
An asymmetric binuclear zinc(II) complex with mixed iminodiacetate and phenanthroline ligands: synthesis, characterization, structural conversion and anticancer properties†
Lubin
Ni‡
a,
Juan
Wang‡
b,
Chang
Liu
b,
Jinhong
Fan
b,
Yun
Sun
*b,
Zhaohui
Zhou
*c and
Guowang
Diao
*a
aCollege of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou 225002, Jiangsu, People's Republic of China. E-mail: gwdiao@yzu.edu.cn
bCollege of Medicine, Yangzhou University, Yangzhou 225001, People's Republic of China. E-mail: ysun@yzu.edu.cn
cState Key Laboratory for Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, People's Republic of China. E-mail: zhzhou@xmu.edu.cn
First published on 5th May 2016
Abstract
Transition metal complexes with substituted high affinity mixed ligands as potential anticancer agents can overcome the drawbacks of platinum-based drugs that are currently being marketed. Here, a new water-soluble asymmetric binuclear iminodiacetato-zinc(II) complex [Zn2(ida)(phen)3(NO3)]·NO3·5H2O (1) with a phenanthroline ligand has been synthesized and fully characterized with a wide range of analytical techniques including single crystal X-ray diffraction as well as spectroscopic techniques, such as FT-IR, UV/Vis, photoluminescence spectroscopy, and furthermore by elemental and thermogravimetric analyses. Moreover, unprecedented (H2O)10 water clusters consisting of a quasi-planar tetramer and six dangling water molecules were observed in the void space of 3D supramolecular assemblies. The conversion behavior of (1) into two monomeric species [Zn(ida)(phen)(H2O)] (2) and [Zn(phen)2(H2O)2]2+ (3) in aqueous solution was first studied by solid-state/solution NMR, ESI-MS, and solution UV/vis spectra. Next, these zinc(II) complexes (1–3), a mixture of (2) and (3) (mole ratio 1/1), ligands (phen and ida) and zinc ions (ZnCl2 and ZnSO4) were further evaluated for the in vitro cytotoxic profile in human hepatoma cell lines (HepG2 and SMMC-7721). We found that complex (1) effectively inhibited the proliferation of hepatocellular carcinoma cells, which is similar to a mixture of (2) and (3) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio, and IC50 values of (1) were almost about 20–50% of (2) or (3). Therefore, this binuclear complex (1) mainly acts as a cooperative inhibitor with complexes (2) and (3) toward tumor growth in solution. We further extended the preliminary research of complex (1) and found that (1) could induce cell cycle arrest at the G0/G1 phase. Additionally, overdosing on (1) exhibited low toxicity of mice (LD50 of (1) in ICR mice = 736 mg kg−1, with 95% confidence interval 635–842 mg kg−1). In conclusion, complex (1) with a high antitumor activity and low toxicity provides a new strategy for the treatment of liver cancer.
:1 molar ratio, and IC50 values of (1) were almost about 20–50% of (2) or (3). Therefore, this binuclear complex (1) mainly acts as a cooperative inhibitor with complexes (2) and (3) toward tumor growth in solution. We further extended the preliminary research of complex (1) and found that (1) could induce cell cycle arrest at the G0/G1 phase. Additionally, overdosing on (1) exhibited low toxicity of mice (LD50 of (1) in ICR mice = 736 mg kg−1, with 95% confidence interval 635–842 mg kg−1). In conclusion, complex (1) with a high antitumor activity and low toxicity provides a new strategy for the treatment of liver cancer.
Introduction
Cancer, as a leading cause of death worldwide, has become a major health problem of global concern.1 As is well known, transition metal based drugs play a central role in antitumor chemotherapy, although the underlying relationship between molecular structures and their biological activities has not yet been completely interpreted so far.2 Platinum-based drugs such as cisplatin, carboplatin, oxaliplatin, etc. have achieved great success in the treatment of various cancers.3,4 However, they induce strong side effects including nephrotoxicity, emetogenesis and neurotoxicity during therapy.5 Therefore, the design and synthesis of novel non-platinum metal complexes with a high antitumor activity and low toxicity are in the focus of global interest.6,7Zinc(II) is an important essential trace element for organisms, and the main building part of a series of enzymatic systems due to its physiological roles. Therefore, one present strategy has been successfully illustrated by new Zn(II) coordination complexes with low toxicity in medicinal therapeutic applications,8 for treating diabetes mellitus9 or cancer.10,11 Another strategy in such ongoing efforts is to synergise the beneficial effect of high affinity and biologically relevant ligands for enhancing anticancer activity of transition metal complexes. Recently, potential biologically active moieties, such as 1,10-phenanthroline (phen) and aminocarboxylic acids, have been extensively involved in a new-type of drug design because of their unique properties.12 Phen is a classic nitrogen-chelating bidentate ligand, which displays strong cooperativity in cation binding to form stable transition metal complexes in solution. Phen and its derivatives are capable of high specific DNA intercalation-binding affinities owing to their rigid electron-deficient heteroaromatic rings.13 Aminocarboxylic acids having good biocompatibility as secondary ligands can further enhance the affinity of mixed-ligand complexes towards DNA through the formation of hydrogen bonding between the ligands and DNA molecules.14
We are therefore interested in investigating an association of Zn(II)-containing structural motifs with some potent biologically active ligands which would result in novel efficient chemotherapeutic agents against human cancers. There have been some reported studies on the crystal structure and biochemical properties of phenanthroline zinc(II) complexes with aminocarboxylic acids.12,15 However, phenanthroline dinuclear zinc(II) complexes substituted by an iminodiacetate ligand have never been documented to date. Moreover, no studies on the conversion behavior of phenanthroline dinuclear zinc(II) complexes with aminocarboxylic acids in solution have been reported. Herein we report synthetic routes, characterization, structural conversion and anticancer properties of a novel water-soluble dimeric phenanthroline iminodiacetato zinc(II) complex [Zn2(ida)(phen)3(NO3)]·NO3·5H2O (1) that stabilized the unprecedented decameric water cluster. The conversion behavior of this dimer (1) into two monomeric species [Zn(ida)(phen)(H2O)] (2) and [Zn(phen)2(H2O)2]2+ (3) in aqueous solution was also first thoroughly investigated. Furthermore, complex (1) significantly inhibits the proliferation of human hepatoma cell lines and overdosing on complex (1) in mice exhibits low toxicity in vivo. The monomeric dissolved species (2) and (3) exert a synergistic inhibitory effect on tumour cell growth in a solution of complex (1).
Experimental
Reagents
All chemicals and reagents in this study were of reagent grade and used without further purification. Dulbecco's modified Eagle's medium (DMEM) was purchased from Thermo Scientific (HyClone, Logan, UT, USA). Fetal bovine serum (FBS) was purchased from Wisent (Quebec, Canada). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Sigma Chemical Co. (St Louis, MO, USA). Phosphate-Buffered Saline (PBS) and penicillin–streptomycin solution were from the Beyotime Institute of Biotechnology (Shanghai, China).Materials and physical measurements
Elemental analyses (C, H and N) were performed using an EA 1110 elemental analyzer. Fourier transform infrared (FT-IR) spectra were recorded on a Bruker Optics Vertex 70 spectrometer. Solution 13C {1H} NMR spectra were recorded on a Bruker Avance AV-400 MHz resonance spectrometer with a D2O solvent using DSS (sodium 2,2-dimethyl-2-silapentane-5-sulfonate) as an internal reference at room temperature. Solid 13C NMR spectra were recorded on a Bruker AV 400 NMR spectrometer using cross polarization, magic angle spinning (12 kHz) and hexamethylbenzene (HMB) as the reference. The thermogravimetric analyses (TG) were performed with a Netzsch TG209 F1 instrument at 10 °C min−1 from 30 to 800 °C in a flowing air atmosphere. UV/vis spectra were recorded on a Perkin-Elmer Lambda 650S spectrometer. Photoluminescence measurements were performed on a Perkin-Elmer LS 50B spectrometer. The X-ray powder diffraction (XRD) patterns were recorded on a Philips X'Pert PRO diffractometer, operating at 40 kV and 30 mA using a Cu-target tube. High mass accuracy ESI spectra were recorded on an ultrahigh-resolution ESI-Time-Of-Flight (Bruker Daltonik maxis (Bremen, Germany)). Spectra were obtained in positive-ion mode, with the capillary held at 4000 V. Complex (1) was dissolved in a H2O/MeCN mixture (80:20) to enhance peak intensity.
