
Rapid synthesis of redox-active dodecaborane B12(OR)12 clusters under ambient conditions†
Alex I.
Wixtrom
a,
Yanwu
Shao
a,
Dahee
Jung
a,
Charles W.
Machan
b,
Shaunt N.
Kevork
a,
Elaine A.
Qian
a,
Jonathan C.
Axtell
a,
Saeed I.
Khan
a,
Clifford P.
Kubiak
b and
Alexander M.
Spokoyny
*a
aDepartment of Chemistry and Biochemistry, University of California, Los Angeles, 607 Charles E. Young Drive East, Los Angeles, California 90095-1569, USA. E-mail: spokoyny@chem.ucla.edu; Web: http://www.organomimetic.com
bDepartment of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, California 92093-0358, USA
First published on 3rd March 2016
Abstract
We have developed a fast and efficient route to obtain perfunctionalized ether-linked alkyl and benzyl derivatives of the closo-[B12(OH)12]2− icosahedral dodecaborate cluster via microwave-assisted synthesis. These icosahedral boron clusters exhibit three-dimensional delocalization of the cage-bonding electrons, tunable photophysical properties, and a high degree of stability in air in both the solid state and in solution. A series of closo-[B12(OR)12]2−, hypocloso-[B12(OR)12]1− and hypercloso-[B12(OR)12]0 clusters have been prepared with reaction times ranging from hours to several minutes. This method is superior to previously reported protocols since it dramatically decreases the reaction times required and eliminates the need for inert atmosphere conditions. The generality of the new microwave-based method has been further demonstrated through the synthesis of several new derivatives, which feature redox potentials up to 0.6 V more positive than previously known B12(OR)12 cluster compounds. We further show how this method can be applied to a one-pot synthesis of hybrid, vertex-differentiated species B12(OR)11(OR′) that was formerly accessible only via multi-step reaction sequence.
 Alexander M. Spokoyny | Dr Alexander Spokoyny was born and spent his childhood and teenage years in Moscow, Russia. After his family immigrated to United States in 2001, Alex attended UCLA, where he was introduced to the area of boron cluster chemistry working as an undergraduate researcher in the laboratory of Prof. M. Frederick Hawthorne. He continued his graduate chemistry education at Northwestern University under the guidance of Prof. Chad A. Mirkin. Upon receiving his Ph.D. in inorganic chemistry in 2011, Alex decided to move further east, where he was an NIH Post-Doctoral Fellow working in the laboratories of Profs. Stephen L. Buchwald and Bradley L. Pentelute at MIT. There his research interests involved the development of new bioconjugation strategies for peptides and proteins and regulation of protein–protein interactions. Alex returned to UCLA in 2014 to assume a position as an Assistant Professor in the Department of Chemistry and Biochemistry. His laboratory's research focuses on a broad range of problems in material science, catalysis and chemical biology with a fundamental emphasis on cluster chemistry. |
Introduction
The existence of an icosahedral dodecaborate [B12H12] cluster was first predicted by Lipscomb and co-workers in 1954.1 In a subsequent theoretical molecular orbital-based approach published in 1955,2 Longuet-Higgins and Roberts predicted that such a cluster would only be stable as dianionic [B12H12]2−. A 1959 study by Shapiro and Williams3 suggested the possible formation of a [B12H12]2− icosahedron, and in 1960 this cluster was first successfully isolated and characterized as a triethylammonium salt by Pitochelli and Hawthorne, albeit in a relatively low yield.4 Subsequent pioneering studies by Hawthorne, Knoth, Muetterties, and others initiated a new era in the field of boron cluster chemistry.5–19 Specifically, these groups have shown that [B12H12]2− can be prepared on a large scale in a high yield (>90%)19 and undergo facile functionalization chemistry that parallels some properties of classical organic molecules (e.g. benzene).20–22 This was an exciting discovery, since previously many boron hydride clusters were perceived as highly unstable species prone to fast degradation by heat, acids, and bases. Conversely, [B12H12]2− salts were shown to be stable in acids and bases, and were thermally stable as high as 810 °C with no observable decomposition.12 Knoth and co-workers were the first to demonstrate the persubstitution of [B12H12]2−, producing halogenated [B12F12]2−, [B12Cl12]2−, [B12Br12]2− and [B12I12]2− derivatives.16,23 In the past two decades, persubstitution of [B12H12]2− was improved with new synthetic methods and extended towards other functional groups including [B12Me12]2− and [B12(OH)12]2−.24–28 Among the perfunctionalized derivatives synthesized, closo-[B12(OH)12]2− is particularly appealing, as it is capable of undergoing further functionalization by forming ether, ester, carbonate, and carbamate linkages.27,29–31While controlled oxidation of the parent [B12H12]2− anion leads to an irreversible cluster degradation forming a B–B linked dimer,17 several perfunctionalized variants have been previously observed to undergo reversible redox behavior.32 For example, Rupich reported that [B12X12]2− could undergo a single electron oxidation to form a stable radical [B12X12]1−,33 which was recently isolated and structurally confirmed as the oxidized radical [B12Cl12]1− species by Knapp and co-workers.34 Hawthorne and co-workers reported that the perfunctionalized B12(OCH2Ph)12 cluster can exist in three distinct redox states accessible via two sequential and quasi-reversible one-electron oxidation reactions of the parent dianionic closo species.35 The same group later showed that other benzyl and alkyl functionalized B12(OR)12 clusters can be synthesized, and all of these species exhibit similarly reversible redox behavior.29 Schleid and co-workers subsequently showed that the parent [B12(OH)12]2− cluster can undergo a one-electron oxidation to form a stable radical [B12(OH)12]1− species.36 Interestingly, the redox potential of the ether-linked B12(OR)12 species can be rationally tuned as a function of the O-bound substituent, reminiscent of many metal-based redox-active inorganic complexes.29,32,37 Unfortunately, all of the reported synthetic routes towards B12(OR)12 clusters currently require either extremely long reaction times (weeks) or highly specialized high-pressure equipment. Furthermore, in all cases strict inert atmospheric conditions are also required for their synthesis.27,29,35
Herein we report a rapid, scalable, and robust synthetic route to a wide range of perfunctionalized B12(OR)12 cluster derivatives utilizing a bench-top microwave reactor. This technology has emerged over the past several decades and has been successfully employed in a large number of synthetic schemes38 which necessitate the reaction heating above the boiling point of the solvent. Our work shows that the microwave-based method enables synthesis of perfunctionalized ether-linked boron clusters within minutes and does not require the use of inert atmosphere and rigorously dried solvents. We further show the synthetic utility of our method in the preparation of previously unknown B12(OR)12 derivatives featuring highly oxidizing redox potentials as well as vertex-differentiated molecular architectures. Synthesized cluster species were all isolated and characterized using solution-based NMR and IR spectroscopic tools and mass spectrometry. Electron delocalization of the radical state in these species was further evaluated by electron paramagnetic resonance (EPR) spectroscopy and elucidated by X-ray photoelectron spectroscopy (XPS).
