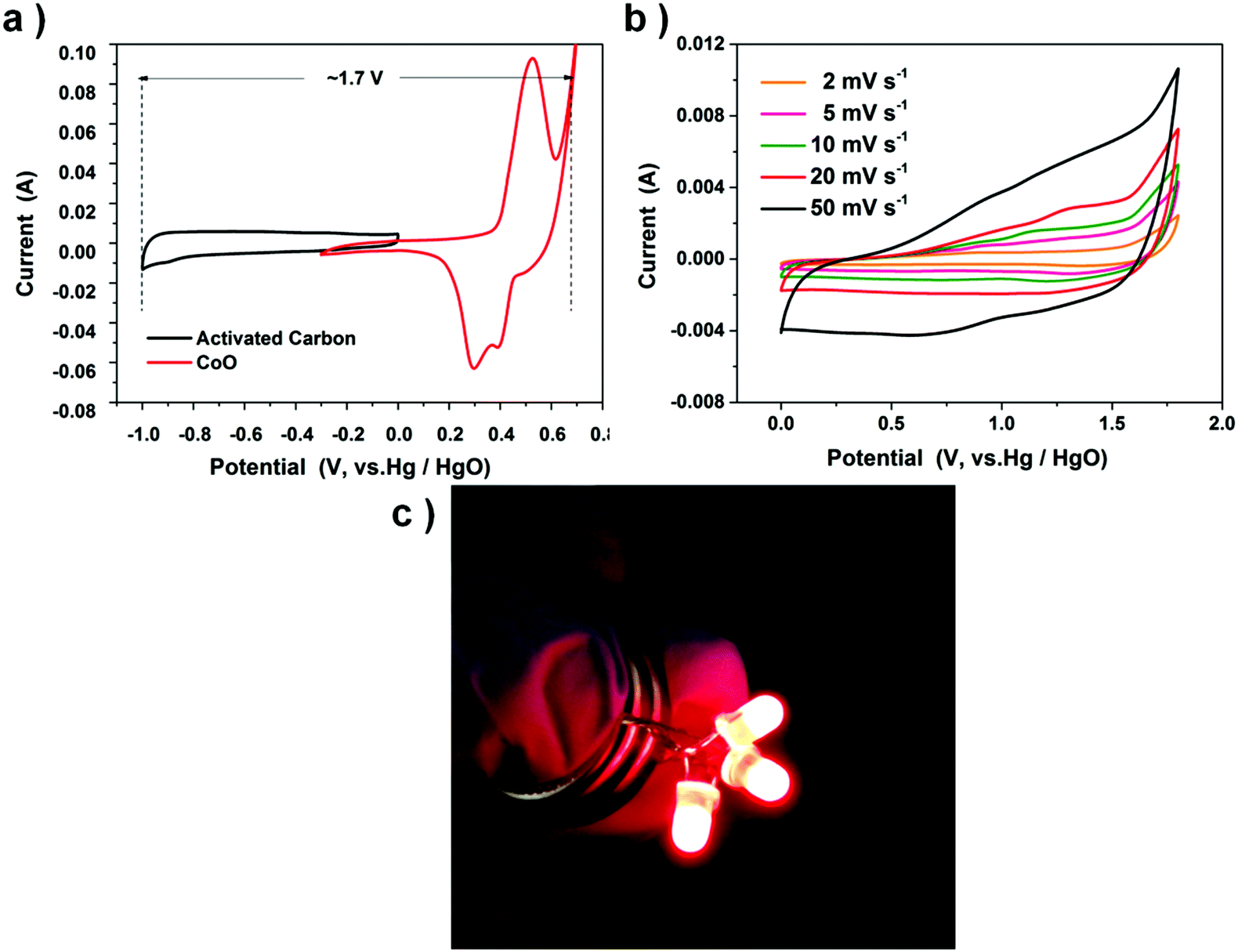
The remarkable electrochemical properties are also ascribed to the original pattern of CoO nano-cubes basically remaining.56,57 To further develop the CoO electrode for real applications, one 1 cm2 asymmetric supercapacitor button is made by employing the CoO cube electrode as the cathode and the activated carbon (AC) as the anode on nickel foam in 2 M KOH, with one piece of cellulose paper. Activated carbon is chosen as the negative electrode to balance the two electrodes’ capacitance due to its outstanding electro-conductibility, low-cost and low electrode potential.
