
Sustainable development of a surface-functionalized mesoporous aluminosilicate with ultra-high ion exchange efficiency†
Pejman
Hadi
a,
Chao
Ning
a,
James D.
Kubicki
b,
Karl
Mueller
cd,
Jonathan W.
Fagan
c,
Zhengtang
Luo
a,
Lutao
Weng
ae and
Gordon
McKay
*af
aChemical and Biomolecular Engineering Department, Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong SAR, China
bDepartment of Geological Sciences, University of Texas at El Paso, El Paso, Texas 79968, USA
cDepartment of Chemistry, The Pennsylvania State University, University Park, PA 16802, USA
dPhysical and Computational Sciences Directorate, Pacific Northwest National Laboratory, Richland, WA 99352, USA
eMaterials Characterization and Preparation Facility, Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China
fDivision of Sustainable Development, College of Science, Engineering and Technology, Hamad Bin Khalifa University, Qatar Foundation, Doha, Qatar. E-mail: kemckayg@ust.hk; Fax: +852 23580054; Tel: +852 23588412
First published on 6th January 2016
Abstract
The present work employs a facile hydroxylation technique to efficiently functionalize the surface of a waste-derived aluminosilicate for ultra-high heavy metal uptake via ion exchange. The functionalization process leads to the transformation of a nonporous hydrophobic waste material to a mesoporous hydrophilic material with a high concentration of ion exchange sites. The modification of the surface and textural characteristics of the mesoporous aluminosilicate has been thoroughly elucidated. The functionalization brings about the partial depolymerization of the aluminosilicate network and the transformation of unreactive bridging oxygens (BO) into non-bridging oxygens (NBO) as active sites as evidenced by 29Si NMR and FTIR. The positively-charged alkali metals bound to the NBO act as facile ion exchange sites. Ultra-high heavy metal uptake capacity of the functionalized material through a combination of ion exchange and physisorption mechanisms has revealed the great potential of this aluminosilicate material for treatment of heavy metal-laden wastewater in a sustainable manner for practical applications.
1. Introduction
The development, structural modification and surface functionalization of aluminosilicate-based materials has sparked great interest in the last few years due to the extensive applications of these materials as catalysts, catalyst supports, reactive fillers and adsorbents.1–7Aluminosilicate polymers generally consist of a continuous network of tetrahedrally-coordinated T–O linkages (where T = Si or Al) connected via bridging oxygens (BO).8,9 The number of these bridging oxygens located at the vertex of each tetrahedron describe the connectivity of these tetrahedra, where Qn (n = 0–4) denotes a tetrahedron with “n” bridging oxygens.10–13 The cleavage of these tetrahedra proceeds via the formation of non-bridging oxygens (NBO) which are favored in most applications due to their high activity and the possibility of interaction with other desired species. Therefore, the Q4 configuration represents the inactive unit, whereas Q3 and Q2 units, surrounded by 1 and 2 non-bridging oxygens, respectively, represent the active entities (T–OH and T–O−) within an aluminosilicate.14–16
One of the useful characteristics of these inorganic polymers is the existence of exchangeable metals (more specifically Na+ or Ca2+) in their network structure.17 These alkali or alkaline earth metals, bound to the AlO4− tetrahedra in the aluminosilicate framework, function as charge compensators.18–20 According to Bouyer et al.21 it is plausible to change the role of these metal ions from charge-balancing species to network modifiers by the hydrolysis of T–O–T′ linkages (where T, T′ = Si and Al). This hydrolysis reaction takes place by the disruption of the bridging oxygen in T–O–T′ tetrahedral unit, generating T–OH and T–O− bonds, depending on stoichiometry or reaction conditions. Because of the opening of the tetrahedral network, the charge-carrying cation can migrate from the framework to the surface and can be replaced by heavy metals if the appropriate conditions are met. Such exchange is augmented if the hydrolyzed materials have an increased surface area (i.e. higher porosity) than the parent material. The resultant aluminosilicate materials that exhibit high ion-exchange capability can have widespread applications in catalysis and wastewater treatment.22
Hydrolysis of siloxane species in aluminosilicate materials has been the subject of many investigations. Many researchers have proposed the depolymerization model of T–O–T′ in the presence of water in aluminosilicate glasses. Nevertheless, Kohn et al.23 have debated this hydrolysis model and have fundamentally ruled out the formation of terminal T–OH groups by the cleavage of T–O–T′ clusters via hydrolysis reactions. Alternatively, they have proposed the formation of protonated bridging oxygen groups (T–O(H)–T′) when the aluminosilicate comes in contact with water.24 It should be noted that there is no universal hydrolytic model that demonstrates the correct set of intermediates and products for all types of hydrolysis and all types of aluminosilicates.25,26 Although hydrolysis reactions, controversially transforming Q4 to Q3 configuration, are central in geochemistry and the glass industry, it cannot be a reliable technique to functionalize the aluminosilicate for ion exchange-based applications that require a large number and homogeneous distribution of terminal active sites (e.g. catalysis and adsorption) considering the trace amount of functional moieties developed via this technique.22
Hydroxylation is an alternative technique that can markedly increase the density of NBOs in aluminosilicates via the transformation of Q4 to Q3 units and, concomitantly, change the position of the alkali metals from charge-balancing species to network modifiers as discussed.27 In an alkaline medium, the T–O–T′ linkages undergo hydroxylation forming T–OH and T–O− species, locally charge-balanced by the cation of the alkaline medium (M), as depicted in reaction (R1).
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) T–O–T′+M–OH → T–OH+T′–O−M+ T–O–T′+M–OH → T–OH+T′–O−M+ | (R1) |
Ion exchange capability for aluminosilicates is a direct function of the metal ions in the position of network modifiers and thus the concentration of NBO sites in the aluminosilicate structure.28 Hence, the aim of this study is to investigate the partial heterolytic cleavage of the T–O–T′ linkages of a waste-derived calcium aluminosilicate material subjected to hydroxylation reactions for enhanced heavy metal deposition via ion exchange. Further, the investigation utilizes nuclear magnetic resonance (NMR) spectroscopy, Fourier-transform infrared (FTIR) spectroscopy, X-ray photoelectron spectroscopy (XPS) and time-of-flight secondary ion mass spectrometry (TOF-SIMS) to determine the changes in the structure of the materials. Moreover, the specific surface areas of the modified aluminosilicate materials, indicative of the tetrahedral unit opening, have been evaluated, because all these properties (i.e. specific surface area, concentration of the NBO and density of the alkali metal ions) are interlinked. Notably, it will be advantageous, in future studies, to study the structural alterations of a homogeneous aluminosilicate synthesized in more controlled conditions to shed light on the functionalization process.
2. Experimental
2.1 Material
Waste printed circuit boards were first shredded into small pieces (1–2 cm size) and then the shredded pieces were ground and pulverized into powder using a crusher. The powder-form material obtained was a mixture of metal (MF) and nonmetal fractions (NMF). The NMF, as the precursor used in this study, was separated from the powder mixture using a high efficiency corona electrostatic separator. The average particle diameter of the NMF was 2 μm with a particle size distribution illustrated in Fig. S1.†2.2 Functionalization of the sample
The surface properties of the original waste-derived aluminosilicate-based material (NMF) were modified using a procedure described elsewhere with slight modifications.29 Briefly, NMF was added to a 1 M KOH solution, as hydroxylating agent, with agitation for 1 h. The impregnation ratio was fixed at 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 w/w, where impregnation ratio is defined as the weight ratio of the activating agent to the precursor. The resultant solution was then activated in a reactor at 573 K for 1 h. A continuous stream of nitrogen gas was maintained throughout the reaction in order to avoid any involvement of external oxygen in the surface properties of the aluminosilicate. The functionalized product (A-NMF) was repeatedly washed with hot and cold deionized water to remove excess activating agent and then freeze-dried to inhibit the re-combination of functional groups and pores that develop during the processing.
:1 w/w, where impregnation ratio is defined as the weight ratio of the activating agent to the precursor. The resultant solution was then activated in a reactor at 573 K for 1 h. A continuous stream of nitrogen gas was maintained throughout the reaction in order to avoid any involvement of external oxygen in the surface properties of the aluminosilicate. The functionalized product (A-NMF) was repeatedly washed with hot and cold deionized water to remove excess activating agent and then freeze-dried to inhibit the re-combination of functional groups and pores that develop during the processing.
