
Water-soluble, heterometallic chalcogenide oligomers as building blocks for functional films†
J. Y.
Chane-Ching
*a,
L.
Perrin
b,
P.
Puech
c,
V.
Bourdon
d,
V.
Foncrose
a,
A.
Balocchi
e,
X.
Marie
e and
P.
Lavedan
d
aUniversité de Toulouse, UPS, CNRS, CIRIMAT, 118 Route de Narbonne, F-31062, Toulouse, France. E-mail: chane@chimie.ups-tlse.fr
bUniversité de Lyon 1, Claude Bernard, ICBMS, UMR 5246, CNRS, INSA, CPE 43 Bd du 11 nov 1918, 69622 Villeurbanne cedex, France
cUniversité de Toulouse, CNRS, CEMES, 29 rue Jeanne Marvig, 31055 Toulouse, France
dUniversité de Toulouse, UPS, Service commun, 118 Route de Narbonne, F-31062, Toulouse, France
eUniversité de Toulouse, INSA-CNRS-UPS, LPCNO, 135 Av. Rangueil, 31077 Toulouse, France
First published on 15th February 2016
Abstract
Monometallic chalcogenide aqueous complexes such as (Sn2S6)4− and (Sn4S10)4− are widely used as functional ligands with applications in nano-electronics and solar cells. We propose a general process route to the formation of all-inorganic, heterometallic chalcogenide oligomers, thus expanding the range of these functional aqueous ligands. From electrospray ionization mass spectrometry, tetramers were shown to be the most predominant oligomers synthesized in Sn(IV)–Zn(II)–S(II), Sn(IV)–Zn(II)–Se(II) and Sn(IV)–Ga(III)–S(II) systems. While tetramers possessing exclusively one Zn cation were identified in the Sn(IV)–Zn(II)–S(II) system, the full range of solid solutions was achieved for (Sna–Gab–Sc)t− oligomers with 1 ≤ a ≤ 4. Using in situ characterization by 119Sn liquid NMR and Raman spectroscopy, supported by DFT calculations, we demonstrate that the various tetramers adopt a compact adamantane-like structure. The charge of the heterometallic oligomers was shown to be controlled by the doping cation valence and the chalcogenide anion deficiency in the tetramers. These water-soluble heterometallic chalcogenide oligomers can serve as ligands, residue-free dispersants or building blocks for functional film fabrication. Using these oligomers, we report here the fabrication of chalcogenide solar cells employing environmentally friendly, all-aqueous, (Sna–Znb–Sc)t−-capped CZTS nanocrystal inks.
Introduction
Soluble metal chalcogenide complexes represent exciting building blocks largely used in various applications such as nanoelectronics,1,2 photovoltaics3 or thermoelectrics.4–6 The use of hydrazinium soluble complexes for semiconducting films was first introduced by Mitzi et al.1 Indeed, the clean temperature decomposition of these precursors yields ultrathin (50 Å), crystalline and continuous metal chalcogenide films for nanoelectronics. Used as electronic linkers, these soluble complexes have been proven to increase electron mobility in nanocrystal arrays, allowing semiconductors to be patterned onto large area substrates for photovoltaics or thermoelectrics.7 More recently, the number of these hydrazinium metal chalcogenide complexes has been increasing. A general synthetic route involving the use of an alkali metal hydride was proposed8 for dissolving a number of metal chalcogenides previously considered to be unreactive. Availability of new complexes such as CdTe2−, Cd2Se32−, PbTe22−, and Bi2Te54− has allowed achieving excellent electron mobility in nanocrystal semiconductor films, widely used in photovoltaics and thermo-electrics. Another strategy is to develop hydrazinium-free, water-soluble, environmentally friendly metal chalcogenide precursors. These water-soluble metal chalcogenide oligomers can be associated with NH4+ counterions to yield highly pure films. While many metal chalcogenide clusters were previously synthesized with an emphasis on the use of organic cations as structure-directing agents,9,10 purely inorganic, hydrazinium-free chalcogenide systems have received less attention. Pioneering studies by Krebs11–15 described isolated water-soluble (Sn2S6)4, (Sn4Se10)4−, and (Ga4Se10)8− chalcogenide complexes with Na+ as the counterion. The reported complexes, usually composed of two or four metal centers of the same metallic cation, were largely developed as ligands for improved electrostatic colloidal stabilization16 or enhanced electronic coupling.17Here, we propose to expand the list of these water-soluble, all-inorganic complexes to heterometallic chalcogenide oligomers. Indeed, association of metal cations that display various valences in a single metal chalcogenide oligomer can improve their electronic and thermal conduction. In addition, their large-scale use in numerous applications such as thermoelectrics and photovoltaics will be facilitated if they are cheap and simple. A nice platform suited to the integration of these heterometallic chalcogenide oligomers is earth abundant copper–zinc–tin chalcogenide (Cu2ZnSnSSe4, CZTSSe) thin films. These films have recently attracted increasing attention18–21 as absorbers for low-cost solar cells. Indeed, solution-based deposition routes22,23 have been recently proposed to potentially provide low-cost scalable routes for the fabrication of CZTSSe solar cells. While solution-based process routes currently involve hydrazine22 or hexane-thiol23 as solvents, less toxic24,25 or safer process routes involving green, stable, all-aqueous inks26 were recently reported for the fabrication of CZTS solar cells. To produce all-aqueous inks, tin chalcogenide complexes including Sn2S64− and Sn2S76− were proposed as ligands to in situ cap the Cu/Zn sulfide nanoparticles,26 resulting in homogeneous and stable dispersions for the formation of CZTS,Se films. Another environmentally friendly approach was developed in our group involving a high temperature nanocrystal synthetic route27 producing low electronic-defect concentration Cu2ZnSnS4 (CZTS) building blocks. The surfaces of these CZTS nanocrystals were further purified by S2− exchange, yielding deagglomerated Cu2ZnSnS4 nanocrystal dispersions. Hence, the design and synthesis of suitable ligands are key steps in the fabrication of high performance films.28 In this context, expanding the range of water-soluble, all-inorganic, highly electrostatically charged metal chalcogenide oligomers represents an interesting challenge. In particular, heterometallic chalcogenide oligomers should be of great interest since they could allow both a better nanocrystal surface charge control and a homogeneous fine-tuning of the nanocrystal film composition. Because of the high potentiality of the CZTSSe solar cell as a low-cost solar cell,18 we focus on the design and synthesis of (Sna–Znb–Sc)t− oligomers. Indeed, Sn and Zn cations over-stoichiometries are required for the fabrication of high performance CZTSSe solar cells,22 rendering (Sna–Znb–Sc)t− oligomers very attractive as all-inorganic dispersants for the design of high purity CZTS dispersions. Heterometallic (Sna–Gab–Sc)t− oligomers including Ga(III) were also explored. We selected Ga(III) as a model trivalent dopant capable of promoting the electronic conductivity of CZTS29 for thermoelectric applications.
A general method for the synthesis of highly water-soluble, discrete, heterometallic, chalcogenide oligomers is proposed involving a facile, low-cost process route and ammonium as the residue-free counterion. Our strategy is based on the incorporation, under mild conditions, of a high-valent cation, introducing stacking faults or symmetry loss into the chalcogenide framework, resulting in lower molecular mass, highly water-soluble oligomers. The chemical composition of the oligomers was determined by electrospray ionization mass spectrometry, demonstrating their heterometallic character. The structure of the oligomers was probed by liquid 119Sn NMR and Raman spectroscopy and supported by quantum chemical calculations at the DFT level. These heterometallic chalcogenide oligomers could prove effective as building blocks for the design of an aqueous, environmentally friendly process route to the fabrication of Cu2ZnSnS,Se4 (CZTS,Se) solar cells. We will show that these building blocks can serve as highly charged, all-inorganic, aqueous ligands. Preliminary results describing a solar cell fabricated from an all-aqueous, (Sna–Znb–Sc)t− capped CZTS nanocrystal dispersion and displaying an encouraging efficiency of 2.6% are reported.
Experimental section
Chemicals
(NH4)2S (solution, 20% d = 1.1 Sigma Aldrich), ZnCl2 (Sigma), SnCl4 5H2O (Fisher Chemicals), CH3NaO3S·xH2O (or hydroxymethanesulfinate hydrate or Rongalite, Aldrich) and Se metal (Acros) were used as received. GaCl3 solution was prepared by dissolution of Ga2O3 (99.99%, Rhodia) in 6 M HCl in an autogeneous reactor at 200 °C for 16 h.Heterometallic chalcogenide oligomer synthesis
Two main process routes have been proposed11,12 for the synthesis of water-soluble chalcogenide oligomers. The first route involves the metal sulfide dissolution in highly basic sulfide solution. In the second route, the oligomers are formed from homogeneous ionic solutions. Because the metal sulfide dissolution process involves highly basic pH, we have explored the formation of heterometallic chalcogenide oligomers from homogeneous solutions since this route allows investigation of larger [S2−] and pH ranges. Oligomers were prepared from solutions defined by two key parameters, s and h (or oh), s = [S2−]/∑[M], h = [H+]/∑[M], and (oh) = [OH−]/∑[M], with M = Sn (IV) or Zn(II) or Ga(III).Oligomers characterization
While the identification of metal chalcogenide solid clusters is generally performed by single-crystal X-ray diffraction, large broad peaks were recorded by X-ray investigation on precipitated solid oligomers. Insights into the speciation of solution species and the structure of the oligomers were gained using in situ liquid techniques including electrospray ionization mass spectrometry (ESI-MS), Dynamic Light Scattering (DLS), and Raman and NMR spectroscopy.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 scans.