:2. Slow evaporation of the solution afforded colorless single crystals of (3) (yield 209 mg, 32%) after approx. two days. The crystal structure of complex (3) has been first reported by Liu et al. in 1991.16
X-ray crystallography
Data collections of (1) were performed on an Oxford Gemini Sultra system with graphite mono-chromate (MoKα radiation, λ = 0.71073 Å) at 173 K. Routine Lorentz and polarization corrections were applied, and an absorption correction was performed using the program CrysAlis (multi-scan).17 The structure was solved using the WinGX package18 and refined by full-matrix least-squares procedures with anisotropic thermal parameters for all the non-hydrogen atoms using SHELXL-97.19 Hydrogen atoms were included and located from difference Fourier maps but not refined anisotropically.Cell culture
Human hepatoma cell lines HepG2 and SMMC-7721 were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, P. R. China). The cells were maintained at 37 °C in a humidified 5% CO2 atmosphere with DMEM medium containing 10% FBS, and 1% penicillin–streptomycin solution.Cell viability
Cell viabilities were determined by the MTT assay as described previously.20 Cells were plated at a density of 1 × 104 per well. After cultured overnight, cells were exposed to either a vehicle control or various concentrations of (1–3), a mixture of (2) and (3) (molar ratio 1/1), ligands (phen and ida), ZnCl2 and ZnSO4 for 24 or 48 h. Cells each well were added to 10 μl of MTT solution (5 mg ml−1) and incubated for 4 h. After incubation with MTT, 100 μl DMSO was used to dissolve purple precipitates. Absorbance was detected at 570 nm by using a microplate spectrophotometer (Bio-tek, Inc.). The effects of (1–3), a mixture of (2) and (3), ligands (phen and ida), ZnCl2 and ZnSO4 on cell proliferation were assessed as the percentage of cell growth (vehicle-treated cells were taken as 100% viable).Cell cycle analysis
Cells were plated at a density of 2 × 106 per well in 150 mm culture dishes, and treated with a vehicle control or various concentrations of (1) for 48 h. Cells were harvested and fixed in 70% ice-cold methanol for 12 h at 4 °C. After being trypsinized, cells were washed with PBS and fixed in 95% ethanol at 4 °C overnight followed by 10 mg ml−1 RNase and 1 mg ml−1 propidium iodide (Beyotime, Jiangsu, P. R. China). Flow cytometric analysis was performed using a flow cytometry system (BD FACSCalibur, USA). The data were analyzed using the ModiFitLT V2.0 software (Verity Software House, Topsham, ME).Ethics statement
All animal experiments in this study were approved by the Institutional Animal Care and Use Committee of Yangzhou University and handled following the International Animal Ethics Committee Guidelines, ensuring minimum animal suffering.Animal
Thirty male ICR mice and 30 female mice, aged 5 weeks, weighing 18 ± 2 g, were purchased from the Yangzhou University Comparative Medicine Center (license no. SCXK (Su) 20120009) and acclimatized under standard conditions for laboratory animals: a temperature of 23 ± 2 °C, a relative humidity of 50 ± 5%, and a 12 hour light/dark cycle.Single-dose acute toxicity testing
All mice were fasted for 6 h prior to conducting the experiment. This study was conducted over 15 days (days 0–14). Briefly, all mice were randomly divided into six groups of ten each (five males and five females). On day 0, the mice were intragastrically (i.g.) administered (1). The doses were 1500, 1125, 844, 632, or 474 mg kg−1. In the control group, ten mice (five males and five females) were intragastrically administered with double distilled water. The mice were observed individually for signs of acute toxicity and behavioral changes for 4 h post dosing and at least once daily for 14 d. The mortality rate of the mice was recorded to calculate the LD50 values. Histopathological and hematological examination, and determination of ALT and BUN levels on mice were performed for all mice alive on day 14. The main processes in this study are as follows (Scheme 1). | ||
| Scheme 1 Illustrations of single-dose acute toxicity testing. | ||
Statistical analysis
All values are expressed as means ± standard error of the mean (S.E.M). Statistical analysis was performed using one-way analysis of variance for multiple comparisons, followed by a Student–Newman–Keuls test to evaluate the significance of differences between the two groups. A p-value less than 0.05 is considered statistically significant.Results and discussion
Synthesis and crystal structure
Complex (1) was successfully synthesized by reacting zinc nitrate together with iminodiacetic acid (H2ida) and 1,10-phenanthroline (phen) ligands at a low pH (2.0–3.0) in a molar ratio of 1:1:1 (Fig. 1a). In our preceding studies, the monomeric phenanthroline iminodiacetato zinc(II) complex [Zn(ida)(phen)(H2O)]·2H2O (2) was obtained at a higher pH of 7.0 (Fig. 1b).15 Therefore, these results exhibit the pH-dependent assembly of two phenanthroline iminodiacetato zinc(II) complexes.
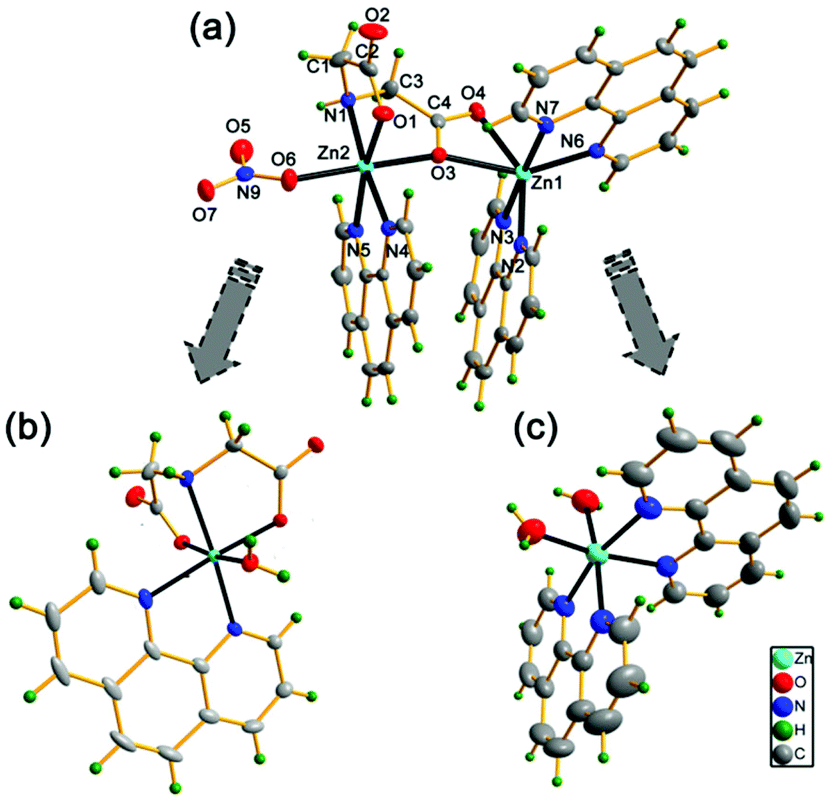 | ||
| Fig. 1 X-ray crystallographic structures of (a) zinc(II) dimeric complex [Zn2(ida)(phen)3(NO3)]+ cation (1), (b) zinc(II) monomeric complex [Zn(ida)(phen)(H2O)]·2H2O (2) and (c) phenanthroline zinc(II) complex [Zn(phen)2(H2O)2]SO4·6H2O (3) with ellipsoid set at 50% probabilities. One counter nitrate anion in (1), one counter sulfate anion in (3) and solvent water molecules in (1)-(3) have been omitted for clarity. | ||
The composition of (1) was determined from X-ray crystallography data in combination with elemental and thermogravimetric analyses (Table S1†). The phase purity of (1) obtained was also confirmed with powder X-ray diffraction (PXRD) measurements (Fig. S2†). Complex (1) was further thoroughly characterized by infrared (FT-IR) and UV/vis spectroscopy, electrospray ionization mass spectrometry (ESI-MS), 1H and 13C NMR spectroscopy, and photoluminescence spectroscopy (PL). Selected bond lengths and angles are listed in Table S2.†
The crystallographic asymmetric unit of (1) contains one [Zn2(ida)(phen)3(NO3)]+ monocation, one nitrate ion as a counterion and five solvent water molecules. The molecular structure of [Zn2(ida)(phen)3(NO3)]+ cation (1) along with the atomic numbering scheme is shown in Fig. 1a. Each Zn(II) ion has a distorted octahedral geometry. The Zn(1) atom is six-coordinated by four nitrogen atoms from two phen ligands and two oxygen atoms from the same carboxy group of an ida ligand, with N(3), N(6), N(7) and O(3) atoms in the equatorial plane and N(2) and O(4) atoms at the apical positions. The Zn(2) atom is surrounded by the third phen ligand, one unidentate nitrate and an O,N,O′-tridentate chelating ida ligand. The N(4) and N(5) atoms from the phen ligand, the imine N(1) atom and one carboxy O(1) atom of the ida ligand constitute the equatorial plane, while the O(6) atom of a nitrate and the bridging atom O(3) from the carboxy group of the ida ligand occupy the axis points. It is noticeable that the distances of Zn(1)–O(3) and Zn(1)–O(4) [2.314(1) and 2.210(1) Å] are much longer in the iminodiacetato zinc complexes reported [2.010(2)–2.209(3), Tables S2 and S3†].15,21 The two different coordinated moieties [Zn(phen)2]2+ and [Zn(phen)(NO3)]+ are thus connected via a bridging ida dianion, while the O(3) atom as another bridge directly links to two Zn(II) ions [Zn(1)⋯Zn(2) 4.394(1) Å], forming an asymmetric binuclear zinc complex. Thus, the ida ligand plays a dual role of double-bridging and a tetradentate agent in complex (1).