2.3 Heavy metal uptake
Studies of the uptake of heavy metals were performed by placing a known amount of the functionalized material (A-NMF) in a batch reactor containing various concentrations of heavy metal species and stirring for 6 h at a fixed temperature of 25 °C. The pH of the heavy metal solutions was adjusted to 5 using 0.5 M HNO3 and NaOH to remove any possibility of metal species precipitation. Also, the pH change during the whole adsorption experiments has been monitored to ascertain that the pH values are below the precipitation pH. The driving force for the heavy metal uptake is the chemical potential difference between the light and heavy metals at the atomic level. The degree of uptake was determined by the mass balance of the heavy metal in the solution phase and the solid phase. After the uptake was accomplished, the heavy metal-doped material (Me-A-NMF) was washed several times with deionized water to remove any unbound species and then freeze-dried for further characterization.2.4 Characterization
X-ray fluorescence spectroscopy (XRF) was performed on a JSX-3201Z (JEOL). Specific surface areas and pore size distributions of the samples were recorded on a Beckman Coulter SA3100 surface area analyzer using the Brunauer–Emmett–Teller (BET) and Barret–Joyner–Halenda (BJH) equations, respectively. The X-ray photoelectron spectroscopy (XPS) analyses were carried out on a PHI 5600 spectrometer equipped with a monochromatic Al Kα X-ray source of 1486.6 eV excitation energy; the binding energy of C 1s at 284.6 eV was used as the reference to correct the spectra. Fourier transform infrared (FTIR) spectra were obtained using a Vertex 70 Bruker FTIR spectrometer with a resolution of 4 cm−1 by thin KBr pellet method. Inductively-coupled plasma atomic emission spectroscopy (ICP-AES), Optima 7300 DV Perkin-Elmer, was used to determine the concentrations of various metal ions in the aqueous medium. ToF-SIMS measurements were conducted with an ION-TOF V spectrometer with a pulsed Bi3+ ion beam (25 keV). The sputter etching was performed using a beam of 3 keV Cs+ to obtain the desired depth profile. Scanning electron micrographs of the samples, coated with a thin layer of gold via sputtering technique, were recorded by a JEOL JSM-6700F scanning electron microscope (SEM) at an operating voltage of 20 kV equipped with an energy dispersive X-ray spectrometer.For solid-state 29Si magic angle spinning (MAS)-NMR spectroscopy, NMF and A-NMF samples were packed into 5 mm zirconium oxide rotors. Direct polarization (DP-) and cross polarization magic angle-spinning (CP) MAS 29Si NMR spectra were obtained at ambient temperature using an NMR spectrometer comprising a Tecmag Apollo console and a wide-bore superconducting NMR magnet with a static field strength of 9.4 T. Spectrometer frequencies of 399.999 MHz and 79.466 MHz were utilized for 1H and 29Si irradiation. The MAS rates were 10 kHz and 7 kHz for the DP- and CP-MAS experiments, respectively. Acquisitions for the DP experiments were accomplished with a standard DP sequence using a 7 μs 29Si 90° pulse and a 60 s recycle delay for 1k transients. The CP experiments were accomplished with a standard CP sequence using a 7 μs 1H 90° pulse, a 4 ms contact pulse, and a 6 s recycle delay for 2k transients. 29Si chemical shifts are reported relative to tetramethylsilane [(CH3)4Si] by assignment of the central silane in tetrakis(trimethylsilyl)silane [(CH3Si)4Si] at −135.5 ppm as an external secondary standard.
3. Results and discussion
3.1 Hydroxylation of the aluminosilicate
The waste-derived NMF is an amorphous material, as revealed by XRD analysis (see Fig. S2†), and is composed of randomly-oriented calcium aluminosilicate fibers partially covered with a carbonaceous material. The inhomogeneous structure of the NMF has been illustrated in Fig. 1. Also, the chemical composition of the NMF is provided in Table S1.† The structure of this material mainly consists of a network of Q4 units. As discussed earlier, Q4 species imply a tetrahedrally coordinated polymeric framework of alternating silicon and BO.30 Although the BOs have lone pairs of electrons and can exert weak electrostatic attraction with positively-charged ions, they cannot participate in covalent or ionic bonding. Therefore, a functionalization method has been carried out in order to increase the density of the NBO and thus “network modifying” cations for enhanced ion exchange capability. This functionalization approach has resulted in the conversion of the hydrophobic nonporous material into a hydrophilic porous material via the cleavage of the Q4 into Q3 and Q2 species (see Fig. 2). | ||
| Fig. 1 SEM images of the (a) parent and (b) functionalized materials. The SEM images of the parent material show that the NMF is composed of randomly-oriented aluminosilicate fiber and is partially covered with a layer of a carbonaceous material that makes it an inhomogeneous material. Upon functionalization, the carbonaceous material is removed from the material and a porous material is obtained. | ||
 | ||
| Fig. 2 Hypothesized surface functionalization process for the transformation of hydrophobic nonporous aluminosilicate into a hydrophilic porous ion exchange material. It is hypothesized that the transformation of the BO into NBO clusters will result in the transformation of “charge-balancing” cations into “network modifying” cations that can more readily be exchanged with heavy metal ions. This process will simultaneously increase the porosity of the aluminosilicate in addition to the surface functionalization. | ||
FTIR spectra of the parent and hydroxylated materials in the frequency region 4000–400 cm−1 are presented in Fig. 3(a). The band within the range of 3800–3000 cm−1 is associated with the stretching vibrations of the hydroxyl groups in Si–OH species31 and has a very low intensity for the parent material (NMF). This result is consistent with the abundance of Q4 species compared with the Q3 units in this fully polymerized material and thus its unreactive surface properties.
 | ||
| Fig. 3 (a) Surface functional groups of the parent (NMF) and functionalized (A-NMF) materials revealed by FTIR spectroscopy. The significant increase in the intensity if the band at 3800–3000 cm−1 reveals the depolymerization of the siloxane bonds and desired hydroxylation of the fully-polymerized parent material; (b) stretching vibrations of the O–H moieties in A-NMF. The existence of an inflection point at lower wavenumbers suggests the existence of two types of hydroxyl groups: those within silanol species (Si–OH) and the hydroxyl groups of the water molecules. (c) The band associated with the T–O linkages (where T = Si or Al) in A-NMF. The existence of an inflection point at higher wavenumber indicates the existence of bridging and non-bridging oxygens. | ||
The FTIR spectrum of the functionalized material (A-NMF), on the other hand, suggests that hydroxylation can considerably alter the properties of the NMF. Significantly higher intensity of the O–H stretching band for A-NMF compared with NMF reflects the favorable depolymerization of the aluminosilicate material and wide-scale transformation of Q4 species into Q3 and Q2 units. The deconvolution of the doublet band using Gaussian peak-fitting at 3471 cm−1 for A-NMF, illustrated in Fig. 3(b), yields two distinct peaks at 3525 cm−1 and 3380 cm−1 that can be ascribed to the stretching vibrations of the H-bonded vicinal hydroxyl groups32 and absorbed water molecules33 on the aluminosilicate surface, respectively. The presence of water molecules can be further confirmed by the presence of a bending vibration band at 1630 cm−1.33,34 The envelope at a frequency range of 1200–800 cm−1 is associated with the T–O linkages attached to either bridging or non-bridging oxygens.35 The deconvolution of this peak allows distinguishing between the bridging T–O–T′ and non-bridging T–O bonds. As depicted in Fig. 3(c), the sharp peak at 999 cm−1 represents the non-bridging T–O species, whereas the Gaussian component at 1097 cm−1, originally appearing as an inflection point, represents the T–O units attached to the bridging oxygens (T–O–T′). Considering the higher relative intensities of the peak attributed to the non-bridging T–O groups in A-NMF, transformation of Q4 → Q3, Q2 clusters can be confirmed.