30 package at the DFT level by means of the hybrid density functional B3PW91.31,32 For the Sn,33 Zn,34 Ga35 and Se33 atoms, the Stuttgart–Dresden pseudopotentials were used in combination with their extended valence basis sets.33–36 For the sulfur atoms, the augmented and polarized all electron 6-311++G(d,p)37 basis sets were used. Optimizations were carried out without any symmetry restriction. The nature of the optimized stationary point, minima or transition state has been verified by means of analytical frequency (IR and Raman) calculations at 298.15 K and 1 atm. Compounds have been optimized in water by means of the SMD solvation model implemented in Gaussian 09.38 Final frequency calculations have also been computed within the SMD implicit solvation scheme.
000 scans.
30 package at the DFT level by means of the hybrid density functional B3PW91.31,32 For the Sn,33 Zn,34 Ga35 and Se33 atoms, the Stuttgart–Dresden pseudopotentials were used in combination with their extended valence basis sets.33–36 For the sulfur atoms, the augmented and polarized all electron 6-311++G(d,p)37 basis sets were used. Optimizations were carried out without any symmetry restriction. The nature of the optimized stationary point, minima or transition state has been verified by means of analytical frequency (IR and Raman) calculations at 298.15 K and 1 atm. Compounds have been optimized in water by means of the SMD solvation model implemented in Gaussian 09.38 Final frequency calculations have also been computed within the SMD implicit solvation scheme.
Solution exchange and infiltration of heterometallic oligomer-capped ultrafine ZnS colloids
000 rpm and redispersed into a fresh aliquot of the (Sna–Znb–Sc)t−, xZn = 0.33, oligomer solution. This operation was repeated three times. The solid nanocrystals were finally redispersed into 0.01 M (NH4)2S yielding a nanocrystal ink of ∼35 g L−1.
Solar cell fabrication
Crack-free Cu2ZnSnS4 films were prepared by dip coating the glass/Mo electrode into a (Sna–Znb–Sc)t− oligomer-capped CZTS nanocrystal aqueous dispersion. Typical conditions for dip coating were v = 200 mm min−1. Crack-free films exhibiting a final thickness of about 2.2 μm were achieved after multiple depositions (18 layers) from aqueous Cu2ZnSnS4 dispersions (c = 35 g L−1) under air atmosphere. Complete carbon purification of nanocrystal films was achieved by implementing a purification step (500 °C 30 min, 125 mg Se) in our annealing process. Densification of the CZTS films was performed under selenium partial pressure (125 mg Se) at 580 °C. The surface of the solar cells was S = 2 × 2 mm2.Results and discussion
Monometallic Mn+ (Mn+ = Sn(IV), Ga(III) or Zn(II)) chalcogenide oligomers
Water-soluble monometallic chalcogenide oligomer formation domains (Fig. S1a and S1b†) were determined on reaction mixtures prepared at [M] = 0.1 M, (M = Sn(IV), Ga(III) or Zn(II)). These formation domains were explored using the two key parameters s and h (or oh), s = [S2−]/[M], h = [H+]/[M], oh = [OH−]/[M].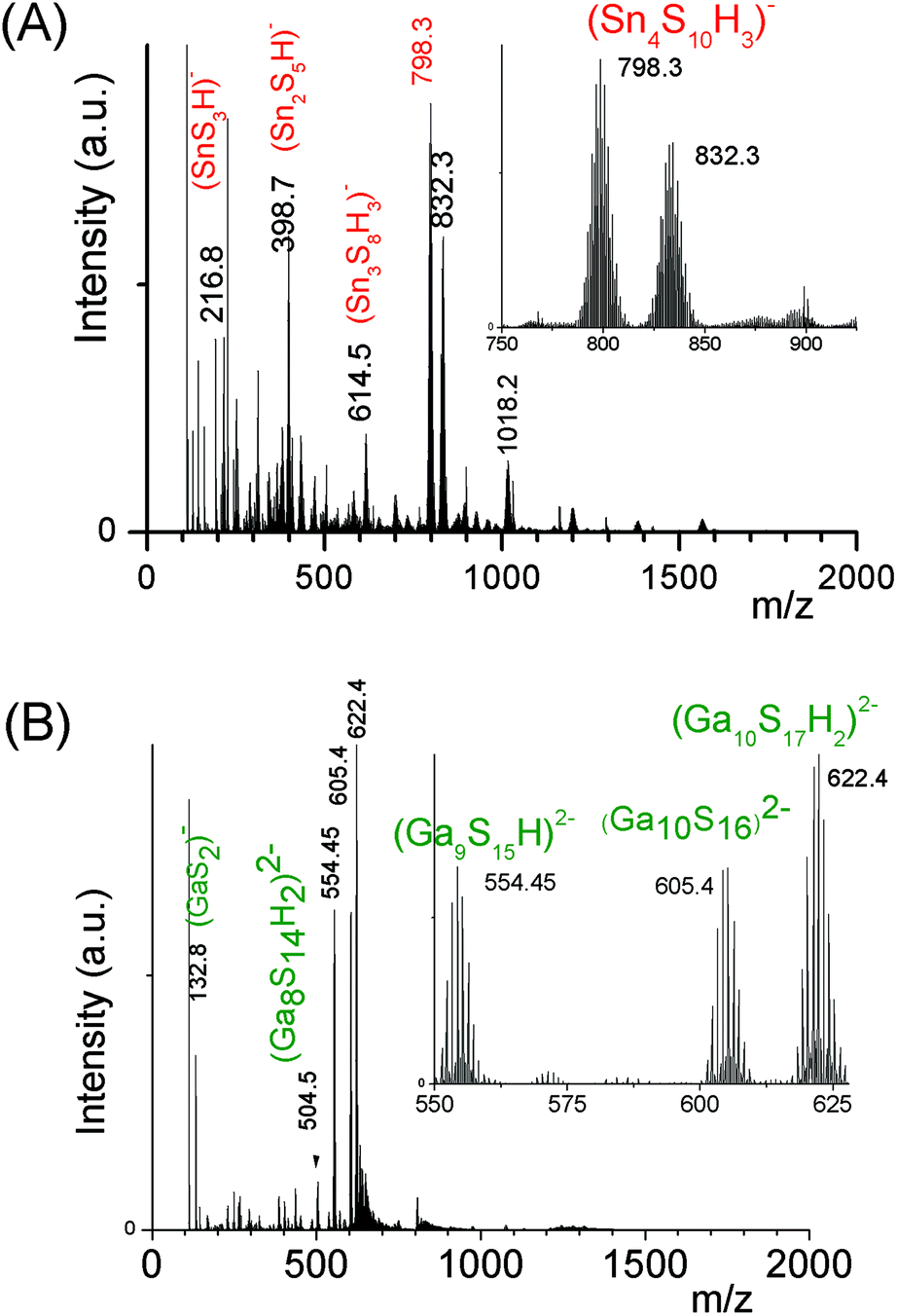 | ||
| Fig. 1 Negative ion ESI-MS spectra of monometallic oligomers. (A) (SnaSb)t− oligomers prepared from a Sn4+ solution ([H+]/[Sn4+] = 1.5), Sn(IV) = 0.1 M and s = 4. Insert: Zoom showing the presence of tetramers. (B) (GaaSb)t− oligomer solution, pH 8.9, showing the presence of doubly-charged oligomers. The oligomer solution is synthesized from a Ga3+ solution ([H]/[Ga3+] = 0), at Ga(III) = 0.1 M and s = 6. This solution is prepared by re-dispersion of the solid in 0.01 M (NH4)2S. Insert: Zoom showing the presence of (GaaSb)t− oligomers including up to 10 Ga(III) cations. | ||
The structures of these (Sna–Sb)t− oligomers were previously reported in the literature.14 Previous data have demonstrated that the (Sn4S10)4− oligomers display an adamantane scaffold.14 Concerning these (Sn4S10)4− oligomers, the relative stability of a linear polytetrahedron structure with respect to the adamantane-like one has been assessed by DFT calculations. Consistent with the experimental data previously reported,14 our calculations indicate that the adamantane T2 structure is more stable than the linear polytetrahedron (T1)4 by 11.8 kcal mol−1 (ΔrG° value).