Fig. 7a shows the CVs of positive and negative electrodes performed in a three part electrode cell and Fig. 7b exhibits the CV curves of the double-electrode supercapacitor button at different scan rates. The button displays a quasi-rectangular CV geometry, unlike the three-electrode electrochemical feature of the CoO nanocube electrode. The button voltage is as large as 1.7 V, almost thrice that of the CoO nanocube electrode (0–0.6 V). Two supercapacitors are connected in series, and after charging at 2 V for 1 min, the device could power three 5 mm diameter red round light-emitting diode (LED) indicators efficiently (Fig. 7c).
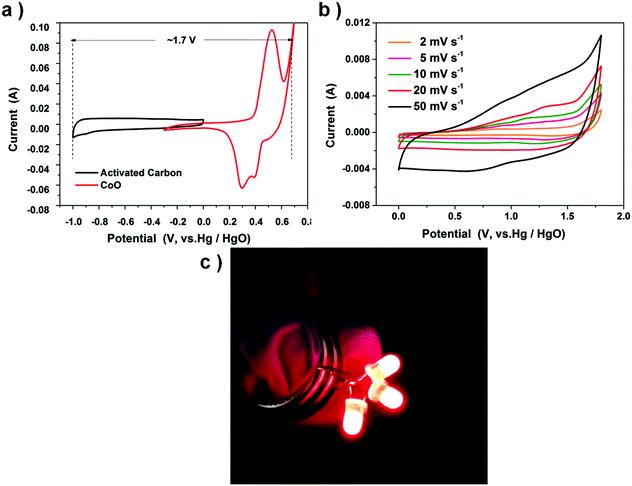 |
| | Fig. 7 (a) Comparative CV curves of CoO cubes and activated carbon (AC) electrodes. (b) CV curves of the CoO cubes and activated carbon asymmetric supercapacitor. (c) Two buttons in series could lighten up three LED indicators. | |
4. Conclusions
In short, we introduce a facile method for preparing metal and metal oxides through decomposition of metal nitrates with OAm and heating at 180 °C. This process is easy to reproduce the dispersed nanoparticles with low-cost metal source and mild reaction temperature. In terms of implemented properties, 22 nm CoO nano-cubes can be employed as an electrode material of pseudocapacitors. This material shows a superior performance in high rate capability and also maintains an excellent stability during 9000 recycles, due to the sustainability of the CoO cubic morphologies. This new synthesis methodology can also be used to synthesize other metals and metal oxides with multiple functionalities.
Acknowledgements
This study was supported by NSFC (51471089), MOE (IRT13R30), 111 Project (B12015), Research Fund for the Doctoral Program of Higher Education of China (20120031110001), Tianjin Sci & Tech Project (10SYSYJC27600).
Notes and references
- S. Jin, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and S. H. Yang, Nat. Chem., 2011, 3, 546–550 CrossRef PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- H. Wang, H. Yang and L. Lu, Electrochim. Acta, 2016, 188, 679–685 CrossRef CAS.
- X. Hu, M. Ma, M. Zeng, Y. Sun, L. Chen, Y. Xue, T. Zhang, X. Ai, R. G. Mendes, M. H. Rümmeli and L. Hu, ACS Appl. Mater. Interfaces, 2014, 6, 22527–22533 Search PubMed.
- S. H. Lee, S.-H. Yu, J. E. Lee, A. Jin, D. J. Lee, N. Lee, H. Jo, K. Shin, T.-Y. Ahn, Y.-W. Kim, H. Choe, Y.-E. Sung and T. Hyeon, Nano Lett., 2013, 13, 4249–4256 CrossRef CAS PubMed.
- S. Zhang, Y. Hao, D. Su, V. V. T. Doan-Nguyen, Y. Wu, J. Li, S. Sun and C. B. Murray, J. Am. Chem. Soc., 2014, 136, 15921–15924 CrossRef CAS PubMed.