Results and discussion
The tetrabutylammonium (TBA) salt of closo-[B12(OH)12]−2 (TBA2[1]) was chosen for use with our microwave synthesis due to its enhanced solubility in organic solvents compared to alkali metal salts of 1 (Fig. 1).29 The synthesis of TBA2[1] was adapted from previously reported protocols by Hawthorne and co-workers (see ESI† for details).29,39,40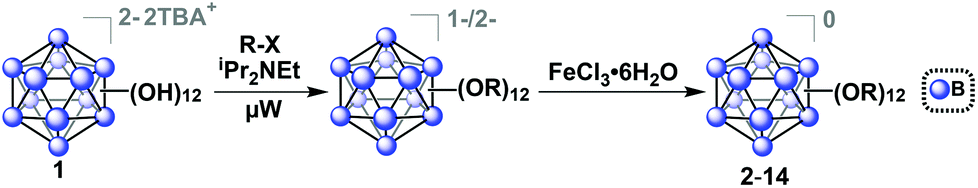 | ||
| Fig. 1 Synthetic route to produce functionalized ether-linked derivatives of the hypercloso boron clusters (2–14) via microwave-assisted synthesis. Precursor synthesis of [1]2− was adapted from previously described methods (see ESI† for details).29,39,40 | ||
Oxygen-free, anhydrous conditions (oven-dried glassware, dried and distilled solvents, nitrogen atmosphere) were initially employed for microwave-assisted syntheses of alkyl- and benzyl-functionalized closo-[B12(OH)12]2− [1] ether-linked derivatives, due to previously described high-pressure and reflux-based methods necessitating stringent air and moisture free conditions. To our surprise, we discovered that microwave reactions utilizing previously reported conditions (benzyl bromide and TBA2[1] in the presence of N,N-diisopropylethylamine (DIEA, Hünig's base) in acetonitrile) are driven at a much higher rate, resulting in quantitative formation of a mixture of charged 1−/2− TBA salts of 2 within 15 minutes at 140 °C, as indicated by in situ11B NMR spectroscopy. Specifically, no parent 11B NMR resonance at δ −18 corresponding to the [B12(OH)12]2− starting material is observed, and a singlet at δ −16 can be seen instead. Concomitant presence of [2]1− radical species in the product mixture can be deduced from the diagnostic pink color of the solution and its measured signature EPR signal (G-factor = 2.008121). Oxidation of the reaction mixture using FeCl3·6H2O in 90/10 ethanol/acetonitrile followed by column chromatography on silica gel produces the pure neutral cluster [2]0 in 63% yield (Fig. 2). Oxidation can be conveniently monitored by 11B NMR, where the fully oxidized cluster [2]0 exhibits a downfield resonance shift at δ 41.8. Overall, this represents a significant reduction in reaction duration from the originally reported 6 days and 4 hours required for reflux and high-pressure reactor methods, respectively, while retaining similar yield.29
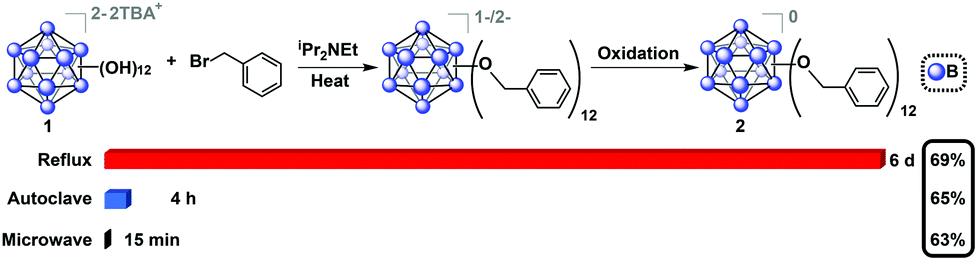 | ||
| Fig. 2 Synthesis of 2 from TBA2[1] using microwave-based method along with two previously reported methods indicating dramatic reduction in reaction time without compromising the isolated yield of [2]0.29 | ||
Encouraged by these results, we then optimized the microwave reaction times with allyl bromide and bromoethane reagents independently, and in both cases observed that complete substitution can be accomplished within 15–30 minutes at 140 °C. These compounds were isolated in their fully oxidized neutral form in a similar fashion to [2]0, (compounds [3]0 and [4]0, respectively; Fig. 3). Notably, previously reported high-pressure reactor synthesis of these species required 3 and 12 hours, respectively, for alkylation to occur at all twelve vertices, suggesting that the microwave-based method can be generally applied to several classes of ether-linked B12(OR)12 clusters and is potentially superior to the previously developed methods.29
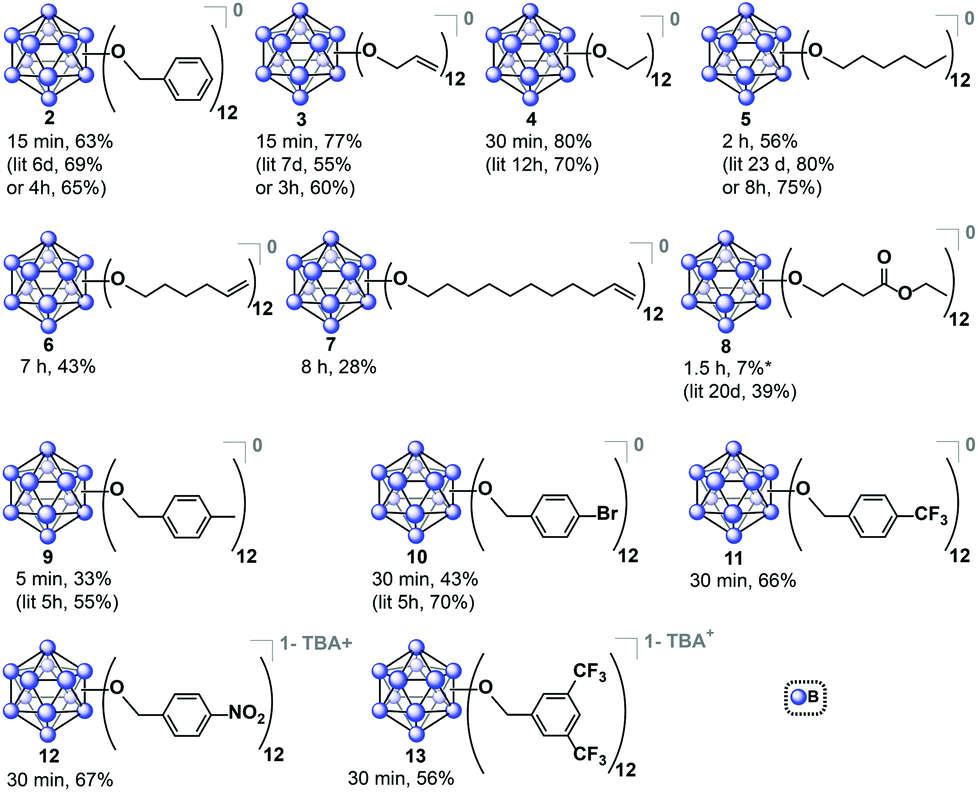 | ||
| Fig. 3 Synthesized B12(OR)12 clusters via microwave-based method. Yields are reported for the species isolated in a designated oxidation state as an average of two independent trials. For previously synthesized species, reported yield is given for comparison.29 *Additional 8% of 1−/2− non-oxidized species collected. | ||