During functionalization, the reaction of the T–O–T′ bridges with the activating agent, KOH, leads to the heterolytic cleavage of the bridging oxygen into two non-bridging oxygens, T–O− and T–OH.22 The potassium ion compensates for the charge deficiency of the former bond to form T–O−K+ (it is notable that according to the concept of the optical basicity of oxide ions proposed by Duffy and Ingram,36 the formation of Al–O−K+ is unfavorable). Also, some calcium ions, originally present as charge compensators of AlO4− units incapable of ion exchanging with other metals, can delocalize from the framework to the surface charge-deficient functional groups. Consequently, the role of some Ca2+ ions may change to network modifier although a major fraction of the calcium ions still prevail as charge balancing species AlO4− in the framework and cannot participate in any reaction. Hence, the calcium and potassium ions doped on the aluminosilicate material can act as bifunctional ion exchangers that can be replaced with more electronegative metal ions, such as heavy metals.
XPS, as another powerful technique to distinguish between the chemical states of the surface elements, has been conducted on the parent and functionalized materials. The low-resolution spectra for these two materials have been presented in Fig. 4. As depicted in this figure, the functionalization brings about significant alterations that are reflected in changes in the XPS spectrum for the functionalized material compared to the spectrum from the parent material. The intensities of the bands at 533.2 eV and 103.0 eV, ascribed to O 1s and Si 2p orbitals, respectively, have been drastically increased upon hydroxylation. This is related to the fact that hydroxylation depolymerizes the aluminosilicate structure by cleaving the tetrahedral cages of the network.
 | ||
| Fig. 4 XPS analysis of the parent and the functionalized materials. The deconvolution of the O 1s peaks for the two materials indicates that the majority of the O atoms (88%) are BO species in the parent material (lower inset figure), while the hydroxylation process results in the transformation of most of the BO species into NBO species (73%) (upper inset figure). | ||
Furthermore, the band at 345 eV, associated with the Ca 2p, has also been intensified during the functionalization. This could be due to higher concentration of the Ca2+ on the surface of the functionalized material compared with the parent material. The existence of potassium atoms is demonstrated by the appearance of a distinct new peak in the binding energy of 293 eV. The disappearance of the band at 285 eV suggests that the carbonaceous components have been either volatilized as a result of relatively high modification temperature or dissolved in the KOH solution upon functionalization.
The textural properties of the parent and modified materials (see Table S2†) are also dramatically different. The parent material has a specific surface area of less than 1 m2 g−1, whereas functionalization significantly increases the surface area and total pore volume to 383 m2 g−1 and 0.85 cm3 g−1, respectively. The isotherm curves for the precursor and the functionalized material are displayed in Fig. S3.†
Several researchers have demonstrated that, assuming a tetrahedral structure for Al3+ in aluminosilicates and assuming no formation of Al–O–Al linkages in the light of a low energetic stability as a result of a strong repulsion between the negative charges on the AlO4− species (Lowenstein's rule37), NBO species will form if the molar ratio of Al/R < 1, where R is a monovalent alkali metal.38 In the case of a divalent metal, such as Ca2+, this ratio must be doubled as one divalent ion can maintain the local charge neutrality of two tetrahedral structures. The X-ray fluorescence (XRF) results (tabulated in Table S1†) indicate an Al/Ca ratio of 0.4 which is lower than the theoretical value of 2 proposed by Smets and Lommen,28 implying the presence of NBO atoms and also validating the dual-role of Ca2+ as network modifier and charge compensator of AlO4− tetrahedra.
According to Brückner et al.39 the analysis of the O peak will provide further information about the type of the BOs and NBOs that are present. The deconvolution of the O peaks for the parent and functionalized materials, illustrated in the inset of Fig. 4, is fit well by two distinct peaks. The peaks at the higher and lower binding energies represent the BO and NBO, respectively, with a chemical shift of around 1 eV between the BO and NBO (ΔEBO–NBO ≅ 1 eV). The ratio of the areas under the BO (ABO) and NBO (ANBO) peaks to the total O peak (Atot) will reveal their relative concentrations. For the parent material, BOs constitute the major fraction of the O atoms  , whereas the functionalization brings about the formation of higher quantities of NBOs
, whereas the functionalization brings about the formation of higher quantities of NBOs 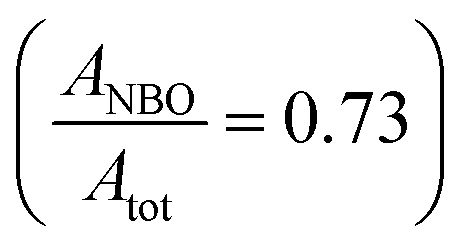 by the cleavage of the T–O–T′ bridges into T–O–NBO functional moieties. These T–O–NBO functional groups can react more readily with other components rather than the unreactive T–O–T′ groups and thus, further explains the higher reactivity of the functionalized material (A-NMF) compared with the parent one (NMF). It can also be argued that the peak at 531.6 eV for the NMF can be partly or wholly ascribed to the O atoms of the carbonate present in the parent material. Even if this hypothesis is valid, it will further decrease the concentration of the NBO species and thus the relative amount of BOs to NBOs will further increase in NMF. Nonetheless, this hypothesis does not apply for A-NMF, because almost all the carbon has been removed during the functionalization process (see Table S1†) and thus no carbonate peaks are expected to be present for the A-NMF in the XPS spectrum.
by the cleavage of the T–O–T′ bridges into T–O–NBO functional moieties. These T–O–NBO functional groups can react more readily with other components rather than the unreactive T–O–T′ groups and thus, further explains the higher reactivity of the functionalized material (A-NMF) compared with the parent one (NMF). It can also be argued that the peak at 531.6 eV for the NMF can be partly or wholly ascribed to the O atoms of the carbonate present in the parent material. Even if this hypothesis is valid, it will further decrease the concentration of the NBO species and thus the relative amount of BOs to NBOs will further increase in NMF. Nonetheless, this hypothesis does not apply for A-NMF, because almost all the carbon has been removed during the functionalization process (see Table S1†) and thus no carbonate peaks are expected to be present for the A-NMF in the XPS spectrum.
The shift in the binding energies of the O core electrons in the NMF and A-NMF is associated with the difference in the electron density on the O atoms by increasing the NBOs.38 According to Alexander et al.40 the bond between the charge compensators and BOs is more ionic than that of the network modifiers and NBOs. Therefore, charge compensators will decrease the electron density on the O atoms and hence the binding energy will be higher when most of the Ca2+ act as charge compensator, as in the case of NMF. As the amount of NBO components increases, the degree of the ionicity of the calcium ions decreases, resulting in higher electron density on the O and lower binding energies. Similar behavior has already been reported for borosilicate glasses,41 where higher concentration of NBO components results in lower binding energies.
NMR results confirm the condensed structure of the NMF. As illustrated in Fig. 5(a), 29Si DP spectrum from NMF shows a broad peak with a maximum at a chemical shift of −99.4 ppm. This peak is clearly related to an aluminosilicate with a range tetrahedral Si sites.42 The sample has a Si/Al ratio of approximately 5 and therefore one would expect a maximum around the shifts of species such as Q4[1Al]. However, there is a distribution of additional Q4[nAl] sites with n = 0–4 that should be present, as well as some sites with non-bridging oxygens (but without hydroxylation, as no signal can be observed in the 1H–29Si CP spectrum of this sample). The lack of a CP signal indicates the absence of detectable numbers of hydroxyl groups using the CP method, but does not rule out the presence of some surface bound hydroxyls on this low surface area sample. These NMR results, together with very low surface area of the sample, agree with the predominance of Q4 structures within the aluminosilicate.