Interestingly, the hydrogen-bonding interactions of five lattice water molecules of crystallization lead to the formation of a new finite decameric water cluster (H2O)10 intercalated in the void spaces of (1) (Fig. 2). Unprecedented (H2O)10 cluster consists of a cyclic quasi-planar tetramer (O1w and O2w), and six dangling water molecules (O3w, O4w, O5w) (Fig. 2b). In the (H2O)10 cluster, O(1w) and O(4w) both act as a hydrogen-bond donor to nearby carboxy O atoms assembling four neighbouring [Zn2(ida)(phen)3(NO3)]+ units around each decameric water cluster (Fig. 2a). The adjacent O(w)⋯O(w) distances (2.679–2.934 Å, 2.82 Å on average, Table S4†) are located in the reported range.22,23 The overall structure is further stabilized by intermolecular π–π interactions between the aromatic rings of phenanthroline entities with an average ring centroid–centroid separation of 3.63 Å. Moreover, C–H⋯O interactions play an important role in stabilizing particular structures of biomolecules such as nucleic acids, proteins, and carbohydrates in biochemistry.24 Complex (1) also displays strong C–H⋯O intermolecular interactions between phen H and carboxy O [C(21)⋯O(4) 3.232(2) Å, ∠C–H⋯O 153.9°, Table S4†], which is further assembled into a 3D supramolecular framework (Fig. S1†).
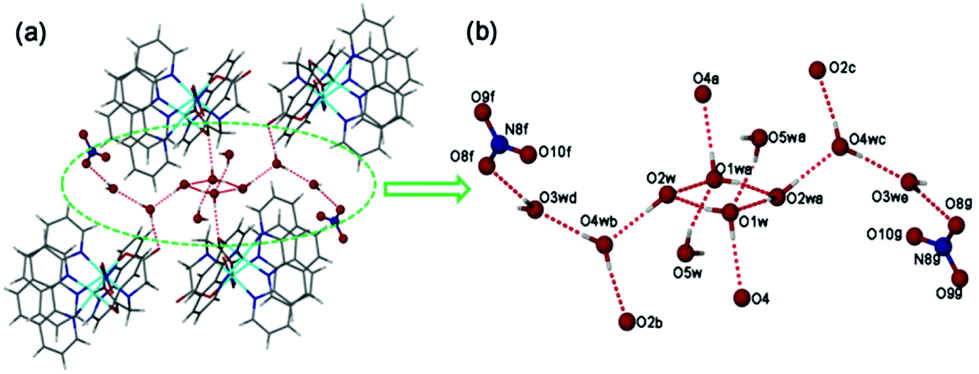 | ||
| Fig. 2 (a) Perspective view of the discrete decameric water cluster (H2O)10; (b) the (H2O)10 cluster gathering four neighbouring units of complex (1) by hydrogen-bonding interactions viewed down the x axis. Symmetry codes: a, 1 − x, 1 − y, 1 − z; b, −x, 1 − y, 1 − z; c, 1 + x, y, z; d, x, 3/2 − y, 1/2 + z; e, 1 − x, y − 1/2, 1/2 − z; f, x, 1/2 − y, 1/2 + z; g, 1 − x, 1/2 + y, 1/2 − z. Color code: red atom (oxygen); blue atom (nitrogen); gray atom (hydrogen); red dash (H-bonding interactions). | ||
General characterization
Thermal gravimetric analysis of compound (1) showed a 7.20% weight loss between 30 and 250 °C, corresponding to five lattice water molecules (calcd value: 8.85%). From 250 °C to 800 °C, the anhydrous compound decomposed with stepwise loss of the organic ida and phen ligands, leading to the decomposition of the 3D frameworks into the residual product zinc oxide (observed 15.9%, calcd 14.5%, JCPDS no. 36-1451) (Fig. S3†). This was also further verified by PXRD patterns of bulk (1) calcined at different temperatures (Fig. S2†). The FT-IR spectrum of (1) exhibited a broad absorption centered at 3425 cm−1 attributed to the O–H stretching frequency derived from the water cluster (Fig. S4†). The sharp vibrations at 854 and 740 cm−1 are due to the δ(C–H) vibrations of the phen ligand.25 Typical carboxy stretching vibrations in (1) appear at 1614, 1587 and 1512 cm−1 for νas(CO2) and at 1426, 1378, and 1320 cm−1 for νs(CO2).26 The separations between these signals (Δν) are 188, 209 and 192 cm−1, respectively, suggesting that ida acts as both bidentate and unidentate ligands between two zinc atoms, which is consistent with the crystallographic structure.Solid-state/solution UV-Vis spectroscopy and fluorescence properties
The UV–vis diffuse reflectance spectrum of complex (1) displays a set of intense absorption bands of 1,10-phen ligand in the UV region (208–326 nm), due to the absorption of ligand-centered π–π* transitions (Fig. 3a).27 However, the electronic spectrum of the complex (1) in water gives slightly different absorption peaks centered at 226 nm, 268 nm with one shoulder at 290 nm (Fig. 3b). The spectrum of the zinc complex (1) is almost overlapping with that of the free phen ligand, revealing allowed π–π* transitions. Nevertheless, the complexation of Zn2+ to the phen ligand in (1) posed a small red shift in the absorbance maximum and a large increase in molar absorption coefficient (Fig. 3b and Table S5†) compared to the free phen ligand, probably due to the steric hindrance within the coordination cage of phen rings around the zinc cations.27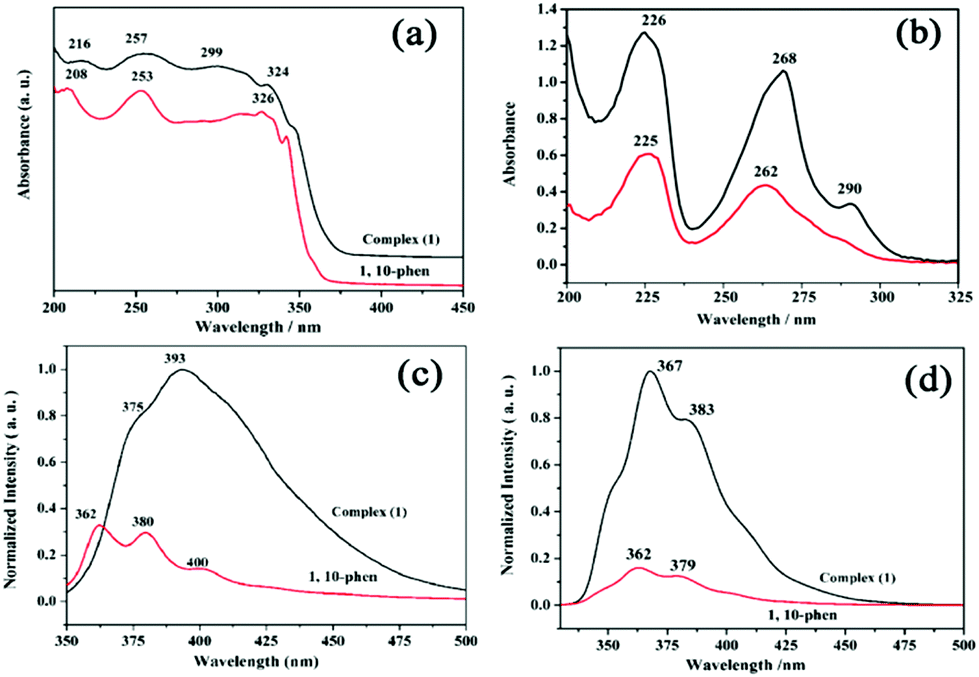 | ||
| Fig. 3 (a) The diffuse reflectance absorption of complex (1) and 1,10-phenanthroline at room temperature; (b) UV/vis spectra of complex (1) and 1,10-phen in aqueous solution at room temperature (C = 0.008 mM); (c) solid-state photoluminescence spectra of complex (1) and 1,10-phen at room temperature. The corresponding excitation wavelengths, λex are 331 and 340 nm, respectively; (d) photoluminescence spectra of complex (1) and 1,10-phen in aqueous solution at room temperature. λex are 312 and 297 nm, respectively (C = 0.1 mM). | ||
Because Zn2+ is spectroscopically silent due to its d10 electron configuration, the design of fluorescent chemosensors for the detection of Zn2+ has been studied intensively.28 To further investigate the potential fluorescence properties of complex (1), the photoluminescence spectra of (1) and the free phen ligand were recorded in the solid-state and in aqueous solution at ambient temperature. Complex (1) exhibits intense photoluminescence in comparison with the free phen ligand (Fig. 3c and d). The emission spectrum displays two characteristic emission bands of two coordinated phen ligands at 375 and 393 nm (λex = 331 nm in the solid state), at 367 and 383 nm (λex = 312 nm in water). The two emission bands in 1 are neither metal-to-ligand charge transfer (MLCT) nor ligand-to-metal charge transfer (LMCT) in nature, since the closed d10 electronic shell configuration of Zn2+ cations leads to the absence of d–d transitions in the photoemission spectra. Therefore, it can be assigned to the π–π* intraligand fluorescence of the 1,10-phenanthroline ligand.29 Compared with the emission peaks of two free ligands, a red-shifted emission for (1) could be attributed to the cooperative effect of diverse weak interactions controlled by π–π stacking and hydrogen bonds, leading to decrease the HOMO–LUMO energy gaps.30 The coordination of phen ligands to the zinc(II) ions effectively reduces the loss of energy via radiationless thermal vibrations by increasing the rigidity of the chromophore, thus enhancing photoluminescence emissions.30