The relatively short perfunctionalization reaction times made possible by this new microwave technique prompted us to investigate whether it would be possible to utilize ambient synthetic conditions. Specifically, we hypothesized that the fast rate of product formation would outcompete the rate of degradation stemming from the presence of adventitious air and moisture during the synthesis. We therefore tested the synthesis of 2 using as-received non-dried acetonitrile (see ESI†) with the reagents added to a reaction vessel open to air. The open-air synthesis of 2 proceeded with full conversion in 15 minutes as indicated by 11B NMR spectroscopy on the crude mixture. Following the normal work-up procedure (see ESI†), [2]0 was isolated in a 63% yield, suggesting that rigorous exclusion of air and moisture is not necessary for this transformation. This open-air synthesis method was successfully used for all subsequent ether-based cluster syntheses reported in this work. We decided to further explore the scope of this transformation by using longer-chain alkyl substituents. Hexyl chain substitution required increased reaction times compared to the shorter ethyl substituent, yet persubstitution was still achieved within two hours, as opposed to 8 hours when using a high-pressure reactor29 (isolated as [5]0, Fig. 3). This increased reaction time likely stems from the increase in the length and size of the alkyl reagent affecting the kinetics of the reaction.41,42 To further probe the limits of the microwave-based method we tested hexene- and undecene-based electrophiles, and even with the requisite increase in reaction time to 7 and 8 hours respectively, persubstitution proceeded to full conversion. Neutral [6]0 and [7]0 were isolated in 43% and 28% yields, respectively, after oxidation and normal purification procedures (Fig. 3). These derivatives have not been synthesized prior to this report, and their preparation illustrates how one can dramatically increase the size of these ether-based dodecaborate clusters via a direct linkage of large substituents featuring terminal olefins onto [B12(OH)12]2−.
Based on the success of the long-chain olefin-containing moieties, we investigated the compatibility of this system with less stable reagents by using ethyl 4-bromobutyrate with TBA2[1]. Perfunctionalization with ethyl butyrate has been challenging using prior synthetic methods, requiring multiple-step sequential additions of the alkyl halide and Hünig's base for 20 days while being handled under inert atmosphere conditions.43 However, utilizing microwave-assisted synthesis, the same product (Fig. 3, [8]0) was obtained via a single 1.5 hour reaction, followed by oxidation with FeCl3·6H2O overnight and purification with column chromatography on Sephadex™ and silica gel.
Benzyl-substituted ether-based clusters can feature a high degree of electrochemical tunability as a function of the substituents attached to the aromatic ring.37 Our method allows for the efficient synthesis of clusters containing both electron-rich (9) and electron-withdrawing (10) benzyl derivatives in yields matching previous methods but with significantly reduced reaction times (Fig. 3). We were further intrigued by the possibility of extending the accessible electrochemical window for this class of compounds by utilizing benzyl halide precursors containing highly electron-withdrawing substituents. The perfunctionalized cluster featuring a trifluoromethyl (CF3) group attached to the para position of the benzyl moiety was prepared using our method in 30 minutes, and following oxidation the isolated neutral compound [11]0 was obtained in 66% yield (Fig. 3). The oxidation potential of [11]1−/[11]0− (E1/2 = 0.56 V vs. Fc/Fc+) measured by cyclic voltammetry (CV) is particularly notable since it is higher than any reported B12(OR)12 cluster to date (previously 0.09 V vs. Fc/Fc+).37 Plotting the Hammett constants of various benzyl substituents42versus the redox potentials of [B12(OR)12] clusters perfunctionalized with these groups37 (Fig. 4) indicates the oxidation potential of these clusters can be rationally extended beyond the previously reported electrochemical window.
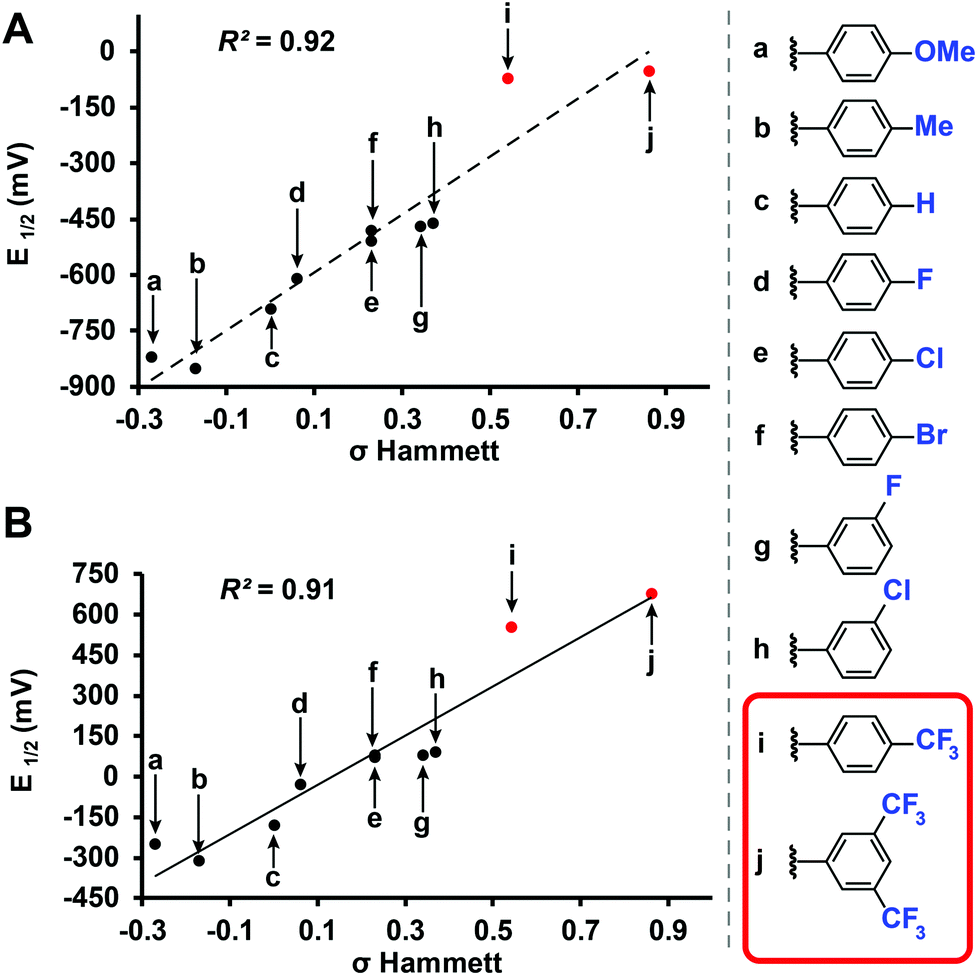 | ||
| Fig. 4 (a) Redox potential of [B12(OR)12]2−/1− and (b) [B12(OR)12]1−/0 substituted with various benzyl substituents (vs. Fc/Fc+) plotted vs. Hammett constants.22,42 Previously characterized37 (black) and new (red) [B12(OR)12]0 clusters are shown. | ||
