
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amphiphilic and double hydrophilic block copolymers containing a polydehydroalanine block†
Mark
Billing
ab,
Grit
Festag
ab,
Peter
Bellstedt
a and
Felix H.
Schacher
 *ab
*ab
aLaboratory of Organic Chemistry and Macromolecular Chemistry, Friedrich-Schiller-University Jena, Humboldtstraße 10, D-07743 Jena, Germany. E-mail: felix.schacher@uni-jena.de
bJena Center for Soft Matter (JCSM), Friedrich-Schiller-University Jena, Philosophenweg 7, D-07743 Jena, Germany
First published on 22nd December 2016
Abstract
We present the synthesis and characterization of amphiphilic and double hydrophilic block copolymers containing a polydehydroalanine (PDha) block. First, polystyrene-block-poly(tert-butoxycarbonylaminomethyl acrylate) (PS-b-PtBAMA) and poly(n-butylacrylate)-block-poly(tert-butoxycarbonylaminomethyl acrylate) (PnBA-b-PtBAMA) were prepared by atom transfer radical polymerization (ATRP). Subsequent deprotection led to the corresponding PS-b-PDha and PnBA-b-PDha block copolymers. All intermediate stages were characterized by 1H-, 13C- or solid state MAS 13C-NMR. Afterwards, the solution behavior of PAA-b-PDha was investigated by a combination of dynamic light scattering, zeta potential measurements, and potentiometric titration – revealing that aggregation occurs upon reaching pH values of 3 or lower. In case of PS-b-PDha, core-corona micelles are formed in aqueous solution, as revealed by a combination of DLS and cryogenic transmission electron microscopy.
Introduction
Polymers carrying charged groups are generally referred to as polyelectrolytes1,2 and, depending on the nature of the functional groups, can be further subdivided into strong and weak polycations and polyanions.3 Whereas strong polyelectrolytes remain permanently charged irrespective of the pH value of the surrounding medium, weak polyelectrolytes exhibit pH-dependent charge characteristics. A further subclass, polyampholytes, features both cationic and anionic charges, either as copolymers or as charged segments in ampholytic block copolymers,4 whereas polyzwitterions are typically characterized by both charges being located within the same monomer unit.3,5–7 All these materials attract continuing interest in different fields of science, as their solution behavior can be influenced through external stimuli such as ionic strength, changes in solvent quality, temperature, or nature and valency of the respective counterions.8–10 As a result, a wide range of applications has been reported for polyelectrolytes and related materials, including sewage treatment, as metal binders (e.g. Cd) in oil recovery, water purification, or as flocculants and emulsifiers.5 In case of polyzwitterions, specific applications in antifouling coatings11 or membranes12 are constantly discussed. Even their use as protective layers for sensors and membranes to extend their lifetime in medical applications or to improve their sensitivity has been reported.12,13Examples for polyampholytes synthesized by free radical polymerization are copolymers of quaternized poly((N,N-dimethylaminoethyl acrylate)-co-(sodium styrene sulfonic acid)) P(DMAEAq-co-NaSS)14 or systems composed of N-(3-aminopropyl) methacrylamide hydrochloride and acrylic acid P(APM-co-MAA).15 In another example, polyampholyte membranes composed of 2-acrylamide-2-methyl propane sulfonic acid (AMPS) and [2-(methacryloyloxy)ethyl] trimethylammonium chloride (DMC) were reported.16 Reversible deactivation radical polymerization (RDRP) techniques have also been used to synthesize well-defined polyampholytes, as shown for poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA)/poly(methacrylic acid) (PMAA) block copolymer brushes using ARGET ATRP,17 or the copolymerization of DMAEMA and MAA18 or 4-vinylbenzoic acid and 4-vinylbenzyl(triphenylphosphonium) chloride via reversible addition fragmentation chain transfer polymerization (RAFT).19
Regarding block polyampholytes, first examples consisting of poly(2-vinylpyridine) (P2VP) and different acrylates/methacrylates were presented by Kamachi et al.20 Giebeler and Stadler later studied the aqueous solution behavior of polyampholytic triblock terpolymers containing both a PMAA and a P2VP segment.21 Such materials were afterwards used for the preparation of multicompartment micelles and, starting from that, the synthesis of nanoscopic hybrid materials,22,23 compartmentalized interpolyelectrolyte complexes,24 and as non-viral vectors in gene delivery applications.25 The temperature responsive self-assembly of zwitterionic block copolymers was recently investigated by the group of Yoshida.26 Here, block copolymers containing N,N-dimethyl-N-(3-(methacrylamido)propyl)aminopropanesulfonate and N-isopropylacrylamide were synthesized via RAFT polymerization and their self-assembly was investigated. In another example, zwitterionic poly(2-methoxyethyl acrylate)-block-poly(3-(N-(2-methacryloyloxyethyl)-N,N-dimethylamino)propane sulfonate) (PMEA-b-PDMAPS) block copolymers were synthesized using ATRP.27