Transformation in aqueous solution
The structural information and the conversions of (1) in aqueous media were investigated by solid/solution NMR spectroscopy, electrospray ionization-mass spectrometry (ESI-MS), and time-resolved UV-vis absorption spectroscopy. The solid 13C NMR spectrum of (1) is shown in Fig. 4a. The presence of two peaks at 183.4 and 177.2 ppm with a downfield shift Δδ of 6.2 ppm can be ascribed to the monodentate and μ2-bridging carboxy carbons [C(2) and C(4), see Fig. 1a] of the coordinated ida ligand, respectively. Moreover, two peaks at 55.7 and 53.8 ppm correspond to two methylene groups of ida.15 The other resonances in the range 128–151 ppm are attributed to the carbons of three coordinated phen rings. These results are in good agreement with the solid-state structure from X-ray crystallography data. However, the solution 13C NMR spectrum of (1) is quite different from that in solid complex (1). Only one set of 13C resonances observed at 182.3 and 56.4 ppm is corresponding to the carboxy and methylene carbons of ida, respectively (Fig. 4b). In comparison with the free ida ligand in solution at pH 2.0, both the 13C NMR signals of carboxy and methylene carbons in (1) have large downfield shifts at (Δδ) 8.9 and 4.9 ppm, respectively (Fig. S6†). The solution 1H NMR spectrum of (1) contained two CH2 signals from the ida ligand observed at 3.98 and 3.40 ppm.15 The signals in the range of 7.98–9.21 ppm are assigned to phen hydrogen atoms (Fig. S5†).31 Based on the molecular structure of (1) in the solid state combined with solid 13C NMR signals, there exist two types of coordination modes of carboxy groups, but only one peak was found for the carboxy groups in solution. This may result from the transformation of the dimeric complex (1) to the monomeric units in solution. To gain further insight into the conversion pathway of (1) in solution, we also investigated the ESI-MS spectrum of (1) in water (Fig. 4c). The spectrum showed a base peak at m/z 537.05 that is equivalent to the monomeric phenanthroline iminodiacetato zinc(II) complex [Zn(ida)(phen)(H2O)(2) + 8H2O]+, which might involve the removal of one electron from the phenanthroline ligand to form a positive molecular ion M+. The other monovalent cationic peak centered at m/z 486.05 corresponds to monomeric phenanthroline zinc(II) species [Zn(phen)2 (3) + (NO3−)]+. Therefore, solution 13C NMR and ESI-MS results are consistent with a conversion pathway reaction of the dimeric complex (1) in aqueous solution. All these results indicate that (1) undergoes transformation in solution at the relatively weak bonded μ2-bridging carboxyl group, which can be easily attacked and substituted by water molecules arising from the bond cleavage of Zn(1)–O(3) and Zn(1)-O(4) associated with the leaving process of the unidentate nitrate ion, leading to form two monomeric species [Zn(ida)(phen)(H2O)] (2) and [Zn(phen)2(H2O)2]2+ (3) (Fig. 4d).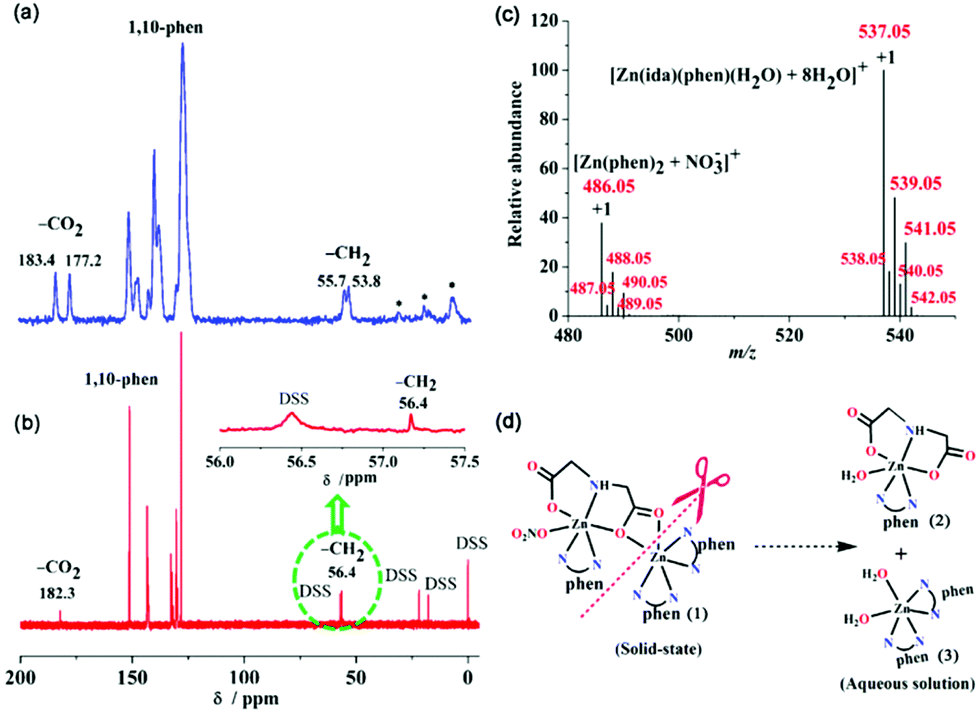 | ||
| Fig. 4 (a) Solid 13C NMR spectrum of (1), (* for spinning sidebands); (b) solution 13C NMR spectrum of (1), (inset: magnification of the methylene carbons (–CH2) region); (c) ESI-MS spectrum of (1) in positive ion mode; (d) conversion of (1) into two different monomers in water. | ||
Moreover, the time-resolved UV-vis absorption spectra of complex (1) in phosphate-buffered saline (PBS) buffer solution (pH = 7.2) were recorded at 2 min intervals (Fig. S7a†). The absorption spectra of two monomeric complexes [Zn(ida)(phen)(H2O)] (2), [Zn(phen)2(H2O)2]SO4 (3), and their mixed species at a 1:1 molar ratio in PBS buffer were also studied as shown in Fig. S7b.† The absorbance maximum of (1) is at the same wavelength as that of monomeric complex (2) or (3) but exhibits a significantly higher intensity (Fig. S7b†). However, the mixed species of (2) and (3) at a 1:1 molar ratio can show almost equivalent absorbance peak intensities, compared with complex (1) (Fig. S7b†). In addition, almost no decrease in absorbance of complex (1) could be observed during 2 hours in PBS buffer (Fig. S7a†), suggesting that the conversion of complex (1) into monomers (2) and (3) probably proceed very rapidly.
In vitro cytotoxicity
To evaluate the cytotoxic activity of complexes ligands (phen and ida) and zinc ions (ZnCl2 and ZnSO4), human hepatoma cell lines (HepG2 and SMMC-7721) were incubated with various concentrations of the complexes (1–3), a mixture of (2) and (3) (1:1), ligands (phen and ida), ZnCl2 and ZnSO4 for 24 or 48 h, respectively. We observed that all the complexes and ligands inhibited the proliferation of hepatoma cell lines in a dose-dependent manner (Fig. 5a–d). A significant decrease of HepG2 cell viability has been detected at low concentrations, especially in the presence of complex (1) and the mixture ((2) and (3), 1:1 molar ratio). Interestingly, (1) and the mixture showed a relevant cytotoxicity with IC50 of 10.01 ± 1.08 and 13.75 ± 3.39 μM after 48 h, respectively, whereas (2) and (3) exhibited 50% of toxicity at higher concentrations (IC50 of 41.63 ± 1.75 and 22.34 ± 0.80 μM after 48 h, respectively) (Fig. 5e). Similarly, the SMMC-7721 cell viability was significantly decreased in incubation with (1) and the mixture at low concentrations (IC50 of 11.75 ± 1.75 and 12.58 ± 2.20 μM after 48 h, respectively). However, (2) and (3) were much less active against SMMC-7721 (IC50 = 44.36 ± 5.27 μM and 27.02 ± 2.78 μM after 48 h, respectively). IC50 values of (2) or (3) were almost about 2–3 fold stronger than that of (1) or a mixture of (2) and (3) at a 1:1 molar ratio. Complex (1) and the mixture exerted a similar 50% inhibition rate from hepatoma cell lines further confirming that complex (1) undergoes dissociation from dimer to two monomers (2) and (3) in solution. Hence, we inferred that binuclear (1) mainly acts as an essential cooperative inhibitor with (2) and (3) toward tumor growth in solution.