For example, according to the trend suggested by this Hammett plot, a para-nitrobenzyl-substituted cluster should exhibit a higher 1−/0 oxidation potential than 11 (Fig. 3 and 4).42 The perfunctionalized cluster 12 featuring para-nitro (NO2) substituent attached to the benzyl was successfully synthesized using our method in 30 minutes, however, oxidation of the reaction mixture containing [12]2−/1− with FeCl3·6H2O did not produce any hypercloso-neutral species [12]0. Instead, the radical cluster species [12]1− was isolated as the only product in 67% yield (Fig. 3). This is not surprising, given the predicted oxidation potential for the [12]1−/[12]0 redox couple is more positive than the oxidizing strength of FeCl3·6H2O.44 Attempts to use stronger chemical oxidants (e.g. ceric ammonium nitrate) resulted in cluster degradation. Furthermore, insufficient solubility of [12]1− as a TBA salt precluded us from obtaining CV measurements for this derivative. Nevertheless, convinced we could expand the electrochemical window for B12(OR)12 species featuring benzyl-based substituents, we turned our attention to a potentially more soluble compound containing a 3,5-bis(trifluoromethyl)benzyl group instead.
Using the microwave method described above, cluster 13 was synthesized in 30 minutes. Interestingly, unlike all of the synthesized clusters reported thus far, the post-microwave reaction mixture was colorless, which is a characteristic feature of the pure dianionic state [13]2− for these clusters. The lack of color persisted even after column chromatography purification on silica gel in air. The identity of the pure isolated TBA salt of [13]2− (73% yield) was validated by full spectroscopic characterization and mass spectrometry (see ESI†). Oxidation of [13]2− with FeCl3·6H2O did not produce the neutral cluster, rather the pure [13]1− radical species was isolated in 78% yield, resulting in 56% net yield for [13]1− species starting from TBA2[1]. Use of stronger chemical oxidants such as ceric ammonium nitrate (CAN) degraded the boron cage, producing a diagnostic 11B NMR resonance at δ 20 characteristic of borates.47 Nevertheless, we were able to observe neutral cluster [13]0 electrochemically via infrared spectroelectrochemistry (IR-SEC) and cyclic voltammetry (CV) in CH2Cl2 (Fig. 5B and C). CVs of [13]1− as the TBA salt in CH2Cl2 showed two quasi-reversible redox features at −0.05 V and 0.68 V vs. Fc/Fc+, corresponding to the 2−/1− and 1−/0 transitions, respectively. IR-SEC experiments where the applied potential was increased to more positive potentials incrementally on the TBA salt of [13]2− showed subtle changes in the IR stretching modes for all three oxidation states of [13] around 1130–1140 cm−1 and 1200–1220 cm−1. The shift in these IR bands assigned to the B–O bond48 to higher wavenumbers from [13]2−/[13]1−/[13]0 is consistent with those observed in the same region for the analogous para-CF3 compound [11]0 which can be isolated in its neutral form via direct synthesis (see ESI†). The high oxidation potential for the [13]1−/[13]0 redox couple observed from cyclic voltammetry (E1/2 = 0.70 V vs. Fc/Fc+) is notable since it is the highest observed 1−/0 oxidation potential for the B12(OR)12 class of clusters reported to date37 and is ∼130 mV higher that the para-CF3 benzyl cluster 11. 19F NMR spectroscopy provides another diagnostic handle on the oxidation state of this compound (Fig. 5D). 11B NMR spectra for these clusters typically show a singlet around δ −14 to −16 for the 2− state, though the 1− state is silent due to the presence of the paramagnetic radical (confirmed by EPR, Fig. 5D (inset)). However, with 19F NMR spectroscopy, a shift from a singlet resonance in [13]2− at δ −63.36 to a broad singlet at δ −63.24 for [13]1− was observed. This broadening is consistent with the paramagnetic nature of [13]1−, where F atoms are located far enough from the unpaired electron-carrying B12-based cluster core to be resolved by 19F NMR spectroscopy.