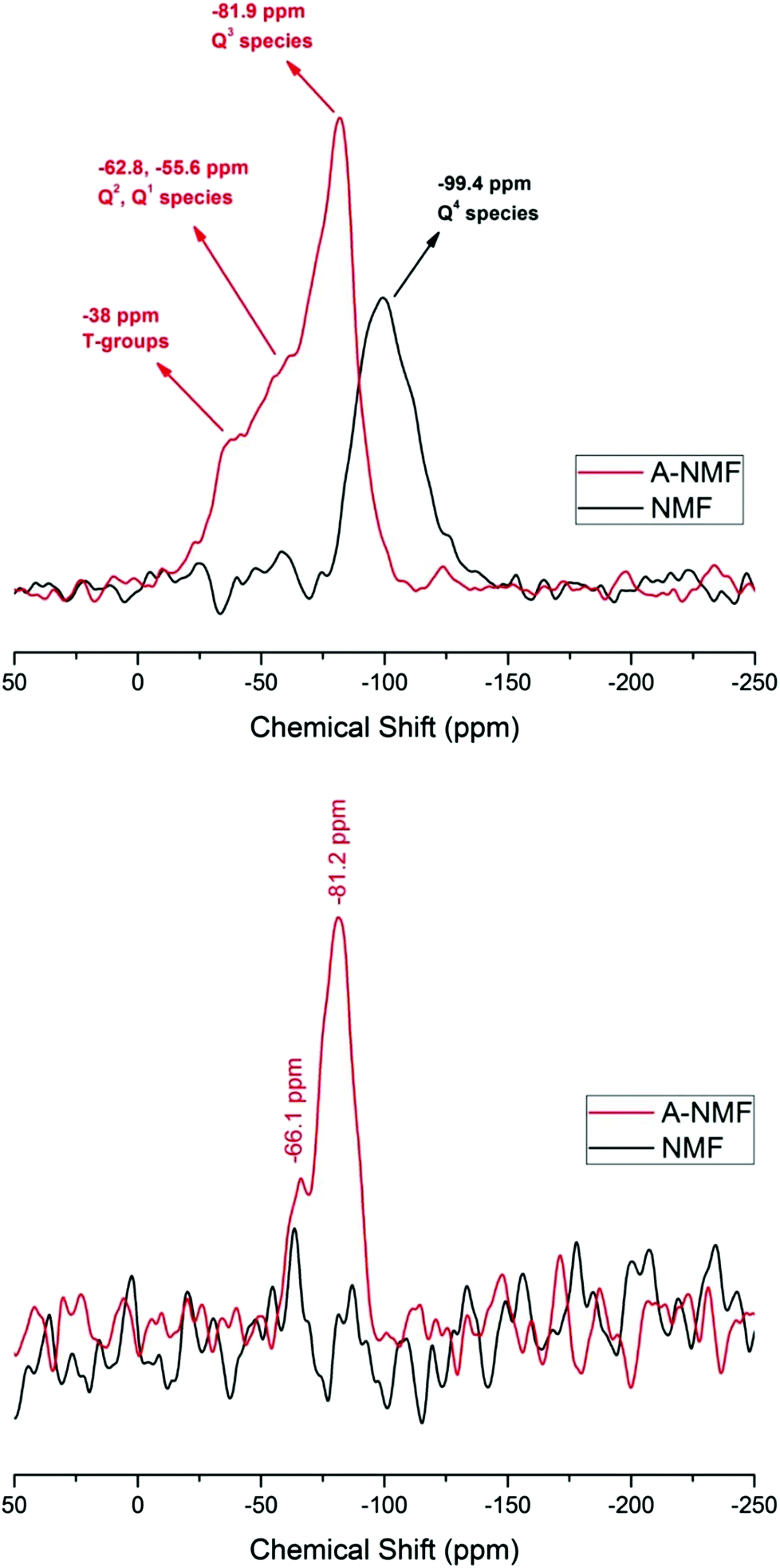 | ||
| Fig. 5 NMR analysis of the parent and the functionalized materials. (a) DP spectra. The 29Si DP NMR spectrum of the precursor shows that this material consists of Q4[nAl] structure, where n can range from n = 0–4. The functionalization brings about the conversion of the Q4 species into non-Q4 groups and thus results in the shift of the maximum peak towards 0 ppm. (b) CP spectra. The absence of any peak in the CP spectrum further confirms the absence of any –OH group in the NMF, whereas A-NMF has a strong CP peak at around −81 ppm, confirming the existence of –OH moieties and thus non-Q4 species. | ||
In the case of the functionalized aluminosilicate, the major peak in the 29Si DP spectrum is observed at −82 ppm. If we ascribe at least a portion of this peak to arise from non-Q4 species with 1 or 2 Al next-nearest neighbors (NNN) to move the resonance to lower chemical shifts, a peak at the same chemical shift (around −82 ppm) should also exist in the 29Si CP spectrum. This is confirmed in Fig. 5(b). The additional spectral intensity with 29Si chemical shift values observed closer to 0 ppm is believed to be caused by some modified T-groups, i.e. direct bonding of some of the C with the aluminosilicate network, with additional presence of Al or metal cations in the NNN coordination sphere. The CP spectrum agrees with this observation, as no peak exists near −30 to −50 ppm. The peak at −66 ppm can be either some T groups associated with –OH or other Q groups (Q3, Q2) with multiple Al or NBOs nearby.
3.2 Metal uptake on the functionalized aluminosilicate
The maximum uptake capacities of the functionalized aluminosilicate for various heavy metals have been summarized in Table 1 and have been compared with several well-structured zeolites and silicates. While the non-functionalized material has no affinity for the heavy metals and the metal uptake does not occur on it, the functionalization of the aluminosilicate considerably facilitates the deposition of heavy metals. The remarkably high heavy metal uptake capacity of A-NMF is attributed to the increased number of NBO sites as well as the existence of two distinct ion exchange sites. The K+ and Ca2+ ions attached to the non-bridging T–O end groups can readily be exchanged with the heavy metals. Also, the mesoporous structure of the functionalized A-NMF permits more rapid transport of the heavy metal ions into the interior pore surfaces in the aluminosilicate.| Material | Maximum metal deposition (mmol g−1) | Ref. | |||||
|---|---|---|---|---|---|---|---|
| Cu2+ | Pb2+ | Zn2+ | Co2+ | Ni2+ | Cd2+ | ||
| a Metal depositions lower than 0.2 mmol g−1 for all the metals have not been included in the table. b The metal deposition capacities of A-NMF is on a wet basis with a moisture content of approximately 20 wt%. The capacities on a dry basis will be higher than the tabulated amounts. | |||||||
| NMF | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 | This work |
| A-NMFb | 2.82 | 3.21 | 2.00 | 3.12 | 3.11 | 2.05 | This work |
| Zeolite and silicate heavy metal uptake capacities | |||||||
| Zeolite | 0.44 | 0.21 | 43 | ||||
| Zeolite 4A | 0.84 | 0.48 | 0.19 | 0.13 | 44 | ||
| Clinoptilolite | 0.4 | 0.13 | 0.26 | 0.04 | 45 | ||
| Zeolite-activated carbon composite | 1.72 | 2.65 | 1.20 | 1.44 | 46 | ||
| Na–X zeolite | 1.16 | 1.68 | 47 | ||||
| Cancrinite zeolite | 2.08 | 2.13 | 1.15 | 1.24 | 1.53 | 48 | |
| Modified zeolite | 1.43 | 2.03 | 0.87 | 49 | |||
| Fly ash zeolite | 1.30 | 0.72 | 50 | ||||
| Functionalized silica | 0.60 | 0.20 | 0.20 | 0.25 | 51 | ||
| Amine-functionalized silica | 0.83 | 52 | |||||
| Modified SBA-15 | 0.92 | 0.4 | 0.32 | 0.36 | 53 | ||
| Silica nano-hollow sphere | 0.47 | 0.53 | 0.36 | 54 | |||
| Thiol-functionalized silica | 0.44 | 55 | |||||
| Commercial ion exchanger heavy metal uptake capacities | |||||||
| Dowex 50W | 0.35 | 0.23 | 0.30 | 0.12 | 0.25 | 71 | |
| Amberlite IR-120 | 0.34 | 0.41 | 1.3 | 0.82 | 0.90 | 72 | |
| Amberlite IRC-748 | 1.13 | 1.06 | 73 | ||||
| Lewatit CNP 80 | 0.16 | 0.35 | 0.31 | 0.32 | 0.04 | 74 | |
| Lewatit TP207 | 1.98 | 1.78 | 1.63 | 1.97 | 1.78 | 75,76 | |
| Amberjet 1500H | 0.39 | 77 | |||||
| Ambersep 252H | 0.18 | 77 | |||||
| Suqing D401 | 2.00 | 1.88 | 1.70 | 2.10 | 1.88 | 75,76 | |
| MCM-41 | 0.71 | 0.68 | 0.41 | 0.71 | 0.67 | 75,76 | |
The ion exchange mechanism of the heavy metal uptake onto the functionalized material is verified by the liberation of K+ and Ca2+ into the medium as the heavy metal ions are deposited on the aluminosilicate. Fig. 6 illustrates the liberation of the K+ and Ca2+ into the solution by Cd2+ deposition. As higher amounts of Cd2+ are deposited onto the hydroxylated aluminosilicate, higher concentrations of K+ and Ca2+ are liberated into the medium. Considering that each heavy metal ion, M2+, can be exchanged with either one Ca2+ or two K+, the theoretical mass balance between the liberated and deposited metals does not thoroughly conform with the experimental findings. In other words, more heavy metal ions are being deposited than the liberated alkali metals. Electroneutrality may be maintained by the incongruent dissolution and leaching of Al3+, verified by the liberation of Al3+ species into the heavy metal containing solution.56 This reveals that although the major mechanism of the heavy metal uptake proceeds via the ion exchange with the lighter metal ions, some heavy metal ions can also be deposited via other routes. This route has been later elucidated to be electrostatic interaction between the heavy metal ions and the lone pair of electrons on the BO.