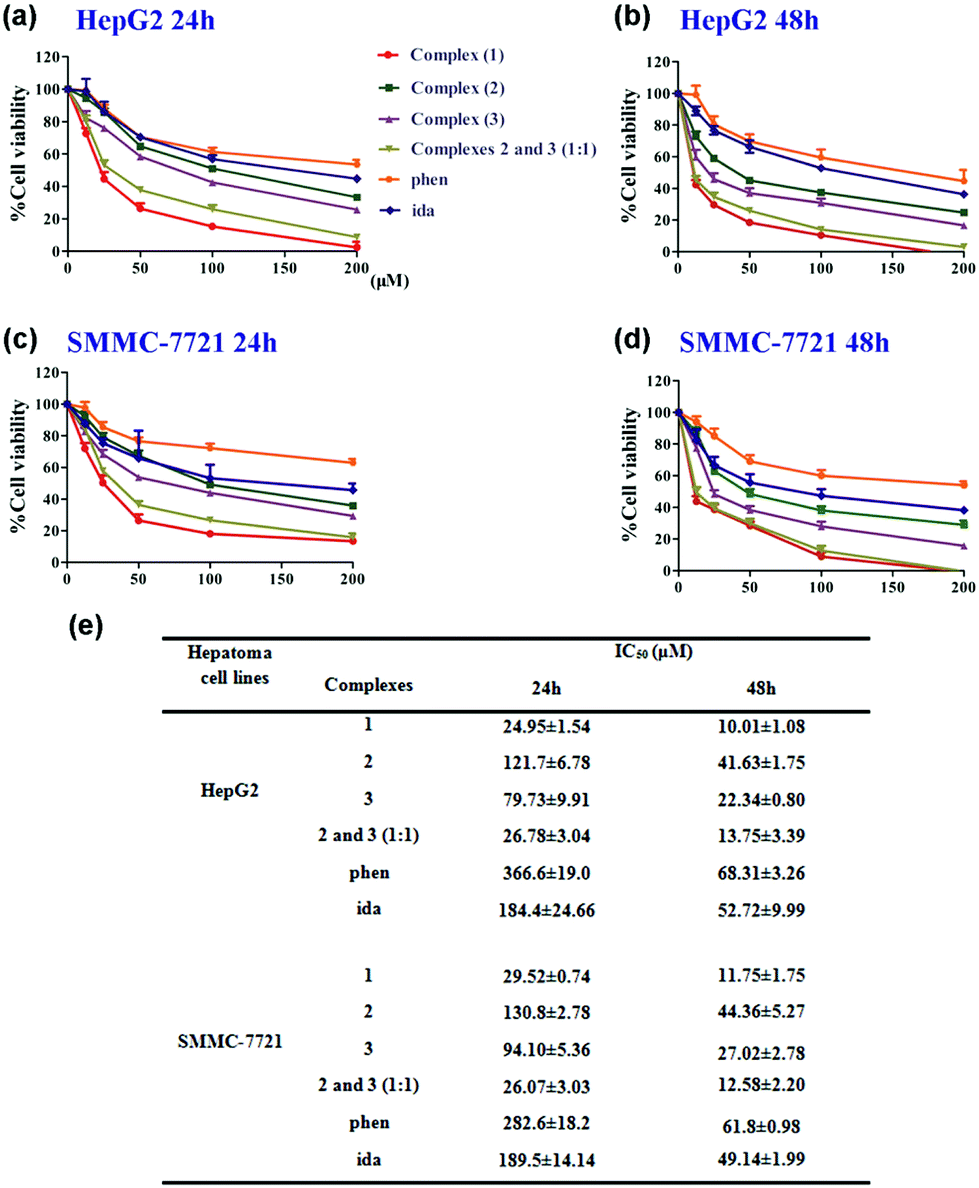 | ||
| Fig. 5 Complexes and ligands inhibited the proliferation of hepatoma cell lines. (a–d) HepG2 and SMMC-7721 cells were incubated with various concentrations of complexes (1–3), mixture of 2 and 3 (1:1), and ligands (phen and ida) for 24 or 48 h, respectively. (e) IC50 values of complexes and ligands calculated on average values of % inhibition at various molecule concentrations. The results are presented as the means ± S.E.M. of three independent experiments. | ||
Furthermore, ligands (phen and ida) had lower inhibition rates against hepatoma cell lines (Fig. 5e), compared to complexes (1–3). Notably, zinc ions (ZnCl2 and ZnSO4) showed lower cytotoxic activity (IC50 values of ZnCl2 and ZnSO4 incubated with HepG2 and SMMC-7721 for 48 h were all >200 μM) (Fig. S8†). Combining the above results, relative cytotoxic activities of complexes and ligands against human hepatoma cell lines are in the following order: (1) ≈ mixture ((2) and (3), 1:1) > (3), (2) > ida, phen > ZnCl2, ZnSO4. All in all, effects of transition metal ion coordination, and the numbers and types of ligand can exert key influence on the evaluation of cytotoxicity. In short, complex (1) exerts similar antitumor activity (Table S7†) compared with the marked platinum-based drugs.32
Cell cycle analysis
To further elucidate the possible mechanism that complex (1) inhibits cell growth, we tested the cell cycle arrest of hepatoma cell lines via flow cytometry after (1) treatment (Fig. 6a). Our results showed that untreated HepG2 and SMMC-7721 cells exhibited a cell cycle profile after 48 h in culture.33 As shown in Fig. 6b, complex (1) could strongly induce HepG2 cells to undergo G0/G1 arrest after 48 h, with a dose-dependent increase in the mean percentage (78.02 ± 3.25% at 12.5 μM, 79.58 ± 2.26% at 25 μM, 80.35 ± 4.65% at 50 μM, 86.59 ± 1.68% at 100 μM) of cells compared with 57.70 ± 3.36% of cells in the control group in G0/G1. In addition, HepG2 cell population at the S phase after (1) treatment significantly decreased (16.13 ± 1.09% at 12.5 μM, 15.12 ± 1.91% at 25 μM, 14.27 ± 1.19% at 50 μM, 11.15 ± 1.26% at 100 μM) compared with 22.68 ± 1.91% of cells in the control group at the S phase. Complex (1) also blocked cells into the G2/M phase (5.86 ± 3.22% at 12.5 μM, 5.31 ± 3.07% at 25 μM, 5.38 ± 4.36% at 50 μM, 2.26 ± 1.55% at 100 μM) compared with 19.63 ± 2.97% of cells in the control group at the G2/M phase. Similarly, the SMMC-7721 cell population at G0/G1 after (1) treatment increased from 72.56 ± 5.80% (at 12.5 μM) to 84.50 ± 4.18% (at 100 μM) in a dose-dependent manner compared to that of the control group which had a corresponding population of 58.78 ± 4.50% at G0/G1 (Fig. 6c). Moreover, S and G2/M phases of SMMC-7721 cells dropped significantly after treatment of complex (1), respectively. The data indicated that complex (1) blocks cell progression into S and G2/M phases, and thus a certain proportion of cells is arrested at G0/G1.34–36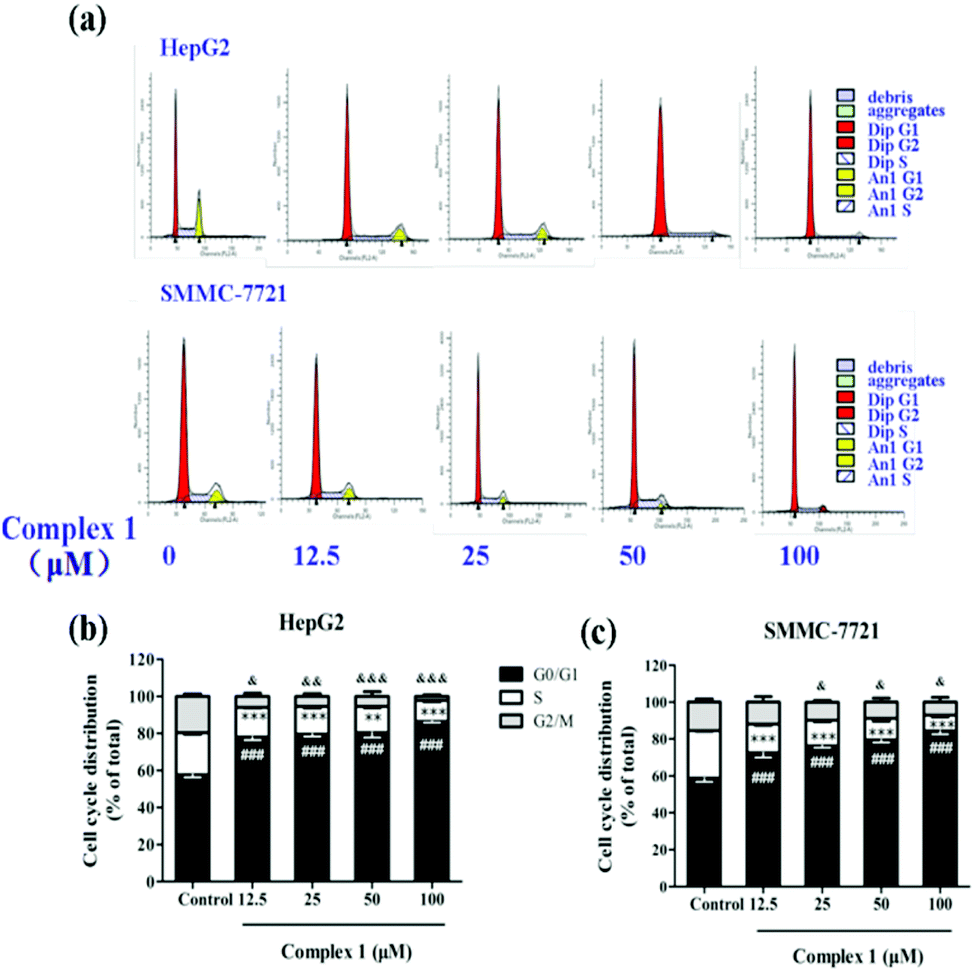 | ||
| Fig. 6 Complex (1) affected cell cycle distribution of hepatoma cell lines. (a) HepG2 and SMMC-7721 cells were treated with 0, 12.5, 25, 50 and 100 μM of complex (1) for 48 h followed by staining with propidium iodide for flow cytometric analysis. (b and c) Graphs depict the cell distribution in the different phases of the cell cycle determined by flow cytometry, and bar charts present the percentage of cells in the indicated phase of cell cycle. The results are presented as the means ± S.E.M. of three independent experiments. ###p < 0.001 compared with the control group at the G0/G1 phase; ***p < 0.001 compared with the control group at the S phase; &p < 0.05, &&p < 0.01, &&&p < 0.001 compared with the control group at the G2/M phase. | ||
Acute toxicity
Safety is another primary concern when considering new drug candidates intended for use in humans.37 Acute toxicity studies may also aid in the selection of starting doses for phase I clinical studies and provide information relevant to acute overdosing in humans.6 We thus examined the in vivo toxicity profile of complex (1). As shown in Fig. 7a, the median lethal dose (LD50) value of (1) in ICR mice is 736 mg kg−1 by intragastric administration, with the 95% confidence limits of 635–842 mg kg−1. Notably, complex (1) is administered by i.g. and has lower toxicity than the marked platinum-based drugs that exert antitumor activity only by i.p. (Table S8†).38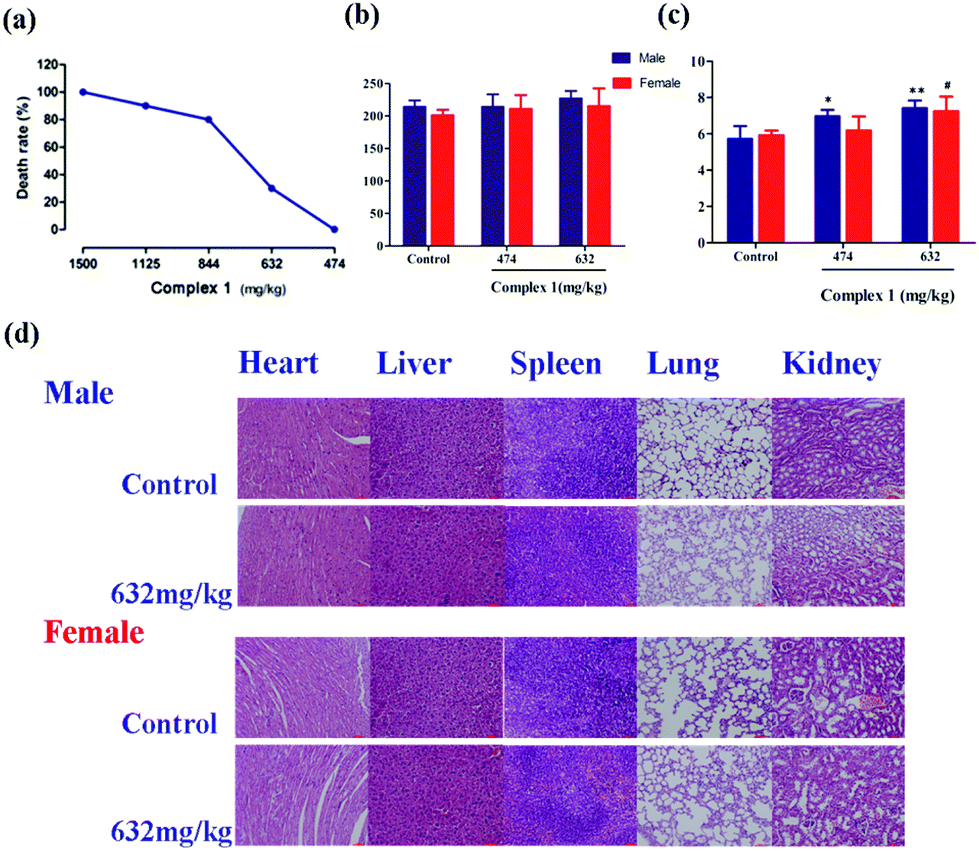 | ||
| Fig. 7 (a) Dose-dependent manner of the complex (1) effects on the mice death. LD50 of complex (1) = 736 mg kg−1 (95% confidence interval 635–842 mg kg−1). Serum ALT (b) and BUN (c) levels in mice treated with complex (1) at various concentrations. (d) Histological examinations included heart, liver, spleen, lung and kidney sections in mice alive after administration with a single dose of (1) 632 mg kg−1 (200×). The results are presented as the means ± S.E.M. *p < 0.05, **p < 0.01 compared with the male control group; #p < 0.05 compared with the female control group. | ||