Heterometallic Sn(IV)–Zn(II) chalcogenide oligomers
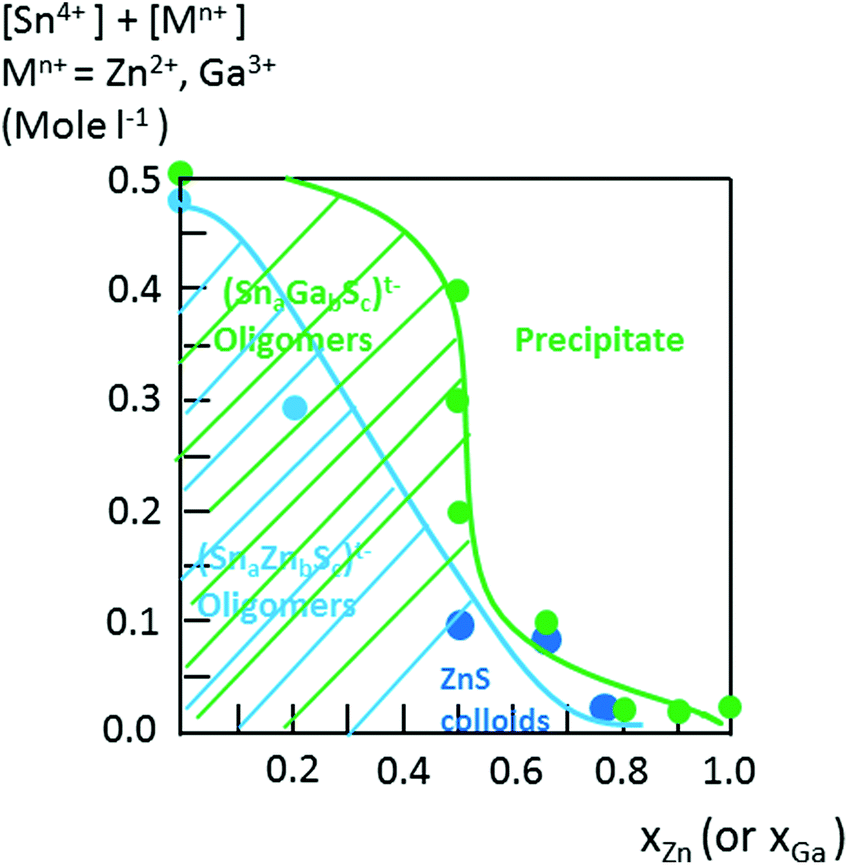 | ||
| Fig. 2 Oligomer formation diagrams, ([Sn4+] + [Mn+]) = 0.1 M, M = Zn2+ or Ga3+. The hatched blue area represents the soluble (Sna–Znb–Sc)t− oligomers domain synthesized by the addition of (Zn2+, Sn4+) solution (H+/(Zn + Sn) = 0.5) onto a (NH4)2S solution at s = 6. Dark blue circles represent the formation of ZnS colloid dispersions. The hatched green area represents the domain of (Sna–Gab–Sc)t− soluble oligomers prepared from (Ga3+, Sn4+) solutions, ([H+]/[Sn] + [Ga] = 0) and S2− solutions at s = 6. | ||
An ESI-MS investigation performed on these limpid solutions (Fig. 3a) without any dilution synthesized at different xZn values, xZn = 0.33, 0.50, and 0.66, revealed the presence of oligomers displaying similar compositions but in different proportions as shown by the relative intensities of the ESI-MS peaks. In these solutions, monometallic Sn chalcogenide oligomers including (SnS3)2−, (Sn2S5)2−, (Sn3S7)2−, (Sn3S8)4−, and (Sn4S10)4− were detected in association with heterometallic oligomers (Fig. 3b). Indeed, mass spectrometry data clearly reveal the presence of heterometallic oligomers including (SnZnS4)2−, (Sn2.0Zn1.0S6)2−, (Sn2.0Zn1.0S7)4−, (Sn3.0Zn1.0S9)4−, and (Sn3.0Zn1.0S8)2− with a larger proportion of the four metal center oligomers.
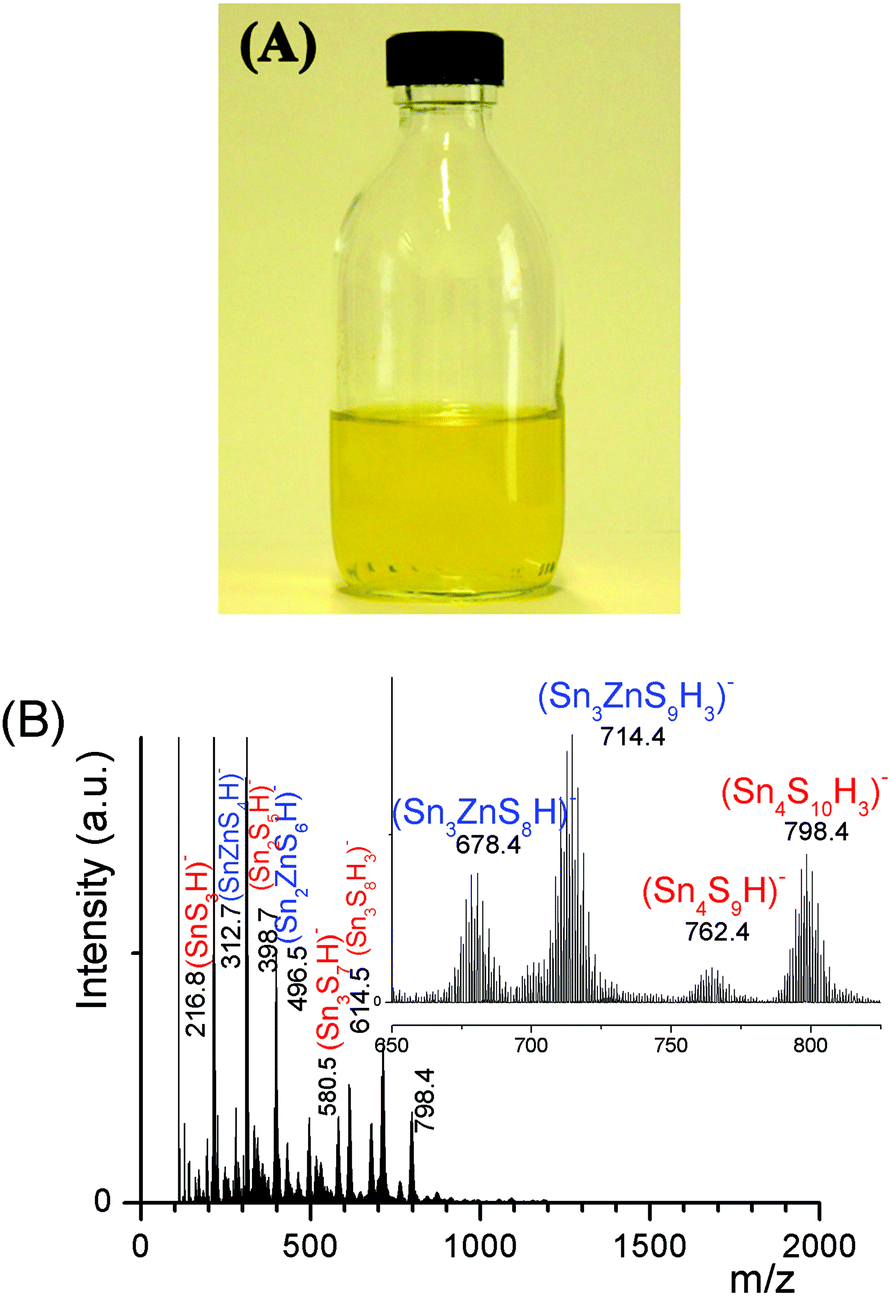 | ||
| Fig. 3 (A) Photograph of a (Sna–Znb–Sc)t− oligomer solution. (B) Negative ion ESI-MS spectrum of (Sna–Znb–Sc)t− oligomer solutions prepared from (Sn4+ + Zn2+) solutions, ([H+]/([Sn4+] + [Zn2+]) = 1.5), [S2−]/([Sn4+] + [Zn2+]) = 6 and ([Sn] + [Zn]) = 0.1 M. xZn = 0.33. Insert: Zoom showing the presence of heterometallic tetramers. | ||
Interestingly, as shown by the isotope patterns reported in Fig. S3c,† two different heterometallic oligomers characterized by a different number of sulfide anions and possessing four metal centers including a sole Zn cation, (Sn3.0 Zn1.0 S8)2− and (Sn3.0 Zn1.0 S9)4−, were clearly identified. From quasi-elastic light scattering investigation, a hydrodynamic average size of 2 nm, consistent with the oligomer low molecular mass, was determined on samples prepared at xZn ≤ 0.33. An increase of xZn results in larger mean hydrodynamic diameter values (Fig. S4†). Indeed, dispersions prepared at xZn, 0. 33 ≤ xZn ≤ 0.50, show a mean size of 5 nm while dispersions synthesized at xZn = 0.66 possess a hydrodynamic diameter of around 18.5 nm. Although all these dispersions are limpid to the naked eye, these data suggest that the (Sna–Znb–Sc)t− heterometallic oligomers are mainly obtained at low xZn values, xZn ≤ 0.33, while dispersions formed for xZn > 0.40 probably contain predominantly ZnS colloids. Interestingly, the size monodispersity of the nano-objects observed at xZn = 0.5 shows the formation of a monodisperse population of ZnS colloids stabilized by (Sn4−xZnxSz)t− oligomers. Liquid 119Sn NMR spectra recorded on the various dispersions show traces at δ = 55 ppm and δ = 77 ppm previously observed on the monometallic (SnaSb)t− oligomers and respectively attributed to (Sn2S6)4− and SnS44−/SnS32− species.16 More importantly, additional peaks of higher intensities are clearly observed at δ = 63 ppm and δ ∼ 82/85 ppm for the entire range of xZn investigated (Fig. 4a). Because a unique heterometallic tetramer oligomer possessing a lonely Zn cation was observed whatever the xZn value, liquid 119Sn NMR spectra recorded on the various dispersions display identical additional peaks. In addition, the larger intensity of these additional peaks observed on the xZn = 0.33 dispersion is consistent with the largest heterometallic oligomer proportion in this dispersion. Therefore, the experimental chemical shifts observed at δ = 63 ppm and δ ∼ 82/85 ppm result from the incorporation of the Zn cation into the four metal center (Sn3.0Zn1.0Sz)t− oligomers. Raman spectra recorded on the various dispersions exhibit a main broad peak centered at 345 cm−1 and a secondary peak at 370 cm−1 (Fig. 4b). Although deconvolution of the main broad peak shows a slight increase of the frequency of this peak observed with increased xZn (Fig. 4b), full assignment of these peaks remains inconclusive due to the overlapping of the different peaks in this region. Indeed, ν1(A1) and ν3(T2) Raman frequencies13 of SnS44− were reported respectively at 348 and 344 cm−1 while the Raman frequencies of 1-longitudinal-optical phonon mode of cubic ZnS41 and quasi longitudinal mode of Wurtzite ZnS42 were respectively reported to be 343 cm−1 and 350 cm−1.