- L. Zhang, W. Niu and G. Xu, Nano Today, 2012, 7, 586–605 CrossRef CAS.
- S. Peng, Y. Lee, C. Wang, H. Yin, S. Dai and S. Sun, Nano Res., 2008, 1, 229–234 CrossRef CAS.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J.-M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- J. Park, J. Joo, S. G. Kwon, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 4630–4660 CrossRef CAS PubMed.
- X. Yang, Y. He, Y. Bai, J. Zhang, L. Kang, H. Xu, F. Shi, Z. Lei and Z.-H. Liu, Electrochim. Acta, 2016, 188, 398–405 CrossRef CAS.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- D. Wang, X. Liang and Y. Li, Chem. – Asian J., 2006, 1–2, 91–94 CrossRef PubMed.
- W. S. Seo, J. H. Shim, S. J. Oh, E. K. Lee, N. H. Hur and J. T. Park, J. Am. Chem. Soc., 2005, 127, 6188–6189 CrossRef CAS PubMed.
- H. Zeng, P. M. Rice, S. X. Wang and S. Sun, J. Am. Chem. Soc., 2004, 126, 11458–11459 CrossRef CAS PubMed.
- S. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204–8205 CrossRef CAS PubMed.
- J. Park, E. Kang, C. J. Bae, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, H. M. Park and T. Hyeon, J. Phys. Chem. B, 2004, 108, 13594–13598 CrossRef CAS.
- Z. Niu, Q. Peng, M. Gong, H. Rong and Y. Li, Angew. Chem., Int. Ed., 2011, 50, 6315–6319 CrossRef CAS PubMed.
- X.-Y. Yu, L. Yu, L. Shen, X. Song, H. Chen and X. W. Lou, Adv. Funct. Mater., 2014, 24, 7440–7446 CrossRef CAS.
- X. Xu, X. Zhang, H. Sun, Y. Yang, X. Dai, J. Gao, X. Li, P. Zhang, H.-H. Wang, N.-F. Yu and S.-G. Sun, Angew. Chem., Int. Ed., 2014, 53, 1–7 CrossRef.
- P. F. Damasceno, M. Engel and S. C. Glotzer, Science, 2012, 337, 453–457 CrossRef CAS PubMed.
- J. He, P. Kanjanaboos, N. L. Frazer, A. Weis, X.-M. Lin and H. M. Jaeger, Small, 2010, 6, 1449–1456 CrossRef CAS PubMed.
- X. Liang, X. Wang, J. Zhuang, Y. Chen, D. Wang and Y. Li, Adv. Funct. Mater., 2006, 16, 1805–1813 CrossRef CAS.
- K. M. Nam, J. H. Shim, H. Ki, S.-I. Choi, G. Lee, J. K. Jang, Y. Jo, M.-H. Jung, H. Song and J. T. Park, Angew. Chem., Int. Ed., 2008, 47, 9504–9508 CrossRef CAS PubMed.
- M. B. Gawande, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371–3393 RSC.
- J. Wang, W. Liu, J. Chen, H. Wang, S. Liu and S. Chen, Electrochim. Acta, 2016, 188, 210–217 CrossRef CAS.
- H. Fan, L. Quan, M. Yuan, S. Zhu, K. Wang, Y. Zhong, L. Chang, H. Shao, J. Wang, J. Zhang and C.-N. Cao, Electrochim. Acta, 2016, 188, 222–229 CrossRef CAS.
- M. B. Gawande, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371–3393 RSC.
- Z. L. Wang, ACS Nano, 2008, 2, 1987–1992 CrossRef CAS PubMed.
- P. Li, C. Nan, Z. Wei, J. Lu, Q. Peng and Y. Li, Chem. Mater., 2010, 22, 4232–4236 CrossRef CAS.
- W. S. Seo, H. H. Jo, K. Lee, B. Kim, S. J. Oh and J. T. Park, Angew. Chem., Int. Ed., 2004, 43, 1115–1117 CrossRef CAS PubMed.