Alanine transaminase (ALT) and blood urea nitrogen (BUN) are two important factors used to evaluate liver and kidney functions, respectively.39 The serum ALT level showed a slight, dose-dependent increase in the groups treated with (1) (Fig. 7b). However, treatment of (1) even at a dose of 474 mg kg−1 or 632 mg kg−1 manifested no significant differences when compared with the control group. As shown in Fig. 7c, treatment of (1) significantly increased the serum BUN level in male mice compared with the control male mice. Similarly, compared with control female mice, treatment of (1) at 632 mg kg−1 significantly increased the serum BUN level in female mice, which implied that (1) at a dose of 632 mg kg−1 has possible kidney toxicity in mice.
Histopathology is an important factor for determining the treatment performance and effects, especially the negative effects.40 To further explore potential toxicity of (1), we conducted the histopathological evaluation with H&E staining. These results showed no significant evidence of tissue toxicity in alive mice treated with (1) at a dose of 632 mg kg−1 (Fig. 7d). Notably, there was no significant kidney change in mice treated with (1) at a dose of 632 mg kg−1. These data documented that treatment of (1) has no significant toxicity on the critical organs of mice.
In the hematology parameters, the oral administration of (1) did not remarkably affect the blood hematologic parameters of mice, because most parameters are within the physiological range (Table S6†). However, a statistical decrease of hemoglobin was observed in the group treated with (1) at a dose of 474 mg kg−1 and 632 mg kg−1 both in male and female mice (p < 0.01, p < 0.01, p < 0.05, and p < 0.01, respectively). Additionally, the white blood cells in mice treated with (1) were significantly increased in comparison with that of the control group (p < 0.01, p < 0.01, p < 0.01, and p < 0.001, respectively). Therefore, we inferred that overdosing on complex (1) might possess lower bone marrow toxicity in mice.
Conclusions
In summary, we have synthesized and fully characterized a novel water-soluble asymmetric binuclear zinc(II) complex [Zn2(ida)(phen)3(NO3)]·NO3·5H2O (1) with mixed ligands of iminodiacetate and 1,10-phenanthroline. The interesting result is the discovery of a novel decameric water cluster (H2O)10, which was further connected to a neighboring [Zn2(ida)(phen)3(NO3)]+ cation into a 3D supramolecular network via extensive hydrogen bonding. The structural conversion of (1) into two monomeric species [Zn(ida)(phen)(H2O)] (2) and [Zn(phen)2(H2O)2]+ (3) in water is illustrated. Next, we demonstrated that (1) could significantly inhibit proliferation of hepatoma cell lines, which is related to mainly arrest the cell cycle at G0/G1 phase. Complex (1) and the mixture ((2) and (3), 1:1 molar ratio) exerted a similar 50% inhibition rate from hepatoma cell lines showing that (1) might be a cooperative inhibitor with (2) and (3) toward tumor growth in solution through its hydrolyzation. Interestingly, effects of transition metal ion coordination, and numbers and types of ligand can exert key influence on the evaluation of cytotoxicity. Furthermore, we conducted the study of acute toxicity on the oral administration of (1) in ICR mice and calculated that the LD50 value of (1) is 736 mg kg−1, with the 95% confidence limit of 635–842 mg kg−1. The results showed that overdosing on complex (1) might exhibit a lower toxicity in mice. Therefore, complex (1) provided direct evidence to exploit the design of novel chemotherapeutic drugs with high antitumor activity and low toxicity. In addition, administration of complex (1) can be through the oral/gavage route, which is not only convenient but also safe. The underlying molecular mechanisms for liver cancer treatment are being explored in our following research. We will further develop this comprehensive approach towards informed transition metal based drug design in targeted cancer therapies.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21401162), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (Grant No. 14KJB430024), and Jiangsu Provincial Postdoctoral Sustentation Fund (Grant No. 1402015B). Financial support from the Priority Academic Program Development of Jiangsu Higher Education Institutions and the Natural Science Foundation of Education Committee of Jiangsu Province (No. 12KJB150023) is gratefully acknowledged. The authors also acknowledge the Testing Center of Yangzhou University for ESI-MS measurements.Notes and references
- (a) M. Garcia, A. Jemal, E. M. Ward, M. M. Center, Y. Hao and R. I. Siegel, Global Cancer Facts & Figures 2007, American Cancer Society, Atlanta GA, 2007, pp. 1–46 Search PubMed; (b) S. E. Atawodi, Infect. Agents Cancer, 2011, 6, 2–9 CrossRef PubMed.
- (a) V. Milacic, D. Chen, L. Ronconi, K. R. Landis-Piwowar, D. Fregona and Q. P. Dou, Cancer Res., 2006, 66, 10478–10486 CrossRef CAS PubMed; (b) K. Kehe and L. Szinicz, Toxicology, 2005, 214, 198–209 CrossRef CAS PubMed; (c) J. J. Soldevila-Barreda, I. Romero-Canelón, A. Habtemariam and P. J. Sadler, Nat. Commun., 2015, 6, 6582–6591 CrossRef CAS PubMed.