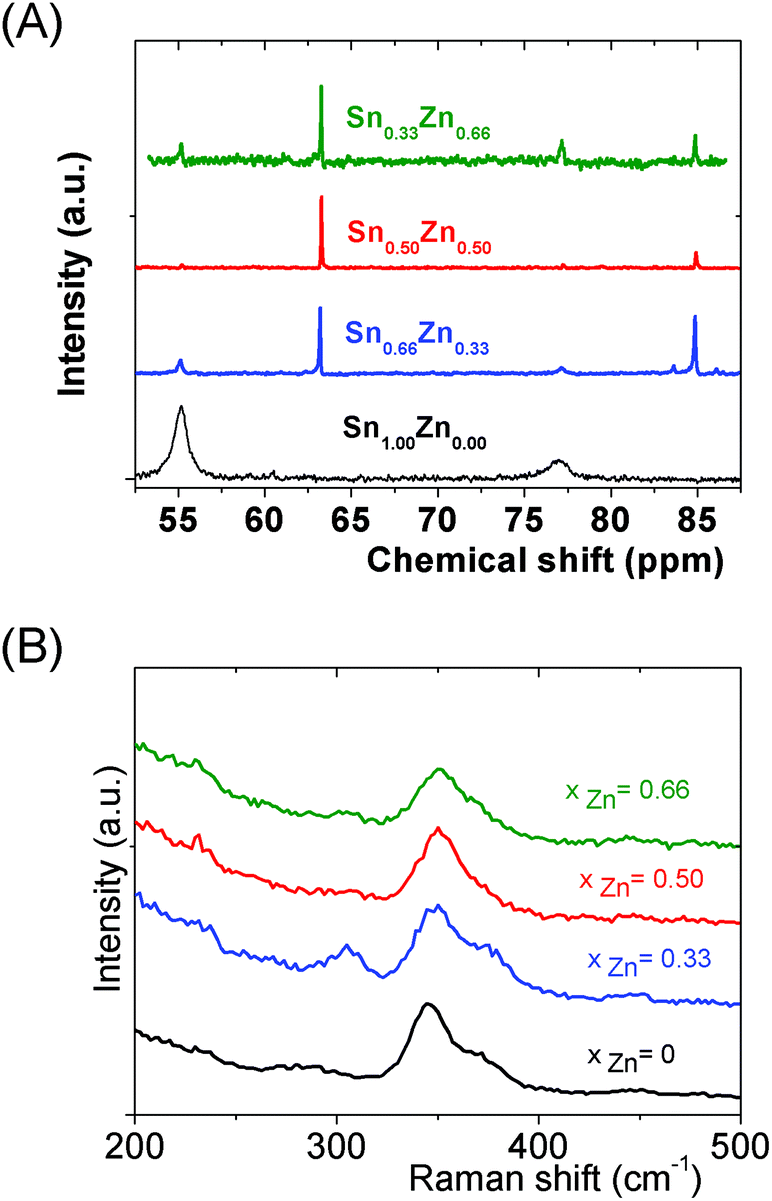 | ||
| Fig. 4 (Sna–Znb–Sc)t− oligomer solutions prepared from (Sn4+ + Zn2+) solutions, ([H+]/([Sn4+] + [Zn2+]) = 1.5), [S2−]/([Sn4+] + [Zn2+]) = 6 and ([Sn] + [Zn]) = 0.1 M. (A) 119Sn Liquid NMR spectra recorded on (Sna–Znb–Sc)t− oligomer dispersions synthesized at various xZn values, xZn = 0.00, 0.33, 0.50 and 0.66. (B) Raman spectra recorded on dispersions prepared at xZn = 0.00, 0.33, 0.50 and 0.66. | ||
The mass spectrum of the (SnaSeb)t− monometallic oligomer solution prepared at [H+]/[Sn] = 0.5, s = 3, [Sn] = 0.1 displays predominant peaks recorded at m/z = 634.4 and 1267.80 (Fig. 5a), which are respectively assigned to (Sn2Se5)2− and (Sn4Se10)4− species. Similarly to the (SnaSb)t− oligomer solution, we thus clearly demonstrate the presence of (Sn4Se10)4− oligomers in slightly basic solutions. Similarly to the Sn–Zn–S system, investigation of the Sn–Zn–Se system at various xZn values, xZn = 0.33, 0.5 and 0.66, reveals the formation of heterometallic oligomers displaying similar compositions, independently of xZn. Simultaneously with the presence of (SnSe3)2−, (Sn2Se5)2−, and (Sn4Se10)4− homometallic oligomers, (SnZnSe4)2−, (Sn3ZnSe8)2−, and (Sn3ZnSe9)4− heterometallic oligomers were identified by ESI-MS in the solutions prepared at various xZn values, xZn = 0.33 (Fig. 5b), 0.50 and 0.66. As illustrated by the isotope patterns, various heterometallic oligomers containing one Zn cation and different numbers of sulfide anions (Sn1.0Zn1.0Se4.0)2−, (Sn3.0Zn1.0Se8.0)2−, and (Sn3.0Zn1.0Se9.0)4− were clearly observed (Fig. 5 and S3c†). Nevertheless, in contrast to the tetramers, no stable structure was achieved from our DFT calculations for the (Sn1.0Zn1.0Se4)2− oligomer. Therefore, the (Sn1.0Zn1.0Se4)2− oligomer experimentally observed by mass spectrometry probably arises from fragmentation in the mass spectrometer. Raman spectra of (Sna–Znb–Sec)t− oligomers were recorded on the solution prepared at xZn = 0.33. Peaks were observed at 208 cm−1, 194 cm−1, 173 cm−1 and 162 cm−1 (Fig. 5c). Note that the adamantane structure of the monometallic tetramer (Sn4Se10)4− was previously and unambiguously determined.14 In addition, experimental Raman peaks recorded on the monometallic (SnaSec)t− oligomer observed in the region of 199 cm−1 (ν1·A1) were previously assigned43 to low molecular species such as the (SnSe4)4− monomer while peaks observed in the region of 178 cm−1 (ν2·A1) were assigned to adamantane-like tetrameric species.43 In contrast to SnS44− and ZnS vibrations, no significant overlapping exists concerning the Raman peaks of the (Sn3Zn1.0Se9)4− heterometallic selenide oligomer. In order to better clarify the structure of the latter heterometallic tetramer, Raman spectra of these oligomers were computed at a DFT level on these selenide oligomers. We report the calculated Raman shifts and scattering activities of (SnaSec)t− and (Sna–Znb–Sec)t− oligomers in Table 1.