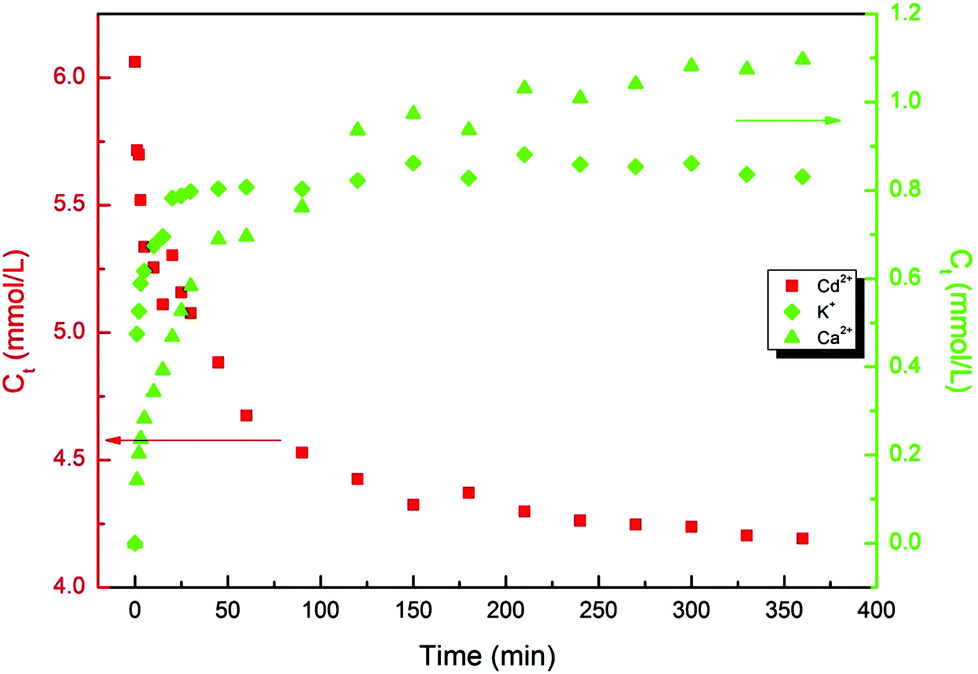 | ||
| Fig. 6 Trend of the potassium and calcium liberation into the medium by the heavy metal uptake (cadmium in this case). These data verify that one of the dominating mechanism for metal uptake is the ion exchange process. However, due to the nonconformity of the mass balance between the monovalent potassium and divalent calcium liberation and divalent heavy metal uptake, the occurrence of an additional phenomenon is hypothesized. | ||
The mechanism of heavy metal uptake on the functionalized material is further elucidated by additional analytical methods. As depicted in Fig. 7, deposition of heavy metals on the aluminosilicate-type materials induces appreciable spectral changes in the fingerprint infrared regions of the Si–O band at ∼1300–800 cm−1.
 | ||
| Fig. 7 FTIR spectra of the A-NMF and the metal-deposited materials in the fingerprint region 1300–800 cm−1. The Gaussian components centered at 1100 cm−1 and 1000 cm−1 are attributed to Q4 and Q3 Si. The deposition of heavy metals brings about the appearance of a new peak at 870 cm−1, attributed to the interaction of the material surface functional groups with the heavy metal ions. | ||
Previous research utilizing Raman spectroscopy of silicate materials has shown that the stretching vibrations of the Si–O band envelope in the range of 1250–850 cm−1 can be fitted with four different components.57 These components have been ascribed to Si–BO species in Q4 units and three Si–NBO stretching vibrations corresponding to various numbers of NBOs (Q3 and Q2). Nevertheless, although it is mathematically feasible to acquire a proper fit for these four components, we believe that it is not plausible to confidently articulate a solid distinction between the Q3 and Q2 components of Si–NBO linkages. Instead, it is more pragmatic to deconvolute the band into two components using Gaussian functions for both Si–BO and Si–NBO species. The component at 1097 cm−1 is assigned to the tetrahedral BOs related to a Q4 configuration of T–O–T′ species of the aluminosilicate, while the component at lower frequency (999 cm−1) corresponds to the NBOs of Q3 or Q2 units.
Upon metal introduction, another component, centered at approximately 870 cm−1, appears in addition to the two components in the A-NMF. Kubicki & Toplis58 performed calculations on the effect of Ca2+ on the T–O–T′ bond length. According to their calculations, the interaction of Ca2+ with this bond will lengthen the T–O–T′ linkage, which in turn, will lower the vibrational frequency. Using an anharmonicity coefficient of 0.893, the interaction between the Ca2+ and T–O–T′ is expected to result in a peak at 922–954 cm−1. Stronger interaction of the heavy metals with the bridging oxygens will further weaken the T–O–T′ bond and result in peaks at frequencies as low as 870 cm−1. Hence, this new Gaussian component is an indication of the interaction of the heavy metals with the aluminosilicate. The type of this interaction is still open to dispute. In another study, Kubicki & Sykes have attributed the peak at 870 cm−1 to the interaction of the BOs with H+ and formation of a T–(OH)–T′ configuration, verified by molecular orbital calculations.24 Kohn et al. have also assigned the shoulder at around 888 cm−1 to the T–(OH)–T′ clusters in sodium aluminosilicate glasses.23 Accordingly, the lone pairs on the O atoms of the BO can induce dipole moment due to the large electronegativity difference between the Si and O atoms which, in turn, can cause the electrostatic interaction between the positively-charged heavy metal ions and the lone pairs.
In order to test this hypothesis, the pH changes in the medium have been closely monitored. We have observed a drastic pH increase when the A-NMF is placed into pure water. The dissociation of water molecules into H+ and OH− and the interaction of the former with the T–O–T′ linkages to form T–(OH+)–T′ could bring about the observed pH increase, because OH− species would be released into solution. Placing the A-NMF in the heavy metal-containing solution increases the aqueous pH level to a much smaller extent. This phenomenon is associated with the stronger electrostatic interaction of M2+ with the O atoms of T–O–T′ clusters rather than the protonation of the BO. In agreement with the findings of Kubicki & Sykes,24 this interaction generates a strong IR band at 870 cm−1 in the metal-doped aluminosilicate material. This hypothesis suggests the existence of an interaction between the BO and heavy metal ions in addition to the ion exchange mechanism. This interpretation accounts for slightly higher metal deposition compared with the liberated ions, as suggested earlier.
Heavy metal deposition also alters the envelope at 3800–3000 cm−1 assigned to the OH stretching vibrations of the H-bonded silanols and the water molecules. Metal uptake brings about the splitting of the band at 3460 cm−1 in A-NMF into three small peaks/shoulders/distortions in metal-doped A-NMF. These new peaks/shoulders are located at approximately-fixed positions for all the metal-doped aluminosilicate samples, appearing around 3415 cm−1, 3465 cm−1 and 3550 cm−1. These bands are attributed to hydrated metal clusters with various coordination numbers. According to O'Brien and Williams,59 partial electron transfer from the water molecules to the cations results in different hydrogen bonding strength in various conformations and thus different vibrational frequencies. Hence, the emergence of several bands in close proximity to each other after the heavy metal uptake is consistent with the hydration of the heavy metals with different coordination numbers and various hydrogen bonding abilities.