- (a) D. J. Higby, H. J. Wallac Jr., D. J. Albert and J. F. Holland, Cancer, 1974, 33, 1219–1225 CrossRef CAS PubMed; (b) B. Rosenberg, L. Van Kamp, J. F. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS PubMed; (c) D. B. Brown, A. R. Khokhar, M. P. Hacker, L. Lokys, J. H. Burchenal, R. A. Newman, J. J. McCormack and D. Frost, J. Med. Chem., 1982, 25, 952–956 CrossRef CAS PubMed; (d) U. Frey, J. D. Ranford and P. J. Sadler, Inorg. Chem., 1993, 32, 1333–1340 CrossRef CAS; (e) T. Boulikas and M. Vougiouka, Oncol. Rep., 2003, 10, 1663–1682 CAS.
- (a) M. D. Hall, H. R. Mellor, R. Callaghan and T. W. Hambley, J. Med. Chem., 2007, 50, 3403–3411 CrossRef CAS PubMed; (b) E. G. Chapman and V. J. DeRose, J. Am. Chem. Soc., 2010, 132, 1946–1952 CrossRef CAS PubMed; (c) S. Chen, D. Xu, H. Jiang, Z. Xi, P. Zhu and Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 1225–12262 CrossRef PubMed; (d) Đ. U. Miodragovic, J. A. Quentzel, J. W. Kurutz, C. L. Stern, R. W. Ahn, I. Kandela, A. Mazar and T. V. O'Halloran, Angew. Chem., Int. Ed., 2013, 52, 10749–10752 CrossRef PubMed.
- (a) R. S. Go and A. A. Adjei, J. Clin. Oncol., 1999, 17, 409–422 CAS; (b) D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed; (c) M. Dobbelstein and U. Moll, Nat. Rev. Drug Discovery, 2014, 13, 1791–1796 CrossRef PubMed.
- (a) M. Galanski, V. B. Arion, M. A. Jakupec and B. K. Keppler, Curr. Pharm. Des., 2003, 9, 2078–2089 CrossRef CAS PubMed; (b) A. M. Kim, S. Vogt, T. V. O'Halloran and T. K. Woodruff, Nat. Chem. Biol., 2010, 6, 674–681 CrossRef CAS PubMed; (c) N. Busto and B. Garcıa, Coord. Chem. Rev., 2013, 257, 2848–2862 CrossRef; (d) M. X. Li, M. Yang, J. Y. Niu, L. Z. Zhang and S. Q. Xie, Inorg. Chem., 2012, 51, 12521–12526 CrossRef CAS PubMed; (e) M. Hajrezaie, K. Shams, S. Z. Moghadamtousi, H. Karimian, P. Hassandarvish, M. Emtyazjoo, M. Zahedifard, N. A. Majid, H. M. Ali and M. A. Abdulla, Sci. Rep., 2015, 5, 12379 CrossRef CAS PubMed.
- (a) D. L. Ma, H.-Z. He, K.-H. Leung, D. S.-H. Chan and C.-H. Leung, Angew. Chem., Int. Ed., 2013, 52, 7666–7682 CrossRef CAS PubMed; (b) S. Phongtongpasuk, S. Paulus, J. Schnabl, R. K. Sigel, B. Spingler, M. J. Hannon and E. Freisinger, Angew. Chem., Int. Ed., 2013, 52, 11513–11516 CrossRef CAS PubMed; (c) A. N. Kate, A. A. Kumbhar, A. A. Khan, P. V. Joshi and V. G. Puranik, Bioconjugate Chem., 2014, 25, 102–114 CrossRef CAS PubMed; (d) A. de Almeida, B. L. Oliveira, J. D. G. Correia, G. Soveral and A. Casini, Coord. Chem. Rev., 2013, 257, 2689–2704 CrossRef CAS; (e) C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed; (f) E.-J. Kim, S. Bhuniya, H. Lee, H. M. Kim, C. Cheong, S. Maiti, K. S. Hong and J. S. Kim, J. Am. Chem. Soc., 2014, 136, 13888–13894 CrossRef CAS PubMed.
- J. A. Drewry and P. T. Gunning, Coord. Chem. Rev., 2011, 255, 459–472 CrossRef CAS.
- Y. Yoshikawa and H. Yasui, Curr. Top. Med. Chem., 2012, 12, 210–218 CrossRef CAS PubMed.
- (a) Q. Jiang, J. Zhu, Y. Zhang, N. Xiao and Z. Guo, BioMetals, 2008, 22, 297–305 CrossRef PubMed; (b) S. Anbu, S. Kamalraj, B. Varghese, J. Muthumary and M. Kandaswamy, Inorg. Chem., 2012, 51, 5580–5592 CrossRef CAS PubMed.
- (a) M. Terenzi, G. Fanelli, G. Ambrosi, S. Amatori, V. Fusi, L. Giorgi, V. Turco Liveri and G. Barone, Dalton Trans., 2012, 41, 4389–4395 RSC; (b) Y. Nakamura, Y. Taruno, M. Sugimoto, Y. Kitamura, H. L. Seng, S. M. Kong, C. H. Ng and M. Chikira, Dalton Trans., 2013, 42, 3337–3345 RSC.
- (a) C.-H. Ng, K. C. Kong, S. T. Von, P. Balraj, P. Jensen, E. Thirthagiri, H. Hamada and M. Chikira, Dalton Trans., 2008, 447–454 RSC; (b) L.-F. Chin, S.-M. Kong, H.-L. Seng, Y.-L. Tiong, K.-E. Neo, M. J. Maah, A. S.-B. Khoo, M. Ahmad, T.-S. A. Hor, H.-B. Lee, S.-L. San, S.-M. Chye and C.-H. Ng, J. Biol. Inorg. Chem., 2012, 17, 1093–1105 CrossRef CAS PubMed; (c) H.-L. Ng, C.-H. Ng and S.-W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m89 CAS; (d) H. L. Seng, H. K. A. Ong, R. N. Z. R. A. Rahman, B. M. Yamin, E. R. T. Tiekink, K. W. Tan, M. J. Maah, I. Caracelli and C.-H. Ng, J. Inorg. Biochem., 2008, 102, 1997–2011 CrossRef CAS PubMed; (e) L.-F. Chin, C.-H. Ng and S.-W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m40 CAS; (f) W.-Y. Huang, Z.-L. Chen, H.-H. Zou, D.-C. Liu and F.-P. Liang, Polyhedron, 2013, 50, 1–9 CrossRef CAS.
- (a) S. Jagadeesan, V. Balasubramanian, P. Baumann, M. Neuburger, D. Häussinger and C. G. Palivan, Inorg. Chem., 2013, 52, 12535–12544 CrossRef CAS PubMed; (b) D. Wesselinova, M. Neykov, N. Kaloyanov, R. Toshkova and G. Dimitrov, Eur. J. Med. Chem., 2009, 44, 2720–2723 CrossRef CAS PubMed; (c) S. Ambika, S. Arunachalam, R. Arun and K. Premkumar, RSC Adv., 2013, 3, 16456–16468 RSC; (d) S. Anbu, M. Kandaswamy, S. Kamalraj, J. Muthumarry and B. Varghese, Dalton Trans., 2011, 40, 7310–7318 RSC; (e) S. Roy, K. D. Hagen, P. U. Maheswari, M. Lutz, A. L. Spek, J. Reedijk and G. P. van Wezel, ChemMedChem, 2008, 3, 1427–1434 CrossRef CAS PubMed.
- (a) B. Selvakumar, V. Rajendiran, P. U. Maheswari, H. Stoeckli-Evans and M. Palaniandavar, J. Inorg. Biochem., 2006, 100, 316–330 CrossRef CAS PubMed; (b) C. Yuan, M. Zhu, Q. Wang, L. Lu, S. Xing, X. Fu, Z. Jiang, S. Zhang, Z. Li, Z. Li, R. Zhu and L. Ma, Chem. Commun., 2012, 48, 1153–1155 RSC.
- L. B. Ni, R. H. Zhang, Q. X. Liu, W. S. Xia, H. X. Wang and Z. H. Zhou, J. Solid State Chem., 2009, 182, 2698–2706 CrossRef CAS PubMed.
- N.-H. Hu and Y.-S. Liu, Acta Crystallogr C., 1991, 47, 2324–2326 CrossRef.
- Oxford Diffraction, CrysAlis CCD and CrysAlis RED, Oxford Diffraction Ltd, Abingdon, UK, 2005 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- G. M. Sheldrick, SHELX97, Programs for Crystal Structure Analysis, Release 97-2, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- H. Yang, C. Liu, Y. Q. Zhang, L. T. Ge, J. Chen, X. Q. Jia, R. X. Gu, Y. Sun and W. D. Sun, Int. Immunopharmacol., 2015, 24, 423–431 CrossRef CAS PubMed.