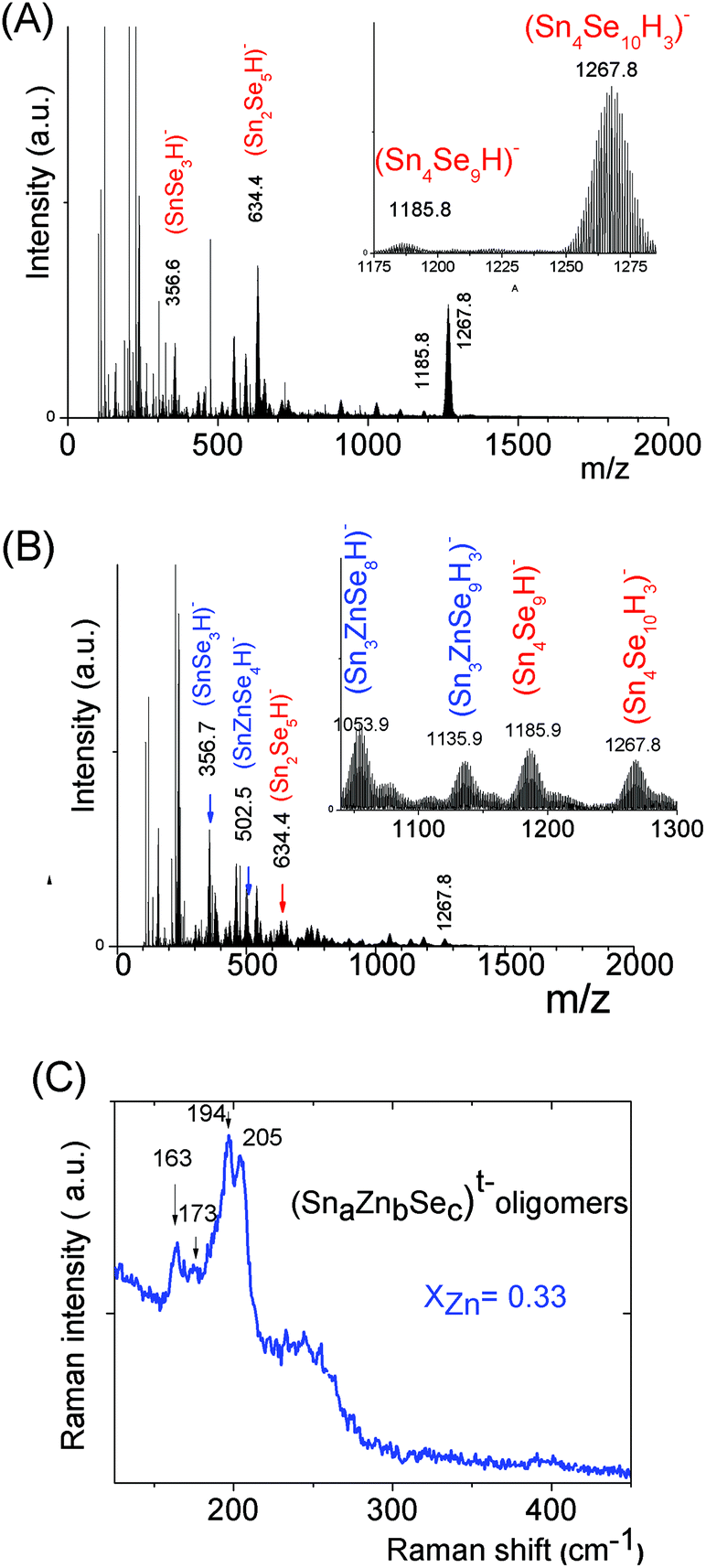 | ||
| Fig. 5 (Sna–Sec)t− and (Sna–Znb–Sec)t− oligomer solutions prepared from (Sn4+ + Zn2+) solutions, ([H+]/([Sn4+] + [Zn2+]) = 0.5), [Se2−]/([Sn4+] + [Zn2+]) = 3 and ([Sn] + [Zn]) = 0.1 M. (A) Negative ion ESI-MS spectrum of a (Sna–Sec)t− solution. (B) Negative ion ESI-MS spectrum of (Sna–Znb–Sec)t− oligomer solutions prepared at xZn = 0.33. (C) Raman spectrum of the (Sna–Znb–Sec)t− oligomer solution, xZn = 0.33. | ||
| Monomers | Frequency, cm−1 (Raman scattering activity >20 Å4 amu−1) |
|---|---|
| Oligomers | |
| (SnSe3)2− | 208(294), 264(34), 264(35) |
| Main peak: 208 | |
| (SnSe4)4− | 184(318), 210(44), 211(44), 212(44) |
| (Sn2Se5)2− | 49.3(21), 58.6(24), 102.1(39), 165.0(81), 211.5(346), 223.3(74), 263.2(32), 297.7(38) |
| Main peak: 212 | |
| (Sn4Se10)4− linear | 201, 256 |
| ada-(Sn4Se10)4− | 43.6(20), 68.4(50), 68.8(50), 69.0(54), 101.5(211), 173.5(589), 220.5(43), 221.0(43), 221.2(43), 247.0(50), 247.2(53), 248.3(50), 253.3(189) |
| Main peak: 173 | |
| (SnZnSe4)2− | Not stable molecular structure; dissociative |
| ada-(Sn3ZnSe9)4− | 73.2(27), 74.8(26), 77.7(46), 95.1(44), 118.5(62), 173.1(499), 208.6(40), 209.6(44), 221.8(25), 227.4(148), 245.3(41), 246.6(39), 247.5(89) |
| Main peak: 173 |
From our calculated Raman shifts, some assignments of the various experimental Raman peaks recorded on the (SnaZnbSec)t− oligomer solutions could be made:
- Main vibrations of low molecular mass species were determined from our calculations in the 205 cm−1–215 cm−1 frequency region: (SnSe3)2− at 208 cm−1 and (Sn2Se5)2− at 212 cm−1. Experimental peaks respectively observed at 194 cm−1 and 205 cm−1 were thus respectively assigned to (SnSe3)2− and (Sn2Se5)2− species, clearly identified on the ESI-MS spectra.
- Vibrations associated with the linear (Sn4Se10)4− structure were determined from our DFT calculations at 201 cm−1 while the main vibration of the (Sn4Se10)4− tetramer adamantane-like structure was determined in the region 170–180 cm−1, corroborating the data previously reported in the literature.43 From these observations, experimental peaks observed in the region 160–180 cm−1 clearly suggest the formation of tetrameric heterometallic oligomers displaying an adamantane-like structure. Although our DFT calculations show that the incorporation of one Zn cation into the adamantane-like tetrameric structure does not result in a significant variation of the main Raman peak frequency, we tentatively assign the peaks experimentally observed at 173 and 162 cm−1 to the non-substituted (Sn4Se10)4− and Zn-substituted (Sn3ZnSe9)4− adamantane-like oligomers. The adamantane-like structures of the (Sn3ZnSe8)2− and (Sn3ZnSe9)4− oligomers as optimized by DFT calculations are reported in Fig. 6.
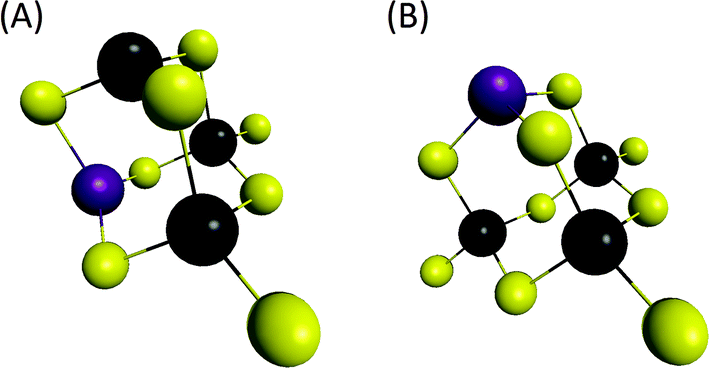 | ||
| Fig. 6 Oligomer computed structures determined by DFT calculations. Yellow: Se, black: Sn, violet: Zn. (A) (Sn3ZnSe8)2−. (B) (Sn3ZnSe9)4−. | ||
Heterometallic Sn(IV)–Ga(III) chalcogenide oligomers
In contrast to the formation of (Sna–Znb–Sc)t− oligomers, soluble (Sna–Gab–Sc)t− oligomer solutions were achieved in the whole range of xGa yielding a larger oligomer formation domain (Fig. 2). Similarly to the Sn(IV)–Zn(II)–S(II) system, more highly concentrated (Sna–Gab–Sc)t− oligomer solutions were observed for compositions close to the most soluble (SnaSc)t− oligomer. Indeed, oligomer solutions with concentrations up to ([Sn] + [Ga]) > 0.4 M were prepared in the presence of NH4Cl by-products for xGa values up to xGa = 0.5 while oligomer solutions prepared at higher xGa values exhibit a much lower ([Sn] + [Ga]) concentration.An ESI-MS investigation performed on various (Sna–Gab–Sc)t− oligomer solutions, 0.33 ≤ xGa ≤ 0.80, clearly indicates that these solutions contain oligomer distributions (Fig. 7). Interestingly, the composition and concentration of these oligomers depend on the xGa molar fraction of the oligomer dispersion. In contrast to the (Sna–Znb–Sc)t− oligomer solutions, a very low proportion of monometallic oligomers containing exclusively Ga(III) cations, i.e. (GaS2)−, (Ga2S4)2− or Sn(IV) cations (Sn4S10)4−, was determined from the relative intensities of the ESI-MS peaks. More importantly, a larger proportion of heterometallic oligomers including (Ga3SnS8)3−, (Ga2Sn2S8)2−, (Ga2Sn2S9)4−, (Ga2Sn2S10)6−, and (GaSn3S10)5− was detected by ESI-MS in these solutions (Fig. 7 and S3d†). It is worth mentioning that these (GaaSnbSc)t− heterometallic oligomers are less polymerized compared to the monometallic (GaaSc)t− oligomers synthesized under similar conditions. Indeed, these heterometallic oligomers are shown to be mainly composed of four metal centers incorporating up to three Ga(III) cations.