XPS can provide information on the oxidation states of the metals deposited on the aluminosilicate. These oxidation states are of high significance in the catalytic behavior of the metal-deposited aluminosilicates. The XPS results from the Cu-A-NMF sample is presented in Fig. 8 and is compared with that of the A-NMF. As depicted in Fig. S4,† there is a remarkable reduction in the peak intensities of the K+ and Ca2+ due to their ion exchange with the Cu2+. Also, the appearance of a new symmetric peak centered at 935.2 eV after the Cu2+ deposition, indicative of the Cu 3p3/2 core region, clearly indicates the incorporation of isolated cupric ions in a spinel-like structure via an ion exchange mechanism.60–62 The high-resolution spectrum of copper (inset of Fig. 8) displays the existence of shake-up satellite features at approximately 944 eV and 964 eV for the 3p spin orbit doublets (Cu 3p3/2 and Cu 3p1/2 core levels).63–65 These satellite peaks form by the electron transfer from a ligand orbital to the metal d orbital (np ligand → 3d copper) which indicates the open 3d9 shell configuration of bivalent copper species. This electronic structure suggests the ability of the Cu-doped aluminosilicate to perform catalytic reactions requiring electron transfer.
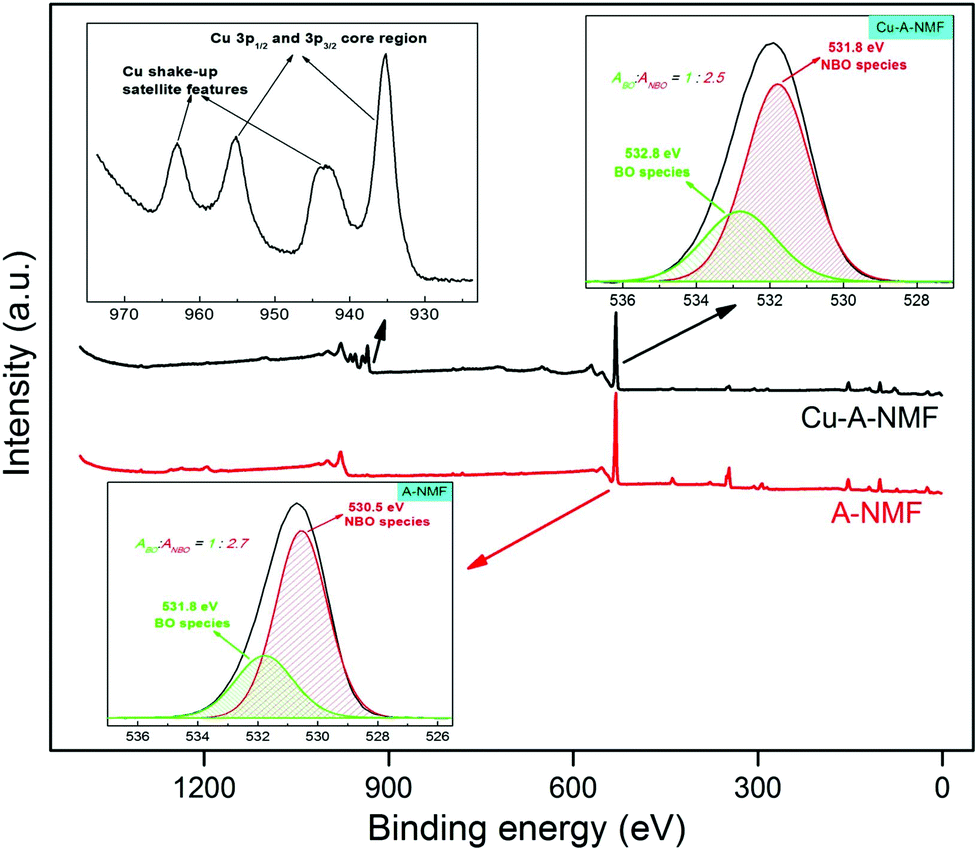 | ||
| Fig. 8 XPS diagram of the A-NMF and the copper-deposited material. The appearance of a peak centered at 935.2 eV indicates the incorporation of isolated cupric ions in a spinel-like structure. Also, the existence of shake-up satellite features suggests the 3d9 shell configuration of bivalent copper species, potentially beneficial for catalytic applications. | ||
The deconvolution of the O peak demonstrates that the concentration of the NBO is not significantly changed by the metal uptake, where the 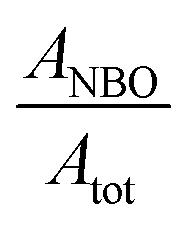 is calculated as 0.73 and 0.72 for the A-NMF and Me-A-NMF, respectively. The constant relative concentration of the non-bridging oxygen is very critical, as it suggests that the structure of the aluminosilicate remains intact during the metal deposition and BOs do not re-form through re-combination of the NBOs. Therefore, the substitution of an alkali or alkaline earth metal with a heavy metal does not change the role of the O atoms in the network. Nonetheless, due to the intrinsic differences in the electronic environments of the light and heavy metals, apparent chemical shifts of the BO and NBO peaks to higher binding energies by the heavy metal deposition can be detected.66 When Ca2+ and K+ are replaced by more electronegative heavy metals, a partial transfer of the electronic charge from the neighboring O to the heavy metal occurs, reducing the electron density on the O atoms which thus results in an increase in the binding energy of the O core element from 530.5 eV for A-NMF to 531.6–531.9 eV for Me-A-NMF.
is calculated as 0.73 and 0.72 for the A-NMF and Me-A-NMF, respectively. The constant relative concentration of the non-bridging oxygen is very critical, as it suggests that the structure of the aluminosilicate remains intact during the metal deposition and BOs do not re-form through re-combination of the NBOs. Therefore, the substitution of an alkali or alkaline earth metal with a heavy metal does not change the role of the O atoms in the network. Nonetheless, due to the intrinsic differences in the electronic environments of the light and heavy metals, apparent chemical shifts of the BO and NBO peaks to higher binding energies by the heavy metal deposition can be detected.66 When Ca2+ and K+ are replaced by more electronegative heavy metals, a partial transfer of the electronic charge from the neighboring O to the heavy metal occurs, reducing the electron density on the O atoms which thus results in an increase in the binding energy of the O core element from 530.5 eV for A-NMF to 531.6–531.9 eV for Me-A-NMF.
The TOF-SIMS depth profile, illustrated in Fig. S5,† shows the distribution of K+ and Ca2+ in the functionalized material and reveals its distribution change by deposition of heavy metals. The concentration of K+ steadily decreases as a function of depth from the surface, but the Ca2+ is approximately constant in the course of sputtering. This behavior is associated with the roles of these two metals in the functionalized material, where all K+ ions play the role of a network modifier, whereas a major fraction of Ca2+ ions acts as charge balancing cations for AlO4− clusters and only a small fraction, migrating from the bulk to the surface, have the same role as K+ ions. When heavy metal is deposited on the aluminosilicate material, the concentration of Ca2+ remains unchanged, whereas the depth profile of the heavy metal is markedly identical to that of the potassium in the functionalized material. The dependence of the heavy metal concentration in different depths to the K+ and Ca2+ concentrations further validate the ion exchange mechanism for the deposition process.