- X. Xu, X. Wang, A. Nisar, X. Liang, J. Zhuang, S. Hu and Y. Zhuang, Adv. Mater., 2008, 20, 3702–3708 CrossRef CAS.
- Y. Sun and Y. Xia, J. Am. Chem. Soc., 2004, 126, 3892–3901 CrossRef CAS PubMed.
- Y. Sun and Y. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS PubMed.
- Ö. Metin, V. Mazumder, S. Özkar and S. Sun, J. Am. Chem. Soc., 2010, 132, 1468–1469 CrossRef PubMed.
- D. Wang, L.-B. Kong, M.-C. Liu, Y.-C. Luo and L. Kang, Chem. – Eur. J., 2015, 21, 17897–17903 CrossRef CAS PubMed.
- Y. Hou, Z. Xu and S. Sun, Angew. Chem., Int. Ed., 2007, 46, 6329–6332 CrossRef CAS PubMed.
- D. Wang, T. Xie, Q. Peng, S. Zhang, J. Chen and Y. Li, Chem. – Eur. J., 2008, 14, 2507–2513 CrossRef CAS PubMed.
- Y. Yu, W. Yang, X. Sun, W. Zhu, X.-Z. Li, D. J. Sellmyer and S. Sun, Nano Lett., 2014, 14, 2778–2782 CrossRef CAS PubMed.
- M. Lattuada and T. A. Hatton, Langmuir, 2007, 23, 2158–2168 CrossRef CAS PubMed.
- S. Mourdikoudis and L. M. Liz-Marzán, Chem. Mater., 2013, 25, 1465–1476 CrossRef CAS.
- J. Rockenberger, E. C. Scher and A. P. Alivisatos, J. Am. Chem. Soc., 1999, 121, 11595–11596 CrossRef CAS.
- Z. Xu, C. Shen, Y. Hou, H. Gao and S. Sun, Chem. Mater., 2009, 21, 1778–1780 CrossRef CAS.
- D. Wang and Y. Li, J. Am. Chem. Soc., 2010, 132, 6280–6281 CrossRef CAS PubMed.
- G. Yu, X. Xie, L. Pan, Z. Bao and Y. Cui, Nano Energy, 2013, 2, 213–234 CrossRef CAS.
- Y. G. Zhu, Y. Wang, Y. Shi, Z. X. Huang, L. Fu and H. Y. Yang, Adv. Energy Mater., 2014, 4, 1301788 CrossRef.
- C. Zheng, C. Cao, Z. Ali and J. Hou, J. Mater. Chem. A, 2014, 2, 16467–16473 RSC.
- A. L. Holmes, J. Hütges, A. Reckmann, E. Muthuswamy, K. Meerholz and S. M. Kauzlarich, J. Phys. Chem. C, 2015, 119, 5671–5678 CrossRef CAS.
- K. Artyushkova, S. Levendosky, P. Atanassov and J. Fulghum, Top. Catal., 2007, 46, 263 CrossRef CAS.
- V. Hadjiev, M. Iliev and I. Vergilov, J. Phys. C: Solid State Phys., 1988, 21, L199 CrossRef.
- K. Cao, L. Jiao, Y. Liu, H. Liu, Y. Wang and H. Yuan, Adv. Funct. Mater., 2015, 25, 1082–1089 CrossRef CAS.
- C. Zhou, Y. Zhang, Y. Li and J. Liu, Nano Lett., 2013, 13, 2078–2085 CrossRef CAS PubMed.
- J. Chen and A. Selloni, J. Phys. Chem. C, 2013, 117, 20002–20006 CrossRef CAS.
- C. Guan, J. Liu, C. Cheng, H. Li, X. Li, W. Zhou, H. Zhang and H. Fan, Energy Environ. Sci., 2011, 4, 4496–4499 Search PubMed.
- J. Zhang, H. Feng, J. Yang, Q. Qin, H. J. Fan, C. Wei and W. Zheng, ACS Appl. Mater. Interfaces, 2014, 6, 22527–22533 Search PubMed.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- T. Brousse, D. Bélanger and J. W. Long, J. Electrochem. Soc., 2015, 162, A5185–A5189 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qi00096g |
|
| This journal is © the Partner Organisations 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.