In this context, we recently reported on tert-butoxycarbonylaminomethyl acrylate (tBAMA) as versatile building block for the preparation of polyampholytes and its polymerization using either free radical polymerization or atom transfer radical polymerization (ATRP).28,29 The use of (in principle) orthogonal protective groups for the amino function (tert-butoxy carbonyl) and the carboxylic acid (methyl ester) allows for selective deprotection of either the two functionalities.29 The formed polymers can be subsequently converted into weak polycations, polyanions, or zwitterionic polydehydroalanine (PDha) featuring high charge densities.29,30 Related to that, earlier attempts by Hermes and Mathias employed free radical polymerization of different capto dative methyl(2-alkanamido) propenoates.31–33
In this contribution, we report on the preparation and solution properties of block copolymers containing a polydehydroalanine block (PDha). Starting from previously synthesized PS-b-PtBAMA and PnBA-b-PtBAMA block copolymers, different deprotection reactions led to amphiphilic or double hydrophilic block copolymers. Thereby, we discovered that our previous assumption that either the –COOH or –NH2 moiety of PtBAMA can be selectively deprotected has to be reconsidered. In both cases, considerable amounts of the other protective group (Boc in the case of –NH2 and methyl ester for –COOH) is cleaved off as well. Nevertheless, we show first investigations of the solution properties of the resulting amphiphilic PS-b-PDha or double hydrophilic PAA-b-PDha block copolymers in aqueous media.
Experimental section
Trifluoroacetic acid (Carl Roth GmbH+Co. KG, ≥99.9%) and lithiumhydroxide monohydrate (Carl Roth GmbH+Co. Kg, ≥55%) were used as received. N-(tert-Butoxycarbonyl)-D-serine methyl ester (Carbolution, 98%) was used as received. Styrene (Sigma Aldrich, ≥99%) contained 4-tert-butylcatechol as inhibitor which was removed by column chromatography over neutral aluminum oxide and subsequent distillation before use. The inhibitor of N-butyl acrylate (Sigma Aldrich, ≥99.9%) was removed by column chromatography over neutral aluminum oxide. Tetrahydrofuran (THF) and dichloromethane (DCM) were purified using a PureSolv-EN™ Solvent purification System (Innovative Technology). Any glassware was cleaned in a KOH/iso-propanol bath and dried at 110 °C. All deuterated solvents were obtained from Deutero. For dialysis, a Spectra/Por®Dialysis membrane with a nominal molecular weight cut-off (MWCO) of 1000 g mol−1 (RC) was used.Nuclear magnetic resonance spectroscopy (NMR)
1H-NMR and 13C-NMR spectra were recorded in CDCl3, CD2Cl2, d-TFA, DMSO-d6, or D2O on a Bruker Fourier spectrometer equipped with a direct observe probehead operating at a proton frequency of 300 MHz. Sample temperature was set to 298 K. Chemical shifts are given in parts per million (ppm, [δ]) and were referenced by using the residual signal of the deuterated solvent.Solid state NMR
13C solid-state magic angle spinning (ssMAS) NMR spectra were either acquired utilizing cross polarization with a contact time of 2 ms and a spinning frequency of 15 kHz or in the case of PAA-b-PtBAA utilizing direct excitation and proton decoupling and a spinning frequency of 5 kHz. All data were collected on a Bruker Avance III HD 400 MHz spectrometer equipped with a 4 mm dual channel probe. Sample temperature was set to 303 K. The carbon chemical shifts were referenced externally, setting the high-frequency (methylene) signal of adamantane to 38.5 ppm.34Size-exclusion chromatography (SEC)
SEC was performed on a Shimadzu system (Shimadzu Deutschland GmbH, Duisburg, Germany) equipped with a SCL-10A system controller, a LC-10AD pump, and a RID-10A refractive index detector using a solvent mixture containing chloroform (CHCl3), triethylamine (TEA), and iso-propanol (i-PrOH) (94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:2) at a flow rate of 1 mL min−1 on a PSS SDV linear S (5 μm) column at 40 °C (Polymer Standards Service GmbH, PSS, Mainz, Germany). The system was calibrated with narrowly distributed PS (310–128000 g mol−1) standards (PSS, Mainz, Germany).