3.3 Uncertainties in the microstructure of the parent and altered material
Q-distributions in amorphous aluminosilicates (glasses and “geopolymers”) have been extensively studied for decades. Previously, it was presumed that the structure of glasses contained only BO when charge of the modifier cation equaled the number of Al atoms. Despite the high abundance of BO, however, NBO were also found in these glasses. For geopolymers, it is well documented that Si/Al in the tetrahedra might have connectivity ranging from 1 to 4. In other words, different amorphous aluminosilicates were characterized by different distributions, and even for the same material, it can be changed, for example, by heating. Generally, the order of abundance of Q2, Q3 and Q4 can be shifted depending on Al/Me ratio, nature of precursors, conversion degree of precursor (for geopolymers) and cooling rate (for glasses). Therefore, Q-distribution is rather a unique fingerprint than a property common for the whole class of material.67–70In this study, NMF is a material of unknown nature, which has been synthesized, processed, and stored with unknown level of control. In fact, NMF is an amorphous Ca-aluminosilicate, a metastable material that represents one of myriad states possible for this chemical composition. In addition, it was neither homogeneous nor pure because it is a waste-derived material. Therefore, the analysis of the NMF structure can create great controversy and be a challenge in this work. Despite the inhomogeneous structure of the NMF, whether the parent material was fully polymerized or it had a certain Q3/Q4 ratio (as suggested by the XPS results) is not the critical issue – at this level of uncertainty, NMF material can be considered as not more than just a control material to study the structural alterations during the functionalization and ion exchange. The key finding is the tendency of this material to decrease the degree of polymerization after aqueous thermo-alkaline reaction. Analytically-proven manifestation of such a decrease in the A-NMF relative to the unaltered NMF is sufficient regardless of the exact state of the starting point. The key to this study is the newly-formed NBO surface sites that improve the ion exchange performance of the parent material. It has been clearly demonstrated by XPS and NMR that this alteration occurs, and the NBO concentration is higher for the altered material.
3.4 Potential application of the functionalized aluminosilicate
The functionalized A-NMF can find applications in various fields of technology, including catalysis and adsorption. Due to the ultra-high uptake capacity of the A-NMF for heavy metal ions, this material can be used as an efficient adsorbent in wastewater treatment. The heavy metal removal capability of the A-NMF in aqueous media has been compared with those of some commercially-available ion exchangers, as summarized in Table 1. The A-NMF is more effective than the commercial ion exchange resins in the removal of heavy metal ions from wastewater due to its multi-functional surface properties. Moreover, some of the ion exchange resins may be effective for the removal of some heavy metals, but may not have a high capacity for others, whereas A-NMF has a high uptake capacity for all positively-charged heavy metal ions used in this study. This high metal uptake capacity makes it possible to use the waste from PCB manufacturing companies to treat their wastewater for “zero waste generation” and “zero wastewater discharge”.4. Conclusion
An aluminosilicate-based material, recovered from waste printed circuit board, was modified by a facile hydroxylation technique. Studies with a variety of carefully-chosen analytical techniques revealed that the modification process results in the depolymerization of Q4 clusters in the tetrahedral structure of the aluminosilicate and formation of Q3 and Q2 components. Some of the calcium ions, originally as charge-compensator for the AlO4− clusters, can migrate from the framework to the surface and act as network modifiers, capable of exchanging with heavy metals. Also, the potassium ions doped on the aluminosilicate structure through the reaction of the aluminosilicate with the activating agent can play the role of ion exchange sites. The heavy metal uptake capacity of the functionalized material has been shown to be promising and comparable with commercial ion exchange resins. Thus, it is proposed that the waste generated by PCB manufacturing companies can be used to treat the heavy metal-laden wastewater generated by them to render “zero waste generation” and “zero wastewater discharge”.Acknowledgements
The authors are grateful for the valuable comments given by Dr Dan Glynn Sykes from the Department of Chemistry, The Pennsylvania State University.References
- S. Pega, C. Boissière, D. Grosso, T. Azaïs, A. Chaumonnot and C. Sanchez, Angew. Chem., Int. Ed., 2009, 48, 2784–2787 CrossRef CAS PubMed.
- G. L. Hill, E. Bailey, M. C. Stennett, N. C. Hyatt, E. M. Maddrell, P. F. Mcmillan and J. A. Hriljac, J. Am. Chem. Soc., 2011, 133, 13883–13885 CrossRef CAS PubMed.
- M. C. Eckersley, P. H. Gaskell, A. C. Barnes and P. Chieux, Nature, 1988, 335, 525–527 CrossRef CAS.
- D.-Y. Kang, N. A. Brunelli, G. I. Yucelen, A. Venkatasubramanian, J. Zang, J. Leisen, P. J. Hesketh, C. W. Jones and S. Nair, Nat. Commun., 2014, 5, 3342 Search PubMed.
- J. Shin, N. H. Ahn, M. a. Camblor, C. M. Zicovich-Wilson and S. B. Hong, Chem. Mater., 2014, 26, 3361–3363 CrossRef CAS.
- J. Kim, L. Lin, J. A. Swisher, M. Haranczyk and B. Smit, J. Am. Chem. Soc., 2012, 134, 18940–18943 CrossRef CAS PubMed.
- T. Li, H. Liu, Y. Fan, P. Yuan, G. Shi, X. T. Bi and X. Bao, Green Chem., 2012, 14, 3255 RSC.
- G. J. Kramer, A. J. M. De Man and R. A. Van Santent, J. Am. Chem. Soc., 1991, 113, 6435–6441 CrossRef CAS.
- Y. Aoki, H. Habazaki and T. Kunitake, J. Am. Chem. Soc., 2009, 131, 14399–14406 CrossRef CAS PubMed.
- J. B. Murdoch, J. F. Stebbins and S. E. Carmichael, Am. Mineral., 1985, 70, 332–343 CAS.
- X. Xue and M. Kanzaki, J. Phys. Chem. B, 1999, 103, 10816–10830 CrossRef CAS.
- P. Onorato, M. N. Alexander, C. W. Struck, G. W. Tasker and D. R. Uhlmann, J. Am. Ceram. Soc., 1985, 68, 148–150 CrossRef.
- M. Roulia, T. Mavromoustakos, A. A. Vassiliadis and G. Mali, J. Phys. Chem. C, 2014, 118, 26649–26658 CAS.
- Y. Xiao and A. C. Lasaga, Geochim. Cosmochim. Acta, 1994, 58, 5379–5400 CrossRef CAS.
- J. P. Hamilton and C. G. Pantano, J. Non-Cryst. Solids, 1997, 222, 167–174 CrossRef CAS.
- X. Pardal, F. Brunet, T. Charpentier, I. Pochard and A. Nonat, Inorg. Chem., 2012, 51, 1827–1836 CrossRef CAS PubMed.
- J. A. Boscoboinik, X. Yu, B. Yang, F. D. Fischer, R. Włodarczyk, M. Sierka, S. Shaikhutdinov, J. Sauer and H.-J. Freund, Angew. Chem., Int. Ed., 2012, 51, 6005–6008 CrossRef CAS PubMed.
- F. Angeli, M. Gaillard, P. Jollivet and T. Charpentier, Chem. Phys. Lett., 2007, 440, 324–328 CrossRef CAS.
- L. Cormier, D. R. Neuville and G. Calas, J. Am. Ceram. Soc., 2005, 88, 2292–2299 CrossRef CAS.
- M. Moesgaard, R. Keding, J. Skibsted and Y. Yue, Chem. Mater., 2010, 22, 4471–4483 CrossRef CAS.
- F. Bouyer, G. Geneste, S. Ispas, W. Kob and P. Ganster, J. Solid State Chem., 2010, 183, 2786–2796 CrossRef CAS.
- V. Murashov, J. Mol. Struct., 2003, 650, 141–157 CrossRef CAS.
- S. C. Kohn, M. E. Smith, P. J. Dirken, E. R. H. van Eck, A. P. M. Kentgens and R. Dupree, Geochim. Cosmochim. Acta, 1998, 62, 79–87 CrossRef CAS.
- J. D. Kubicki and D. G. Sykes, Geochim. Cosmochim. Acta, 2004, 68, 3909–3918 CrossRef CAS.
- R. Hellmann, R. Wirth, D. Daval, J. P. Barnes, J. M. Penisson, D. Tisserand, T. Epicier, B. Florin and R. L. Hervig, Chem. Geol., 2012, 294–295, 203–216 CrossRef CAS.
- A. F. White and S. L. Brantley, Chem. Geol., 2003, 202, 479–506 CrossRef CAS.
- F. Tielens, C. Gervais, J. Franc, F. Mauri and D. Costa, Chem. Mater., 2008, 20, 3336–3344 CrossRef CAS.
- B. M. J. Smets and T. P. A. Lommen, Phys. Chem. Glasses, 1982, 23, 83–87 CAS.
- P. Hadi, J. Barford and G. McKay, Environ. Sci. Technol., 2013, 47, 8248–8255 CAS.
- M. E. Simonsen, C. Sønderby, Z. Li and E. G. Søgaard, J. Mater. Sci., 2009, 44, 2079–2088 CrossRef CAS.