- (a) H. B. Xu, Y. H. Zhao, Z. M. Su, G. H. Li, Y. Ma, K. Z. Shao, D. X. Zhu, H. J. Zhang and S. M. Yue, Chem. Lett., 2004, 33, 446–447 CrossRef CAS; (b) A. C. Morel, D. Choquesillo-Lazarte, C. Alarcón-Payer, J. M. González-Pérez, A. Castiñeiras and J. Niclós-Gutiérrez, Inorg. Chem. Commun., 2003, 6, 1354–1357 CrossRef CAS; (c) S. W. Jin, D. Q. Wang and W. Z. Chen, Inorg. Chem. Commun., 2007, 10, 685–689 CrossRef CAS.
- (a) L. J. Barbour, G. W. Orr and J. L. Atwood, Nature, 1998, 393, 671–673 CrossRef CAS; (b) M. Yoshizawa, T. Kusukawa, M. Kawano, T. Ohhara, I. Tanaka, K. Kurihara, N. Niimura and M. Fujita, J. Am. Chem. Soc., 2005, 127, 2798–2799 CrossRef CAS PubMed; (c) L.-Y. Wang, Y. Yang, K. Liu, B.-L. Li and Y. Zhang, Cryst. Growth Des., 2008, 8, 3902–3904 CrossRef CAS; (d) L. J. Barbour, G. W. Orr and J. L. Atwood, Chem. Commun., 2000, 859–860 RSC.
- (a) M.-L. Wei, C. He, W.-J. Hua, C.-Y. Duan, S.-H. Li and Q.-J. Meng, J. Am. Chem. Soc., 2006, 128, 13318–13319 CrossRef CAS PubMed; (b) P. S. Lakshminarayanan, E. Suresh and P. Ghosh, J. Am. Chem. Soc., 2005, 127, 13132–13133 CrossRef CAS PubMed; (c) D. Sun, D.-F. Wang, N. Zhang, R.-B. Huang and L.-S. Zheng, Cryst. Growth Des., 2010, 10, 5031–5033 CrossRef CAS.
- (a) S. Metzger and B. Lippert, J. Am. Chem. Soc., 1996, 118, 12467–12468 CrossRef CAS; (b) R. A. Musah, G. M. Jensen, R. J. Rosenfeld, D. E. McRee, D. B. Goodin and S. W. Bunte, J. Am. Chem. Soc., 1997, 119, 9083–9084 CrossRef CAS; (c) T. Steiner and W. Saenger, J. Am. Chem. Soc., 1993, 115, 4540–4547 CrossRef CAS.
- (a) J. Morizzi, M. Hobday and C. Rix, J. Mater. Chem., 2001, 11, 794–798 RSC; (b) R. Clarke, K. Latham, C. Rix, M. Hobday and J. White, CrystEngComm, 2004, 6, 42–50 RSC; (c) O. O. Litsis, V. A. Ovchynnikov, V. P. Scherbatskii, S. G. Nedilko, T. Y. Sliva, V. V. Dyakonenko, O. V. Shishkin, V. I. Davydov, P. Gawryszewska and V. M. Amirkhanov, Dalton Trans., 2015, 44, 15508–15522 RSC.
- (a) A. Patra, T. K. Sen, R. Bhattacharyya, S. K. Mandal and M. Bera, RSC Adv., 2012, 2, 1774–1777 RSC; (b) S. K. Papageorgiou, E. P. Kouvelos, E. P. Favvas, A. A. Sapalidis, G. E. Romanos and F. K. Katsaros, Carbohydr. Res., 2010, 345, 469–473 CrossRef CAS PubMed.
- (a) A. D'Aléo, E. Cecchetto, L. D. Cola and R. M. Williams, Sensors, 2009, 9, 3604–3626 CrossRef PubMed; (b) S.-G. Roh, Y.-H. Kim, K. D. Seo, D. H. Lee, H. K. Kim, Y. Park, J.-W. Park and J.-H. Lee, Adv. Funct. Mater., 2009, 19, 1663–1671 CrossRef CAS; (c) Z. Mao, W. Senevirathna, J.-Yu. Liao, J. Gu, S. V. Kesava, C. Guo, E. D. Gomez and G. Sauvé, Adv. Mater., 2014, 26, 6290–6294 CrossRef CAS PubMed; (d) H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259–3302 RSC; (e) W. Senevirathna, J.-Yu. Liao, Z. Mao, J. Gu, M. Porter, C. Wang, R. Fernando and G. Sauvé, J. Mater. Chem. A, 2015, 3, 4203–4214 RSC.
- Z. Xu, J. Yoon and D. Spring, Chem. Soc. Rev., 2010, 39, 1996–2006 RSC.
- (a) X. Shi, G. Zhu, X. Wang, G. Li, Q. Gand, X. Zhao, G. Wu, G. Tian, M. Xue, R. Wang and S. Qiu, Cryst. Growth Des., 2004, 5, 341–346 CrossRef; (b) T. L. Hu, R. Q. Zou, J. R. Li and X. H. Bu, Dalton Trans., 2008, 1302–1311 RSC; (c) Z. Liu, W. He and Z. Guo, Chem. Soc. Rev., 2013, 42, 1568–1600 RSC.
- (a) K. Binnemans, Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS PubMed; (b) R. G. Xiong, J. L. Zuo, X. Z. You, B. F. Abrahams, Z. P. Bai, C. M. Che and H. K. Fun, Chem. Commun., 2000, 2061–2062 RSC; (c) J. Z. Wang, Anorg. Allg. Chem., 2011, 637, 1612–1615 CrossRef.
- (a) G. A. Naganagowda, K. V. Ramanathan, V. Gayathri and N. M. Nanjegowda, Magn. Reson. Chem., 2000, 38, 223–228 CrossRef CAS; (b) D. Sorsche, C. Pehlken, C. Baur, S. Rommel, K. Kastner, C. Streb and S. Rau, Dalton Trans., 2015, 44, 15404–15407 RSC.
- (a) L. F. Qin and I. O. Ng, Cancer Lett., 2002, 175, 27–38 CrossRef CAS PubMed; (b) Z. Feng, Y. Lai, H. Ye, J. Huang, X. G. Xi and Z. Wu, Cancer Sci., 2010, 101, 2476–2482 CrossRef CAS PubMed; (c) G.-Q. Li, X.-G. Chen, X.-P. Wu, J.-D. Xie, Y.-J. Liang, X.-Q. Zhao, W.-Q. Chen and L.-W. Fu, PLoS One, 2012, 7, e48994 CAS; (d) H. Du, W. Yang, L. Chen, M. Shi, V. Seewoo, J. Wang, A. Lin, Z. Liu and W. Qiu, Oncol. Rep., 2012, 27, 143–150 CAS.
- (a) J. F. Kou, C. Qian, J. Q. Wang, X. Chen, L. L. Wang, H. Chao and L. N. Ji, J. Biol. Inorg. Chem., 2012, 17, 81–96 CrossRef CAS PubMed; (b) F. Wang, L. He, W. Q. Dai, Y. P. Xu, D. Wu, C. L. Lin, S. M. Wu, P. Cheng, Y. Zhang, M. Shen, C. F. Wang, J. Lu, Y. Q. Zhou, X. F. Xu, L. Xu and C. Y. Guo, PLoS One, 2012, 7, e50638 CAS.
- P. Heffeter, M. A. Jakupec, W. Körner, S. Wild, N. G. V. Keyserlingk, L. Elbling, H. Zorbas, A. Korynevska, S. Knasmüller and H. Sutterlüty, Biochem. Pharmacol., 2006, 71, 426–440 CrossRef CAS PubMed.
- Q. Xin, Z. Y. Ma, C. Z. Xie, X. Fei, Y. W. Zhang, J. Y. Xu, Z. Y. Qiang, J. S. Lou, G. J. Chen and S. P. Yan, J. Inorg. Biochem., 2011, 105, 728–737 CrossRef PubMed.
- P. Venkatesan, N. Puvvada, R. Dash, K. Prashanth, D. Sarkar and B. Azab, Biomaterials, 2011, 32, 3794–3806 CrossRef CAS PubMed.
- C. Liu, W. Zhang, H. Yang, W. D. Sun, X. D. Gong, J. X. Zhao, Y. Sun and G. W. Diao, PLoS One, 2014, 9, e10176 Search PubMed.
- (a) M. P. Hacker, A. R. Khokhar, I. H. Krakoff, D. B. Brown and J. J. McCormack, Cancer Res., 1986, 46, 6250–6254 CAS; (b) Y. Yu, L. G. Lou, W. P. Liu, H. J. Zhu, Q. S. Ye, X. Z. Chen, W. G. Gao and S. Q. Hou, Eur. J. Med. Chem., 2008, 43, 1438–1443 CrossRef CAS PubMed.
- X. H. Huang, P. C. Xiong, C. M. Xiong, Y. L. Cai, A. H. Wei, J. P. Wang, X. F. Liang and J. L. Ruan, Phytomedicine, 2010, 17, 930–934 CrossRef CAS PubMed.
- P. S. Randhawa, R. Shapiro, M. L. Jordan, T. E. Starzl and A. J. Demetris, Am. J. Surg. Pathol., 1993, 17, 60–68 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data, selected bond lengths and angles, hydrogen bonds, X-ray powder diffraction pattern, FT-IR and 1H NMR spectra, thermogravimetric analysis (TGA) for compound (1), hematological parameters in mice treated with (1) after 2 weeks. CCDC 1401929. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qi00072j |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2016 |