:4:2) at a flow rate of 1 mL min−1 on a PSS SDV linear S (5 μm) column at 40 °C (Polymer Standards Service GmbH, PSS, Mainz, Germany). The system was calibrated with narrowly distributed PS (310–128000 g mol−1) standards (PSS, Mainz, Germany).
Water based SEC
SEC was performed on a Jasco system equipped with a DG-2080-53 degasser, PU-980 pump, and a RI-2031 Plus refractive index detector (Jasco Deutschland Labor- und Datentechnik GmbH, Groß-Umstadt, Germany) using 0.1 M Na2HPO4/0.05% NaN3 pH 9 as eluent at a flow rate of 1 mL min−1 on a column set of PSS SUPREMA 1000 Å and 30 Å (10 μm) at 30 °C (PSS, Mainz, Germany).Zeta-potential measurements
The samples for the zeta-potential measurements were prepared by titration of a 1 g L−1 solution of PAA25-b-PDha50 in 0.1 N HCl with 0.1 N NaOH. For the titration and pH detection, a Metrohm 765 Dosimat titrator with a Greisinger electronic GMH3539 digital pH-/mV-electrode with thermometer was used. 1 mL samples for the measurements were taken at the desired pH values. The ζ-potentials were measured using a Zetasizer Nano ZS from Malvern (Malvern Instruments GmbH Gültstein, Germany) via M3-PALS technique with a He–Ne laser operating at 633 nm. The detection angle was 13°. The electrophoretic mobilities (u) were converted into ζ-potentials via the Smoluchowski equation.35where η denotes the viscosity and ε the permittivity of the solution.

Synthesis of tert-butoxycarbonylaminomethyl acrylate (tBAMA)
N-(tert-Butoxycarbonyl)-D-serine methyl ester (10 g, 45.6 mmol) was dissolved in dichloromethane (200 mL). Methanesulfonyl chloride (Ms-Cl; 6 mL, 77.5 mmol) was added to the solution under vigorous stirring. The reaction mixture was cooled to 0 °C and triethylamine (TEA, 23 mL, 165.9 mmol) was added drop-wise. The solution was stirred at 0 °C for 1 h, and further 2 h at room temperature. Then the reaction mixture was washed with a potassium bisulfate solution (1%) to neutrality. The organic phase was dried over Na2SO4, filtered and the solvent removed under reduced pressure. The product was further purified via column chromatography with silica gel (hexane/ethyl acetate v/v 8/2). The product was dried under reduced pressure obtaining a colorless oil in a yield of 87% (8 g, 39 mmol).
1
H-NMR (300 MHz, CDCl
3, δ): 7.00 (s, 1 H, –NH), 6.13 (s, 1 H, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–), 5.70 (s, 1 H –CCH–), 3.8 (s, 3 H, –O–CH3–), 1.46 (s, 9 H, –COO–C(CH3)3) ppm; 13C-NMR (75 MHz, CDCl3, δ): 164.57 (–COO–CH3), 152.82 (–COO–C(CH3)3), 131.13 (CH2C), 105.28 (CH2C), 80.58 (–COO–C(CH3)3), 52.98 (–COO–CH3), 28.45 (–COO–C(CH3)3) ppm.
CH–), 5.70 (s, 1 H –CCH–), 3.8 (s, 3 H, –O–CH3–), 1.46 (s, 9 H, –COO–C(CH3)3) ppm; 13C-NMR (75 MHz, CDCl3, δ): 164.57 (–COO–CH3), 152.82 (–COO–C(CH3)3), 131.13 (CH2C), 105.28 (CH2C), 80.58 (–COO–C(CH3)3), 52.98 (–COO–CH3), 28.45 (–COO–C(CH3)3) ppm.