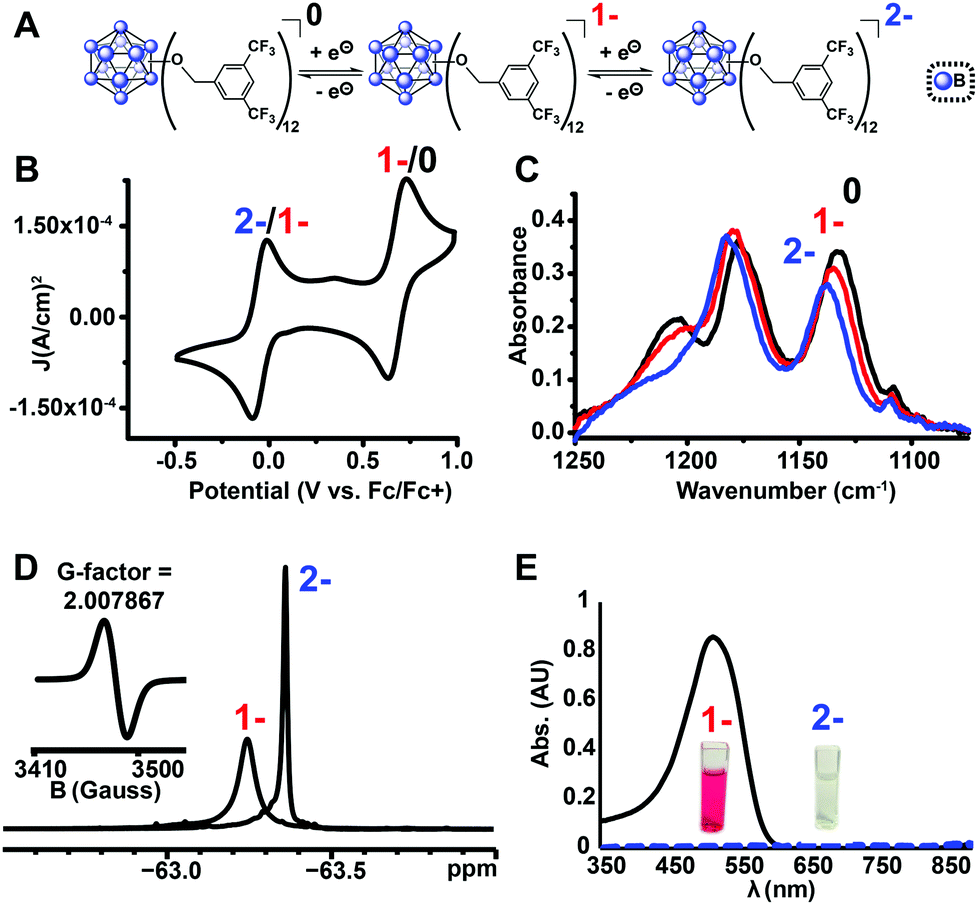 | ||
| Fig. 5 (a) Reversible redox activity of 13; (b) cyclic voltammogram (CV) demonstrating two independent, one-electron oxidation/reduction waves between 2−/1−, and 1−/0 states (1 mM 13 with 0.1 M TBAPF6 in CH2Cl2; glassy carbon working electrode, Pt wire counter electrode, Ag/AgCl pseudoreference electrode behind a CoralPor tip; referenced to an internal ferrocene standard); (c) infrared spectroelectrochemical (IR-SEC)45,46 analysis of 0/1−/2− states of 13; (d) 19F NMR spectra of [13]1− and [13]2− with EPR of [13]1− (inset); (e) UV-Vis spectra of [13]1− and [13]2−. | ||
The structural parameters of the boron clusters featuring persubstituted vertices can exhibit significant distortions in the solid state as a function of the substituent and the redox state as determined by X-ray crystallographic studies. Specifically, [2]2− exhibits nearly identical B–B bond distances (1.781(4)–1.824(4) Å) and angles (B–B–B 107.798° < a < 109.229°) as expected for a perfect icosahedron, yet as the cluster is oxidized to the electron-deficient [2]1− the structure expands and distorts slightly, with further distortion observed in the neutral state.35 Additionally, the B–O bond lengths decrease as the cluster is oxidized from [2]2−, which contains the longest average B–O distances, to the neutral [2]0 state with the shortest B–O distances.35 This observed trend of B–B bond lengthening, B–O bond contraction, and B–B–B angle distortion within the core as a function of cluster oxidation state is supported qualitatively by the crystal structures for neutral [11]0 and radical [13]1− (Fig. 6), which show comparable changes to those observed between [2]1− and [2]0. Selected bond lengths and angles for [11]0 and [13]1− are shown in Fig. 6.
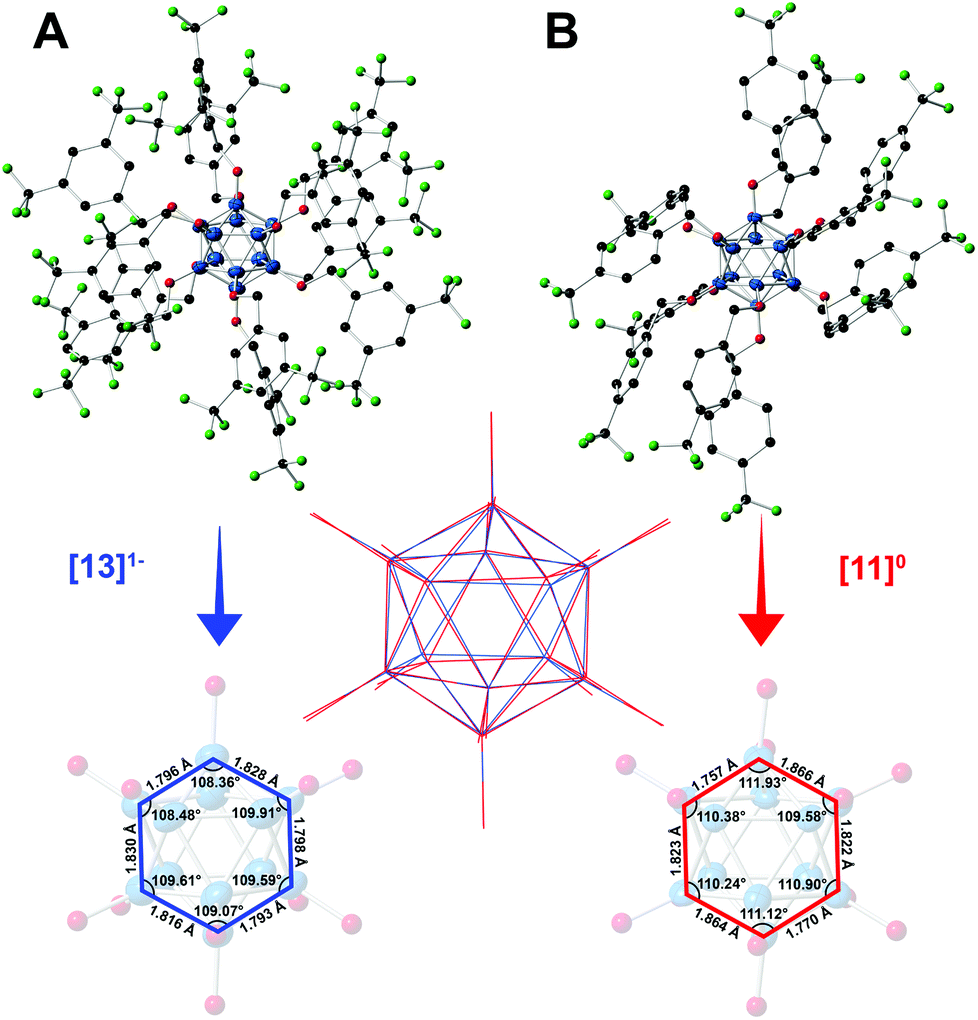 | ||
| Fig. 6 (A–B) Solid state X-ray structures for [13]1− and [11]0 shown with 50% thermal ellipsoid probabilities for the boron atoms (hydrogen atoms omitted for clarity). B atoms are blue, O – red, C – black, and F – green. Selected bond lengths and angles of the boron cores (substituents omitted for clarity) for [11]0 and [13]1− are shown on the bottom. Overlay of the two cores ([13]1− in blue, [11]0 in red) is depicted in the middle. | ||
X-ray photoelectron spectroscopy (XPS) has been widely used to study oxidation states in inorganic compounds.49–52 We therefore decided to utilize this technique to further elucidate the oxidation state and effect of functionalization for these boron clusters. Boron XPS spectra for several representative clusters synthesized in our study (Fig. 7) indicate a clear trend observed in the shift of B–B bond peak energies depending on the redox state of the functionalized cluster. The previously discussed geometric distortion of these boron cluster icosahedra with oxidation from 2− to 1− and further to neutral species corresponds directly to increased B–B binding energy (Fig. 7A). The nature of the substituents also produces a clear trend in these measurements, as compound 13 also exhibits an increase in B–B binding energy as the cluster increases in oxidation state from [13]2− to [13]1− (Fig. 7B), yet both are higher in energy than that of neutral cluster [11]0. However, despite the change in oxidation potential and binding energy from substituent effects, the nature of the electron radical delocalization throughout the boron-based core remains consistent. A single, broad symmetric EPR signal centered between 3450 and 3500 Gauss was observed for all cluster species isolated in the radical form with g values ranging between 2.0079 and 2.0081 depending on the substituent (see ESI†). Due to the 3D delocalization of the single electron across the 12 boron nuclei comprising the cluster, there exist a large number of possible hyperfine couplings.36 Overlap of these hyperfine couplings ultimately gives rise to the single broad line observed in the EPR spectra.