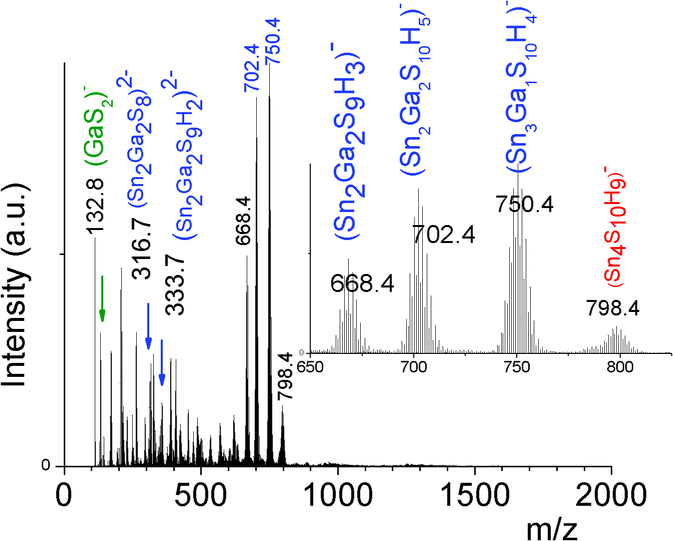 | ||
| Fig. 7 Negative ion ESI-MS spectrum of (Sna–Gab–Sc)t− oligomer solutions prepared from (Sn4+ + Ga3+) solutions, ([H+]/([Sn4+] + [Ga3+]) = 0) at xGa = 0.5, [S2−]/([Sn4+] + [Ga3+]) = 6 and ([Sn] + [Ga]) = 0.1 M showing the presence of heterometallic tetrameric oligomers. | ||
We can note that the large variety of heterometallic tetrameric oligomers (Ga3SnS8)3−, (Sn3GaS10)5−, and (Sn2Ga2S10)6− possessing various Ga(III) cation and sulfide anion contents display various charges, thus allowing charge control by a fine tuning of the xGa value. Liquid 119Sn NMR spectra recorded on the (Sna–Gab–Sc)t− oligomer solutions display similar features to those observed on (Sna–Znb–Sc)t− oligomer solutions. Peaks at δ = 55 ppm and δ = 77 ppm and respectively assigned to the monometallic (Sn2S6)4− and SnS44−/SnS32− species were observed on the sample synthesized at xGa = 0.33. Consistently, a significant decrease of the intensity of these peaks is observed with increased number of Ga(III) atoms in the heterometallic tetramers. More interestingly, as for (Sna–Znb–Sc)t− oligomer solutions, numerous additional peaks which were not observed on monometallic (Sna–Sc)t− oligomers were clearly detected at δ = 82/85 ppm on heterometallic (Sna–Gab–Sc)t− oligomer solutions (Fig. 8a). Compared with the spectra recorded on (Sna–Znb–Sc)t− oligomer solutions, the larger number of peaks appearing in the chemical shift range 82/85 ppm highlights the higher number of Sn environments observed in (Sna–Gab–Sc)t− oligomers. Fig. 8b shows Raman spectra recorded on various (Sna–Gab–Sc)t− oligomer solutions (xGa = 0.00, 0.33, 0.5, 0.66 and 1.00) prepared under slightly basic conditions. Fine inspection of these spectra suggests a solid solution behavior involving two non-interacting vibration modes. Although the Raman behavior of solid solution is quite complicated, polar modes usually transfer their intensities44 while both the position and intensities of transverse and longitudinal modes in solid solution are linked.45Fig. 8b shows a continuous variation of both the Raman intensity and the Raman shifts of the Ga–S vibration (310–320 cm−1) and of the Sn–S vibration (345 cm−1) observed for the whole range of solid solutions. Ge4S104−, Ge4Se104−, Sn4S104−, and Sn4Se104− adamantane-like structures were previously reported as the predominant condensation structures in monometallic oligomer solutions of lower pH values.14 To determine the structure of the heterometallic chalcogenide tetramers in the Sn(IV)–Ga(II)–S(II) system, Raman spectra of (Sn4−xGaxS10)4− adamantane-like species were thus computed at a DFT level. Table S5† reports the calculated Raman frequency of the main peak for various species, 0 ≤ xGa ≤ 1. The frequency of the main peak has been determined by convoluting Lorentzian functions whose amplitudes are proportional to computed Raman activity intensities and a half-band width of 20 cm−1. Although the main peak values are overestimated, these calculations clearly show a lower frequency of the main peak for the (Ga4S10)4− adamantane tetramer compared with the (Sn4S10)4− tetramer. Thus, experimentally determined Raman shifts on the (Ga4S10)4− and (Sn4S10)4− adamantane tetramer solutions are consistent with the DFT results. Although more data including dynamic effective charge and intensity transfer between polar modes are needed to propose a more complete model, the continuous variation of the Raman shifts experimentally observed in the whole range of solid solutions demonstrates that the various (Sna–Gab–Sc)t− heterometallic oligomers preserve the adamantane structure. The structures of the (Sn3GaS10)5− (Sn2Ga2S10)6− and (Sn2Ga2S9)4− oligomers determined by DFT are reported in Fig. 9.
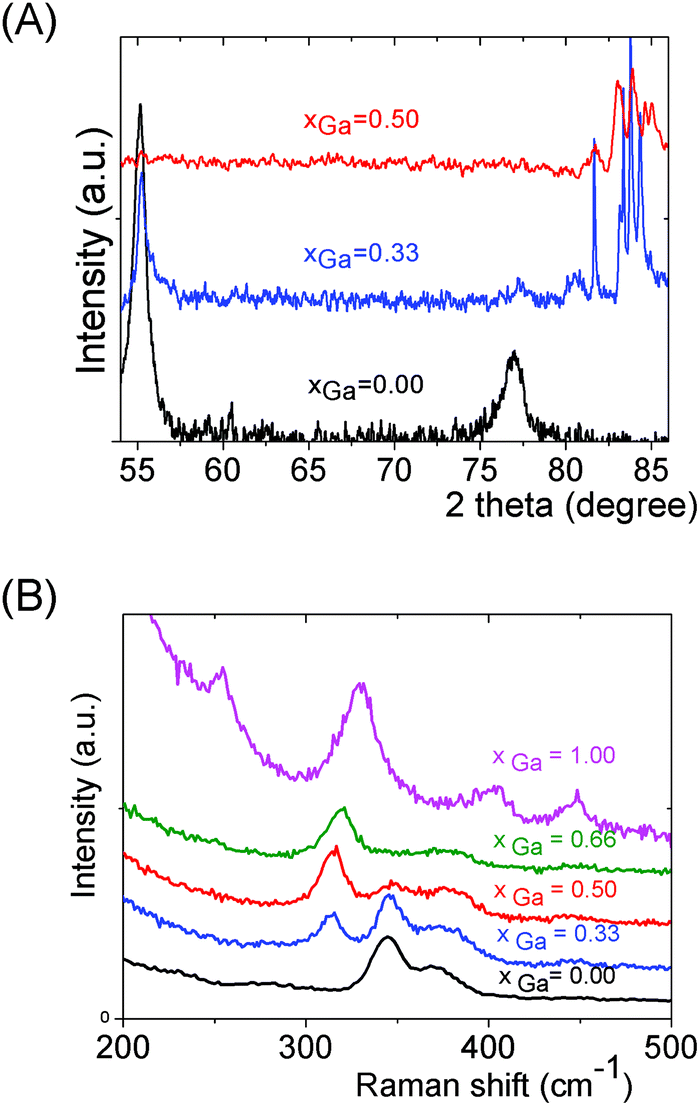 | ||
| Fig. 8 (Sna–Gab–Sc)t− oligomer solutions at various xGa values prepared by the addition of a (Sn4+ + Ga3+) solution ([H+]/([Sn4+] + [Ga3+]) = 0) into a (NH4)2S solution at [S2−]/([Sn4+] + [Ga3 + ]) = 6 and ([Sn] + [Ga]) = 0.1 M. (A) 119 Sn liquid NMR spectra. (B) Raman spectra. Spectra of (Sna–Sc)t− and (Gab–Sc)t− monometallic oligomer solutions are also reported. | ||
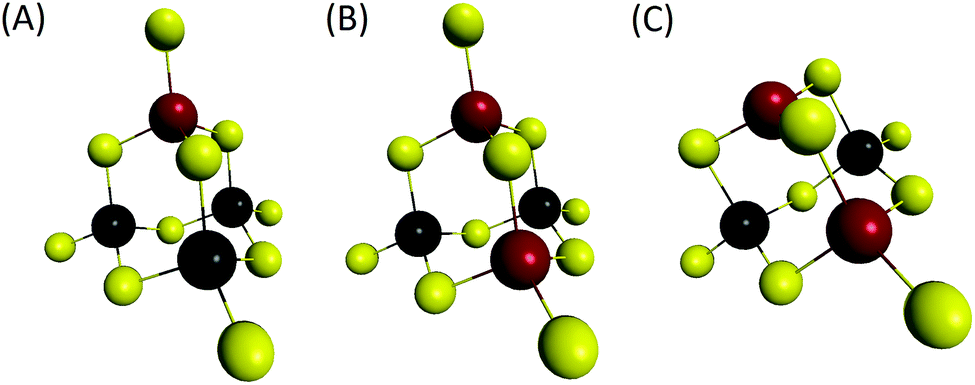 | ||
| Fig. 9 Computed structures. Yellow: S, black: Sn, red: Ga. (A) (Sn3GaS10)5−, (B) (Sn2Ga2S10)6− and (C) (Sn2Ga2S9)4− oligomers. | ||
Heterometallic (Sna–Gab–Sc)t− oligomers were prepared by our mild process route. Incorporation of a tetravalent cation such as Sn(IV) into the Ga(III) oligomers allows the formation of a series of short, discrete, highly charged tetrameric adamantane oligomers. These heterometallic oligomers were fully characterized by ESI-MS, Raman and 119Sn liquid NMR spectroscopy.