- V. Dugas and Y. Chevalier, J. Colloid Interface Sci., 2003, 264, 354–361 CrossRef CAS PubMed.
- F. Garbassi, L. Balducci, P. Chiurlo and L. Deiana, Appl. Surf. Sci., 1995, 84, 145–151 CrossRef CAS.
- C. Zhou, T. Sun, Q. Gao, A. Alshameri, P. Zhu, H. Wang, X. Qiu, Y. Ma and C. Yan, J. Taiwan Inst. Chem. Eng., 2014, 45, 1073–1079 CrossRef CAS.
- Y. T. Fang, T. Liu, Z. C. Zhang and X. N. Gao, Renewable Energy, 2014, 63, 755–761 CrossRef CAS.
- Y. Kong, H. Y. Zhu, G. Yang, X. F. Guo, W. H. Hou, Q. J. Yan, M. Gu and C. Hu, Adv. Funct. Mater., 2004, 14, 816–820 CrossRef.
- J. A. Duffy and M. D. Ingram, J. Non-Cryst. Solids, 1976, 21, 373–410 CrossRef CAS.
- W. Lowenstein, Am. Mineral., 1954, 39, 92 Search PubMed.
- Y. Miura, S. Matsumoto, T. Nanba and T. Akazawa, Phys. Chem. Glasses, 2000, 41, 24–31 CAS.
- R. Brückner, H. U. Chun, H. Goretzki and M. Sammet, J. Non-Cryst. Solids, 1980, 42, 49–60 CrossRef.
- M. N. Alexander, P. I. K. Onorato, C. W. Struck, J. R. Rozen, G. W. Tasker and D. R. Uhlmann, J. Non-Cryst. Solids, 1986, 79, 137–154 CrossRef CAS.
- Y. Miura, H. Kusano, T. Nanba and S. Matsumoto, J. Non-Cryst. Solids, 2001, 290, 1–14 CrossRef CAS.
- J. V. Oglesby and J. F. Stebbins, Am. Mineral., 2000, 85, 722–731 CrossRef CAS.
- L. Curkovic, S. Stefanovic and T. Filipan, Water Res., 1997, 31, 1379–1382 CrossRef CAS.
- K. S. Hui, C. Y. H. Chao and S. C. Kot, J. Hazard. Mater., 2005, 127, 89–101 CrossRef CAS PubMed.
- M. Sprynskyy, B. Buszewski, A. P. Terzyk and J. Namieśnik, J. Colloid Interface Sci., 2006, 304, 21–28 CrossRef CAS PubMed.
- V. K. Jha, M. Matsuda and M. Miyake, J. Hazard. Mater., 2008, 160, 148–153 CrossRef CAS PubMed.
- V. K. Jha, M. Nagae, M. Matsuda and M. Miyake, J. Environ. Manage., 2009, 90, 2507–2514 CrossRef CAS PubMed.
- W. Qiu and Y. Zheng, Chem. Eng. J., 2009, 145, 483–488 CrossRef CAS.
- R. Apiratikul and P. Pavasant, Chem. Eng. J., 2008, 144, 245–258 CrossRef CAS.
- C. Wang, J. Li, X. Sun, L. Wang and X. Sun, J. Environ. Sci., 2009, 21, 127–136 CrossRef CAS.
- L. Bois, A. Bonhommé, A. Ribes, B. Pais, G. Raffin and F. Tessier, Colloids Surf., A, 2003, 221, 221–230 CrossRef CAS.
- J. Aguado, J. M. Arsuaga, A. Arencibia, M. Lindo and V. Gascón, J. Hazard. Mater., 2009, 163, 213–221 CrossRef CAS PubMed.
- M. Mureseanu, A. Reiss, I. Stefanescu, E. David, V. Parvulescu, G. Renard and V. Hulea, Chemosphere, 2008, 73, 1499–1504 CrossRef CAS PubMed.
- M. Najafi, Y. Yousefi and A. A. Rafati, Sep. Purif. Technol., 2012, 85, 193–205 CrossRef CAS.
- G. Li, Z. Zhao, J. Liu and G. Jiang, J. Hazard. Mater., 2011, 192, 277–283 CAS.
- J. P. Hamilton, S. L. Brantley, C. G. Pantano, L. J. Criscenti and J. D. Kubicki, Geochim. Cosmochim. Acta, 2001, 65, 3683–3702 CrossRef CAS.
- A. Quaranta, A. Rahman, G. Mariotto, C. Maurizio, E. Trave, F. Gonella, E. Cattaruzza, E. Gibaudo and J. E. Broquin, J. Phys. Chem. C, 2012, 116, 3757–3764 CAS.
- J. D. Kubicki and M. J. Toplis, Am. Mineral., 2002, 87, 668–678 CrossRef CAS.
- J. T. O'Brien and E. R. Williams, J. Phys. Chem. A, 2008, 112, 5893–5901 CrossRef PubMed.
- M. Brandhorst, J. Zajac, D. J. Jones, J. Rozière, M. Womes, A. Jimenez-López and E. Rodríguez-Castellón, Appl. Catal., B, 2005, 55, 267–276 CrossRef CAS.
- D. V. Cesar, C. A. Perez, M. Schmal, V. Maria and M. Salim, Appl. Surf. Sci., 2000, 157, 159–166 CrossRef CAS.
- C. M. Chanquía, A. L. Cánepa, J. Bazán-Aguirre, K. Sapag, E. Rodríguez-Castellón, P. Reyes, E. R. Herrero, S. G. Casuscelli and G. A. Eimer, Microporous Mesoporous Mater., 2012, 151, 2–12 CrossRef.
- R. S. Vieira, M. L. M. Oliveira, E. Guibal, E. Rodríguez-Castellón and M. M. Beppu, Colloids Surf., A, 2011, 374, 108–114 CrossRef CAS.
- J. Andas, F. Adam and I. A. Rahman, Appl. Surf. Sci., 2013, 284, 503–513 CrossRef CAS.
- L. Wu, H. Wang, H. Lan, H. Liu and J. Qu, Sep. Purif. Technol., 2013, 117, 118–123 CrossRef CAS.
- J. Serra, P. González, S. Liste, C. Serra, S. Chiussi, B. León, M. Pérez-Amor, H. O. Ylänen and M. Hupa, J. Non-Cryst. Solids, 2003, 332, 20–27 CrossRef CAS.
- B. T. Poe, P. F. McMillan, B. Cote, D. Massiot and J. P. Coutures, J. Phys. Chem., 1992, 96, 8220–8224 CrossRef CAS.
- P. S. Singh, T. Bastow and M. Trigg, J. Mater. Sci., 2005, 40, 3951–3961 CrossRef CAS.
- P. Duxson, J. L. Provis, G. C. Lukey, F. Separovic and J. S. J. Van Deventer, Langmuir, 2005, 21, 3028–3036 CrossRef CAS PubMed.
- H. Oudadesse, A. C. Derrien, M. Lefloch and J. Davidovits, J. Mater. Sci., 2007, 42, 3092–3098 CrossRef CAS.
- E. Pehlivan and T. Altun, J. Hazard. Mater., 2006, 134, 149–156 CrossRef CAS PubMed.
- A. Demirbas, E. Pehlivan, F. Gode, T. Altun and G. Arslan, J. Colloid Interface Sci., 2005, 282, 20–25 CrossRef CAS PubMed.
- L.-C. Lin, J.-K. Li and R.-S. Juang, Desalination, 2008, 225, 249–259 CrossRef CAS.
- E. Pehlivan and T. Altun, J. Hazard. Mater., 2007, 140, 299–307 CrossRef CAS PubMed.
- P. Hadi, J. Barford and G. McKay, J. Environ. Chem. Eng., 2014, 2, 332–339 CrossRef CAS.
- P. Hadi, J. Barford and G. McKay, Chem. Eng. J., 2013, 228, 140–146 CrossRef CAS.
- S. Rengaraj, J.-W. Yeon, Y. Kim, Y. Jung, Y.-K. Ha and W.-H. Kim, J. Hazard. Mater., 2007, 143, 469–477 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qi00182j |
| This journal is © the Partner Organisations 2016 |