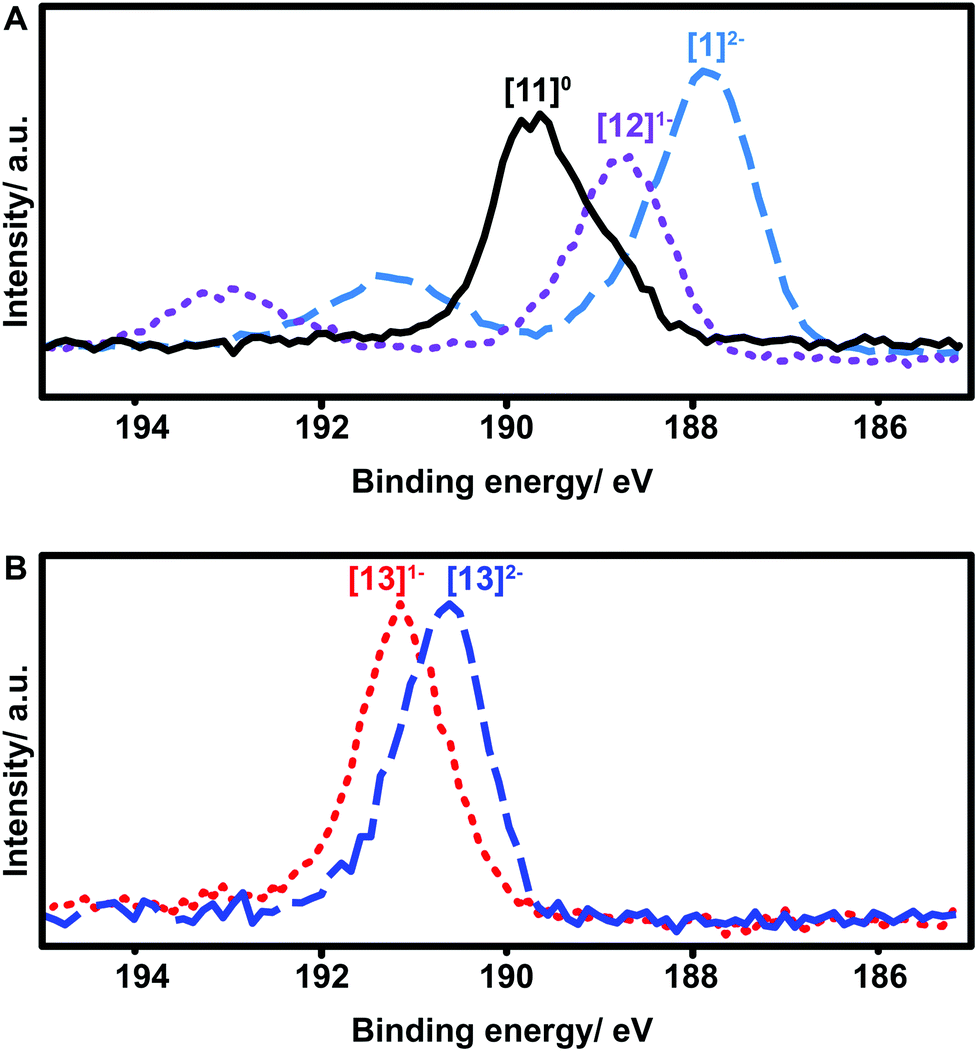 | ||
| Fig. 7 (a) Boron XPS spectra for TBA2[1]2−, TBA[12]1−, and [11]0 showing an increase in B–B bond energy with increasing oxidation state; (b) boron XPS spectra for TBA2[13]2− and TBA[13]1− indicating the higher B–B bond energies of the [13]2− and [13]1− anions compared to the other substituted clusters. | ||
In addition to previously mentioned benefits such as shortened reaction durations and the lack of stringent requirements for inert reaction conditions, microwave-assisted synthesis allows for one-pot, single-step reactions that would otherwise require more elaborate protocols. Monosubstitution of a benzyl ligand followed by persubstitution of the remaining eleven vertices has previously required a lengthy process involving several steps,53 whereas we demonstrate a one-pot approach enabled by our microwave-based method. For example, mixed-substituent B12(OEt)11(OBn) cluster (14, Fig. 8) can be formed in a single step simply by adding a stoichiometric amount of the desired reagents into a single reaction vessel.