Discussion
Heterometallic chalcogenide oligomers as environmentally-friendly building blocks for solar cells
Successful development1,2 of hydrazine-based metal chalcogenide complexes relies on its easy decomposition to the parent metal chalcogenides without any impurities. Charged, water-soluble, organic-free, heterometallic chalcogenide oligomers involving a residue-free, ammonium cation described here are extremely attractive from an environmental perspective (Fig. S6†). Indeed, the (Sna–Znb–Sc)t− heterometallic oligomers or (Sna–Znb–Sc)t−-capped ZnS colloids could provide exciting opportunities as building blocks for the design of an all-aqueous process route for low-cost photovoltaic device fabrication. These all-inorganic heterometallic chalcogenide oligomers or ultrafine colloids could serve as dispersants or composition control additives.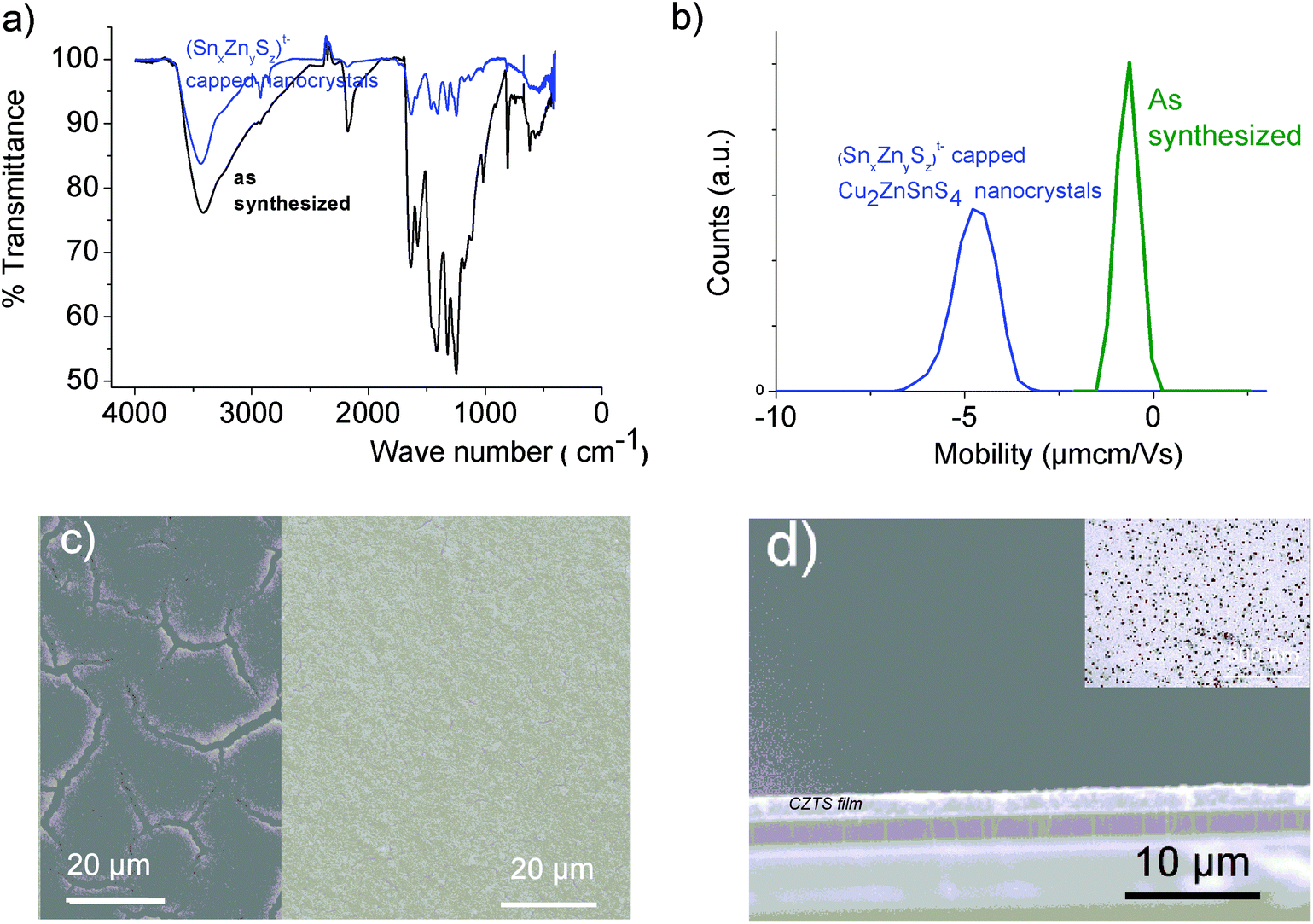 | ||
| Fig. 10 (a) FTIR spectra of 30 nm, SCN− capped (as-synthesized) and (SnaZnbSc)t− oligomers-capped Cu2ZnSnS4 nanocrystals. (b) Electrophoretic mobility measured for an aqueous solution of 30 nm Cu2ZnSnS4 nanocrystals capped with (SnaZnbSc)t− oligomers. (c) Cu2ZnSnS4 nanoparticle film prepared from an aqueous dispersion on the Mo electrode. Left: SEM image of a single-coated film reveals the presence of large cracks. Right: SEM image showing a crack-free film achieved from oligomer-capped CZTS nanocrystals using a multi-coating procedure. (d) Typical cross section SEM image of a crack-free CZTS film showing a thickness of 1.5 μm. Insert: TEM image of the Cu2ZnSnS4 nanocrystal building blocks showing a nanocrystal mean diameter of around 30 nm. | ||
Conclusions
A general synthetic process route under mild conditions is proposed for the preparation of heterometallic chalcogenide oligomers in Sn(IV)–Zn(II)–S(II), Sn(IV)–Zn(II)–Se(II) and Sn(IV)–Ga(III)–S(II) systems. ESI-MS, used as an in situ characterization tool, indicates predominant formation of heterometallic oligomers composed of four metal centers in the Sn(IV)–Zn(II)–S(II) and Sn(IV)–Ga(III)–S(II) systems. 119Sn Liquid NMR and Raman experimental data determined on these heterometallic oligomers show a full range of solid solutions achieved for (Sna–Gab–Sc)t− oligomers, while the incorporation of a sole Zn cation is observed for (Sna–Znb–Sc)t− oligomers. The heterometallic approach favors the formation of short oligomers, resulting in highly water-soluble species displaying an adamantane structure as shown by the Raman results coupled to quantum chemical calculations. Such low-cost, all-inorganic, environmentally friendly oligomers in association with a residue-free ammonium counterion can potentially be used as chalcogenide precursors in environmentally benign synthetic routes to the formation of electronic grade films. Notably, these heterometallic oligomers represent exciting building blocks for CZTS solar cell fabrication. Indeed, (Sna–Znb–Sc)t− oligomers, used as dispersants and composition additives, can greatly help in the design of a nontoxic, environmentally friendly, aqueous process route to the fabrication of CZTS absorbers. Using these heterometallic chalcogenide oligomers, a CZTS solar cell has been successfully fabricated with a preliminary device efficiency of 2.6%.Extension to other systems including Sn(IV)–Ge(IV)–S(II), Sn(IV)–Sb(III)–S(II), and Sn(IV)–Fe(III)–S(II) could open up exciting opportunities for designing new complex chalcogenide materials for applications in electronic devices and thermoelectric materials.