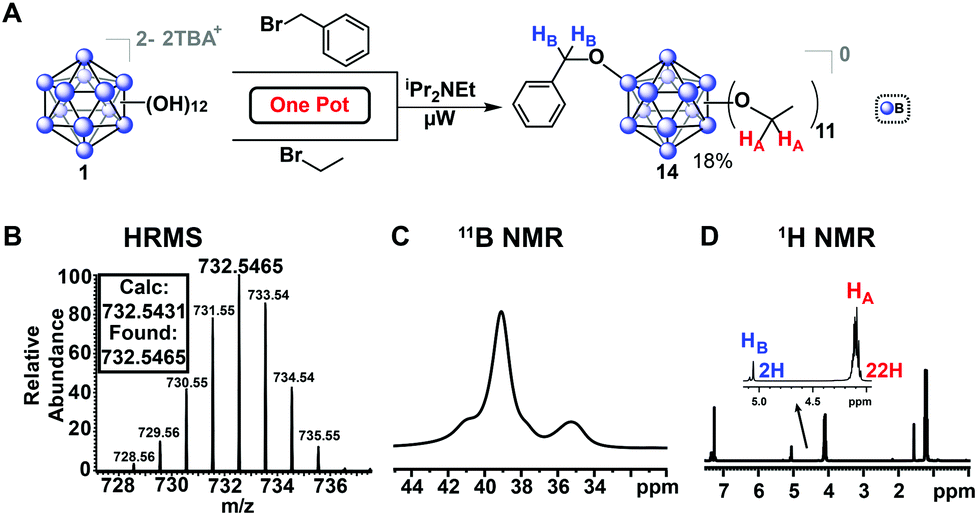 | ||
Fig. 8 One-pot synthesis of vertex-differentiated hypercloso cluster [14]0 from an approximately 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 molar mixture of benzyl bromide:bromoethane:TBA2[1]. Isolated yield of [14]0 was 18% (compound [4]0 was also formed as an additional product of the reaction). 11B NMR spectra indicates loss of the icosahedral symmetry due to vertex differentiation, 1H NMR integrations show 24H (CH2) and 36H (CH3) for the 11 ethyl groups and 2H (CH2) and 5H (Ph) for the single benzyl moiety. :1:1 molar mixture of benzyl bromide:bromoethane:TBA2[1]. Isolated yield of [14]0 was 18% (compound [4]0 was also formed as an additional product of the reaction). 11B NMR spectra indicates loss of the icosahedral symmetry due to vertex differentiation, 1H NMR integrations show 24H (CH2) and 36H (CH3) for the 11 ethyl groups and 2H (CH2) and 5H (Ph) for the single benzyl moiety. | ||
This reaction was completed in 30 minutes, producing three distinct species as a mixture which were oxidized with FeCl3·6H2O as previously described and subsequently isolated via column chromatography on silica: the perfunctionalized ethyl cluster [4]0, a small amount (<5%) of di-substituted [B12(OEt)10(OBn)2]0 clusters, and the desired [14]0 in 18% yield. This method represents a significantly faster route to produce [B12(OR)11(OR′)] mixed-substituent clusters.54
Conclusions
A rapid microwave-assisted synthetic route to perfunctionalized ether-linked B12(OR)12 clusters is disclosed and the robust nature of the technique demonstrated by the synthesis and characterization of multiple derivatives of 1. For previously synthesized compounds, reaction duration was significantly reduced and prior requirements for oxygen-free and anhydrous reaction conditions were eliminated. Our method also allows for a unique one-pot synthesis of mixed-substituent clusters with good selectivity under the same open-air conditions. The cluster species described here maintain the attractive properties of earlier derivatives, behaving as redox-active cores which show delocalization of electrons throughout the entire 3D boron cage, while the new functional groups provide a significant expansion to the available tuneable redox potential window for this class of clusters.Acknowledgements
We gratefully acknowledge Department of Chemistry and Biochemistry at UCLA for start-up funds. Portions of this material are based upon work supported by the National Science Foundation (CHE-1048804). E.A.Q. acknowledges the USPHS of the National Institutes of Heath (NIH) for the predoctoral training fellowship through the UCLA Chemistry-Biology Interface Training Program under the National Research Service Award (T32GM008496). C.W.M. and C.P.K. acknowledge support for this work from the AFOSR through a Basic Research Initiative (BRI) grant (FA9550-12-1-0414). We would like to thank Mr Kent Kirlikovali for assisting with EPR sample preparation and Mr Rafal Dziedzic for assistance with DART mass spectrometry. We thank Dr Liban Saleh for commenting on the manuscript.Notes and references
- B. C. Jr. W. H. Eberhardt and W. N. Lipscomb, J. Chem. Phys., 1954, 22, 989–1001 CrossRef.
- H. C. Longuet-Higgins and M. de V. Roberts, Proc. R. Soc. London, Ser. A, 1955, 230, 110–119 CrossRef CAS.
- I. Shapiro and R. E. Williams, J. Am. Chem. Soc., 1959, 81, 4787–4790 CrossRef CAS.
- A. R. Pitochelli and F. M. Hawthorne, J. Am. Chem. Soc., 1960, 82, 3228–3229 CrossRef CAS.
- E. B. Moore, L. L. Lohr and W. N. Lipscomb, J. Chem. Phys., 1961, 35, 1329 CrossRef CAS.
- R. Hoffmann and W. N. Lipscomb, J. Chem. Phys., 1962, 36, 2179 CrossRef CAS.
- R. Hoffmann and W. N. Lipscomb, J. Chem. Phys., 1962, 37, 520 CrossRef CAS.
- E. L. Muetterties, R. E. Merrifield, H. C. Miller, W. H. Knoth and J. R. Downing, J. Am. Chem. Soc., 1962, 84, 2506–2508 CrossRef CAS.
- R. Hoffmann and W. N. Lipscomb, J. Chem. Phys., 1962, 37, 2872 CrossRef CAS.
- H. C. Miller, N. E. Miller and E. L. Muetterties, J. Am. Chem. Soc., 1963, 85, 3885–3886 CrossRef CAS.
- I. A. Ellis, D. F. Gaines and R. Schaeffer, J. Am. Chem. Soc., 1963, 85, 3885–3885 CrossRef CAS.
- E. L. Muetterties, J. H. Balthis, Y. T. Chia, W. H. Knoth and H. C. Miller, Inorg. Chem., 1964, 3, 444–451 CrossRef CAS.
- W. R. Hertler, Inorg. Chem., 1964, 3, 1195–1196 CrossRef CAS.
- W. R. Hertler and M. S. Raasch, J. Am. Chem. Soc., 1964, 86, 3661–3668 CrossRef CAS.
- H. C. Miller, N. E. Miller and E. L. Muetterties, Inorg. Chem., 1964, 3, 1456–1463 CrossRef CAS.
- W. H. Knoth, H. C. Miller, J. C. Sauer, J. H. Balthis, Y. T. Chia and E. L. Muetterties, Inorg. Chem., 1964, 3, 159–167 CrossRef CAS.
- R. J. Wiersema and R. L. Middaugh, Inorg. Chem., 1969, 8, 2074–2079 CrossRef CAS.
- J. A. Wunderlich and W. N. Lipscomb, J. Am. Chem. Soc., 1960, 82, 4427–4428 CrossRef CAS.
- (a) I. B. Sivaev, V. I. Bregadze and S. Sjöberg, Collect. Czech. Chem. Commun., 2002, 67, 679–727 CrossRef CAS; (b) M. Davidson, Contemporary boron chemistry, Royal Society of Chemistry, 2000, vol. 253 Search PubMed; (c) N. S. Hosmane, Boron Science: New Technologies and Applications, Taylor & Francis, 2011 Search PubMed; (d) C. Knapp, in Comprehensive Inorganic Chemistry II, Elsevier, 2013, pp. 651–679 Search PubMed; (e) D. Olid, R. Núñez, C. Viñas and F. Teixidor, Chem. Soc. Rev., 2013, 42, 3318 RSC; (f) V. Geis, K. Guttsche, C. Knapp, H. Scherer and R. Uzun, Dalton Trans., 2009, 2687 RSC.
- R. N. Grimes, J. Chem. Ed., 2004, 81, 657 CrossRef CAS.