Acknowledgements
This work was mainly supported by funding from the Region Reunion. The authors acknowledge ANR (Agence Nationale de la Recherche, NovACEZ Project) for the development of CZTS solar cells. The authors thank Alain Seraphine for helpful discussions.References
- D. B. Mitzi, L. L. Kosbar, C. E. Murray, M. Copel and A. Afzali, Nature, 2004, 428, 299–303 CrossRef CAS PubMed.
- D. K. Kim, Y. Lai, B. T. Diroll, C. B. Murray and C. R. Kagan, Nat. Commun., 2012, 3, 1216 CrossRef PubMed.
- C. Jiang, J.-S. Lee and D. V. Talapin, J. Am. Chem. Soc., 2012, 134, 5010–5013 CrossRef CAS PubMed.
- D. Yang, C. Lu, H. Yin and I. P. Herman, Nanoscale, 2013, 5, 7290–7296 RSC.
- M. V. Kovalenko, B. Spokoyny, J.-S. Lee, M. Scheele, A. Weber, S. Perera, D. Landry and D. V. Talapin, J. Am. Chem. Soc., 2010, 132, 6686–6695 CrossRef CAS PubMed.
- M. Ibáñez, D. Cadavid, R. Zamani, N. García-Castelló, V. Izquierdo-Roca, W. Li, A. Fairbrother, J. D. Prades, A. Shavel, J. Arbiol, A. Pérez-Rodríguez, J. R. Morante and A. Cabot, Chem. Mater., 2012, 24, 562–570 CrossRef.
- J. S. Lee, M. V. Kovalenko, J. Huang, D. S. Chung and D. V. Talapin, Nat. Nanotechnol., 2011, 6, 348–352 CrossRef CAS PubMed.
- D. S. Dolzhnikov, H. Zhang, J. Jang, J. S. Son, M. G. Panthani, T. Shibata, S. Chattopadhyay and D. V. Talapin, Science, 2015, 347, 425–428 CrossRef CAS PubMed.
- H. Li, A. Laine, M. O. Keefe and O. M. Yaghi, Science, 1999, 283, 1145–1147 CrossRef CAS PubMed.
- T. Wu, X. Bu, P. Liao, L. Wang, S.-T. Zheng, R. Ma and P. Feng, J. Am. Chem. Soc., 2012, 134, 3619–3622 CrossRef CAS PubMed.
- V. B. Krebs, S. Pohl and W. Schiwy, Z. Anorg. Allg. Chem., 1972, 393, 241–352 CrossRef.
- W. Schiwy, S. Pohl and B. Krebs, Z. Anorg. Allg. Chem., 1973, 402, 77–86 CrossRef CAS.
- S. Pohl, W. Schiwy, N. Weinstock and B. Krebs, Z. Naturforsch., 1973, 28b, 565–569 Search PubMed.
- B. Krebs, Angew. Chem., Int. Ed. Engl., 1983, 22, 113–134 CrossRef.
- B. Krebs, D. Voelker and K. O. Stiller, Inorg. Chim. Acta, 1982, 65, L101–L102 CrossRef CAS.
- M. V. Kovalenko, M. I. Bodnarchuk, J. Zaumseil, J.-S. Lee and D. V. Talapin, J. Am. Chem. Soc., 2010, 132, 10085–10092 CrossRef CAS PubMed.
- M. V. Kovalenko, M. Scheele and D. V. Talapin, Science, 2009, 324, 1417–1420 CrossRef CAS PubMed.
- K. Ito and T. Nakazawa, Jpn. J. Appl. Phys., 1988, 27(11), 2094–2097 CrossRef CAS.
- H. Katagiri, N. Sasaguchi, S. Hando, S. Hoshino, J. Ohashi and T. Yokota, Sol. Energy Mater. Sol. Cells, 1997, 49, 407–414 CrossRef CAS.
- Y. S. Lee, T. Gershon, O. Gunawan, T. K. Todorov, T. Gokmen, Y. Virgus and S. Guha, Adv. Energy Mater., 2015, 5, 1401372 Search PubMed.
- S. Ahmed, K. B. Reuter, O. Gunawan, L. Guo, L. T. Romankiw and H. Deligianni, Adv. Energy Mater., 2012, 2(2), 253–259 CrossRef CAS.
- W. Wang, M. T. Winkler, O. Gunawan, T. Gokmen, T. K. Todorov, Y. Zhu and D. B. Mitzi, Adv. Energy Mater., 2013, 1, 1301465 Search PubMed.
- Y. Cao, M. S. Denny, J. J. Caspar, W. E. Farneth, Q. Guo, A. S. Ionkin, L. K. Johnson, M. Lu, I. Malajovich, D. Radu, H. D. Rosenfeld, K. R. Choudhury and W. Wu, J. Am. Chem. Soc., 2012, 134(38), 15644–15647 CrossRef CAS PubMed.
- J. V. Embden, A. S. R. Chesman, E. D. Gaspera, N. W. Duffy, S. E. Watkins and J. J. Jasieniak, J. Am. Chem. Soc., 2014, 136, 5237–5240 CrossRef PubMed.
- G. Larramona, S. Bourdais, A. Jacob, C. Chone, T. Muto, Y. Cuccaro, B. Delatouche, C. Moisan, D. Pere and G. Dennler, J. Phys. Chem. Lett., 2014, 5, 3763–3767 CrossRef CAS PubMed.
- J. Zhong, Z. Xia, C. Zhang, B. Li, X. Liu, Y. B. Cheng and J. Tang, Chem. Mater., 2014, 26(11), 3573–3578 CrossRef CAS.
- J. Y. Chane-Ching, V. Foncrose, O. Zaberca, D. Lagarde, A. Balocchi, X. Marie, T. Blon, P. Puech and R. Bodeux, Sol. Energy Mater. Sol. Cells, 2015, 141, 364–371 CrossRef CAS.
- A. Carrete, A. Shavel, X. Fontané, J. Montserrat, J. Fan, M. Ibáñez, E. Saucedo, A. Pérez-Rodríguez and A. Cabot, J. Am. Chem. Soc., 2013, 135(43), 15982–15985 CrossRef CAS PubMed.
- X. Y. Shi, F. Q. Huang, M. L. Liu and L. D. Chen, Appl. Phys. Lett., 2009, 94, 122103 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter, 1992, 46, 6671–6687 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. Bergner, M. Dolg, W. Kuechle, H. Stoll and H. Preuss, Mol. Phys., 1993, 80, 1431–1441 CrossRef CAS.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- T. Leininger, A. Berning, A. Nicklass, H. Stoll, H.-J. Werner and H.-J. Flad, Chem. Phys., 1997, 217, 19–27 CrossRef CAS.
- J. M. L. Martin and A. Sundermann, J. Chem. Phys., 2001, 114, 3408–3420 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5646 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- X. Bu, N. Zheng and P. Feng, Chem. – Eur. J., 2004, 10, 3356–3362 CrossRef CAS PubMed.
- B. R. Tagirov, O. M. Suleimenov and T. M. Seward, Geochim. Cosmochim. Acta, 2007, 71, 4942–4953 CrossRef CAS.
- Z. Y. Xu and Y. C. Zhang, Mater. Chem. Phys., 2008, 112, 333–336 CrossRef CAS.
- O. Brafman and S. S. Mitra, Phys. Rev., 1968, 171, 931–934 CrossRef CAS.
- J. Campbell, D. P. Diciommo, H. P. A. Mercier, A. M. Pirani, G. J. Schrobilgen and M. Willuhn, Inorg. Chem., 1995, 34, 6265–6272 CrossRef CAS.
- S. M. Shapiro and J. D. Axe, Phys. Rev. B: Condens. Matter, 1972, 6, 2420 CrossRef CAS.
- G. Landa, R. Carles and J. B. Renucci, Solid State Commun., 1993, 86, 351–355 CrossRef CAS.
- F. Lips and S. Dehnen, Inorg. Chem., 2008, 47, 5561–5563 CrossRef CAS PubMed.
- A. Nag, M. V. Kovalenko, J. S. Lee, W. Liu, B. Spokoyny and D. V. Talapin, J. Am. Chem. Soc., 2011, 133, 10612–10621 CrossRef CAS PubMed.
- S. Chen, X. G. Gong, A. Walsh and S. H. Wei, Appl. Phys. Lett., 2010, 96, 021902–021903 CrossRef.
- L. Choubrac, A. Lafond, C. Guillot-Deudon, Y. Moelo and S. Jobic, Inorg. Chem., 2012, 51(6), 3346–3348 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qi00250h |
| This journal is © the Partner Organisations 2016 |