- M. F. Hawthorne, J. Chem. Ed., 2009, 86, 1131 CrossRef CAS.
- A. M. Spokoyny, Pure Appl. Chem., 2013, 85, 903–919 CrossRef CAS PubMed.
- W. H. Knoth, H. C. Miller, D. C. England, G. W. Parshall and E. L. Muetterties, J. Am. Chem. Soc., 1962, 84, 1056–1057 CrossRef CAS.
- W. Gu and O. V. Ozerov, Inorg. Chem., 2011, 50, 2726–2728 CrossRef CAS PubMed.
- D. V. Peryshkov, A. A. Popov and S. H. Strauss, J. Am. Chem. Soc., 2009, 131, 18393–18403 CrossRef CAS PubMed.
- S. V. Ivanov, S. M. Miller, O. P. Anderson, K. A. Solntsev and S. H. Strauss, J. Am. Chem. Soc., 2003, 125, 4694–4695 CrossRef CAS PubMed.
- M. F. Hawthorne, Pure Appl. Chem., 2003, 75, 1157–1164 CrossRef CAS.
- T. Peymann, A. Herzog, C. B. Knobler and M. F. Hawthorne, Angew. Chem., Int. Ed., 1999, 38, 1061–1064 CrossRef CAS.
- O. K. Farha, R. L. Julius, M. W. Lee, R. E. Huertas, C. B. Knobler and M. F. Hawthorne, J. Am. Chem. Soc., 2005, 127, 18243–18251 CrossRef CAS PubMed.
- A. Maderna, C. B. Knobler and M. F. Hawthorne, Angew. Chem., Int. Ed., 2001, 40, 1661–1664 CrossRef.
- S. S. Jalisatgi, V. S. Kulkarni, B. Tang, Z. H. Houston, M. W. Lee and M. F. Hawthorne, J. Am. Chem. Soc., 2011, 133, 12382–12385 CrossRef CAS PubMed.
- W. Kaim, N. S. Hosmane, S. Záliš, J. A. Maguire and W. N. Lipscomb, Angew. Chem., Int. Ed., 2009, 48, 5082–5091 CrossRef CAS PubMed.
- M. W. Rupich, J. Electrochem. Soc., 1985, 132, 119 CrossRef CAS.
- R. T. Boeré, S. Kacprzak, M. Keßler, C. Knapp, R. Riebau, S. Riedel, T. L. Roemmele, M. Rühle, H. Scherer and S. Weber, Angew. Chem., Int. Ed., 2011, 50, 549–552 CrossRef PubMed.
- T. Peymann, C. B. Knobler, S. I. Khan and M. F. Hawthorne, Angew. Chem., Int. Ed., 2001, 40, 1664–1667 CrossRef CAS.
- N. Van, I. Tiritiris, R. F. Winter, B. Sarkar, P. Singh, C. Duboc, A. Muñoz-Castro, R. Arratia-Pérez, W. Kaim and T. Schleid, Chem. – Eur. J., 2010, 16, 11242–11245 CrossRef PubMed.
- M. W. Lee, O. K. Farha, M. F. Hawthorne and C. H. Hansch, Angew. Chem., Int. Ed., 2007, 46, 3018–3022 CrossRef CAS PubMed.
- (a) A. de la Hoz, A. Diaz-Ortiz and A. Moreno, Chem. Soc. Rev., 2005, 34, 164 RSC; (b) R. B. N. Baig and R. S. Varma, Chem. Soc. Rev., 2012, 41, 1559–1584 RSC; (c) M. B. Gawande, S. N. Shelke, R. Zboril and R. S. Varma, Acc. Chem. Res., 2014, 47, 1338–1348 CrossRef CAS PubMed; (d) A. de la Hoz and A. Loupy, Microwaves in Organic Synthesis, 2 Volume Set, Wiley, 2013 Search PubMed; (e) C. O. Kappe, D. Dallinger and S. S. Murphree, Practical Microwave Synthesis for Organic Chemists, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2008 Search PubMed.
- T. Peymann, C. B. Knobler, S. I. Khan and M. F. Hawthorne, J. Am. Chem. Soc., 2001, 123, 2182–2185 CrossRef CAS PubMed.
- M. J. Bayer and M. F. Hawthorne, Inorg. Chem., 2004, 43, 2018–2020 CrossRef CAS PubMed.
- R. W. Taft, J. Am. Chem. Soc., 1953, 75, 4231–4238 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- A. Pushechnikov, S. S. Jalisatgi and M. F. Hawthorne, Chem. Commun., 2013, 49, 3579–3581 RSC.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- I. S. Zavarine and C. P. Kubiak, J. Electroanal. Chem., 2001, 495, 106–109 CrossRef CAS.
- C. W. Machan, M. D. Sampson, S. A. Chabolla, T. Dang and C. P. Kubiak, Organometallics, 2014, 33, 4550–4559 CrossRef CAS.
- C. G. Salentine, Inorg. Chem., 1983, 22, 3920–3924 CrossRef CAS.
- L. A. Leites, Chem. Rev., 1992, 92, 279–323 CrossRef CAS.
- N. V. Alov, J. Anal. Chem., 2005, 60, 431–435 CrossRef CAS.
- W. Temesghen and P. Sherwood, Anal. Bioanal. Chem., 2002, 373, 601–608 CrossRef CAS PubMed.
- X. P. Zhu, T. Yukawa, M. Hirai, H. Suematsu, W. Jiang, K. Yatsui, H. Nishiyama and Y. Inoue, Appl. Surf. Sci., 2006, 252, 5776–5782 CrossRef CAS.
- M.-Y. Xing, W.-K. Li, Y.-M. Wu, J.-L. Zhang and X.-Q. Gong, J. Phys. Chem. C, 2011, 115, 7858–7865 CAS.
- L. N. Goswami, Z. H. Houston, S. J. Sarma, H. Li, S. S. Jalisatgi and M. F. Hawthorne, J. Org. Chem., 2012, 77, 11333–11338 CrossRef CAS PubMed.
- Recent examples of other mixed substituted dodecaborate clusters: (a) O. Bondarev, A. A. Khan, X. Tu, Y. Sevryugina, S. S. Jalisatgi and M. F. Hawthorne, J. Am. Chem. Soc., 2013, 135, 13204–13211 CrossRef CAS PubMed; (b) C. Jenne and C. Kirsch, Dalton Trans., 2015, 44, 13119–13124 RSC; (c) Y. Zhang, J. Liu and S. Duttwyler, Eur. J. Inorg. Chem., 2015, 31, 5158–5162 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and spectroscopic characterization details, X-ray crystallographic data for 11 and 13, NMR spectra, mass spectra, IR spectra, cyclic voltammetry, XPS spectra, and EPR spectra. CCDC 1438967–1438968. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00263j |
| This journal is © the Partner Organisations 2016 |