The block copolymers PS30-b-PtBAMA40 and PnBA25-b-PtBAMA50 were synthesized according to a recently published procedure.28
Synthesis of PS30-b-(PAMA10-co-PDha30)
PS30-b-PtBAMA40 (300 mg) was dissolved in dichloromethane (5 mL). Afterwards, trifluoroacetic acid (1.15 mL, 0.0149 mol, 10 eq. per monomer unit) was added and the reaction was stirred at RT for 19 h. Then, the reaction mixture was precipitated in 40 mL hexane. The isolated block copolymer (257 mg) was dried in vacuo.1 H-NMR (300 MHz, d-TFA, δ): 8.00–6.20 (ArH), 4.45–3.85 (–COOCH3), 3.60–2.40 (overlapping –NH3+, –CH2–CH–, –CH2–CH–, –CH2–C–), 1.50 (–NH–COOC(CH3)3); 13C-NMR (75 MHz, d-TFA, δ): 173.87 (–COO–CH3), 156.16 (–Ar–C, overlapping with solvent signals), 59.12 (–CH2–CH–, –CH2–C–), 50.00 (–COO–CH3), 39.13 (–CH2–CH–, –CH2–C–) ppm.
Synthesis of PS30-b-(PtBAMA3-co-PDha37)
PS30-b-PtBAMA40 (363 mg) was dissolved in dimethyl sulfoxide (6 mL) and water (0.5 mL). Afterwards, LiOH·H2O (1.65 g, 21 eq. per monomer unit) was suspended in the solution and the reaction mixture was heated to 100 °C for 22 h. After cooling down to RT and centrifugation, the precipitated block copolymer was isolated and mixed with water (10 mL), before the solution was neutralized with HClaq. For further purification, the solution was dialyzed against water. Afterwards, the block copolymer was dried in vacuo (210 mg).1 H-NMR (300 MHz, d-TFA, δ): 7.30–6.30 (ArH), 2.40–1.30 (–CH2–CH–, –CH2–CH–, –CH2–C–), 2.20–1.18 (–COO(CH3)3); 13C-NMR (75 MHz, solid state, δ): 179.34 (–COOH, –COOCH3), 157.26 (–NH–COO–(CH3)3), 146.37/128.53 (–Ar–C–), 61.99 (–CH2–C–, –CH2–CH–), 46.27 (–CH2–C–), 42.34 (–CH2–CH–), 30.24 (–COO(CH3)3) ppm.
Synthesis of PS30-b-PDha40
PS30-b-(PtBAMA3-co-PDha37) (100 mg) was dissolved in water (10 mL) and trifluoroacetic acid (1 mL, 0.0129 mol, 26 eq. per monomer unit) was added. The reaction mixture was stirred for 96 h and subsequently neutralized using sodium hydroxide solution (0.2 M). Afterwards, the dissolved block copolymer was further purified by dialysis against water. The isolated block copolymer was dried in vacuo (107 mg).1 H-NMR (300 MHz, d-TFA, δ): 7.30–6.30 (ArH), 1.30–2.40 (–CH2–CH–, –CH2–CH–, –CH2–C–); 13C-NMR (75 MHz, solid state, δ): 179.34 (–COOH, –COOCH3), 146.37/128.53 (–Ar–C–), 61.99 (–CH2–C–, –CH2–CH–), 46.27 (–CH2–C–), 42.34 (–CH2–CH–) ppm.
Synthesis of PnBA25-b-(PAMA15-co-PDha35)
PnBA25-b-PtBAMA50 (300 mg) was dissolved in dichloromethane (5 mL). Afterwards, trifluoroacetic acid (1.15 mL, 0.0149 mol, 10 eq. per monomer unit) was added and the reaction was stirred at 50 °C for 1 h. After cooling down to RT, the reaction mixture was precipitated in 40 mL hexane. The isolated block copolymer (176 mg) was dried in vacuo.1 H-NMR (300 MHz, CD 2 Cl 2, δ): 4.15 (–COOCH2–), 2.39 (–CH2–CH–), 1.98 (–CH2–CH–), 1.65 (–COO–CH2–CH2–), 1.45 (–CH2–CH3), 0.95 (–CH2–CH3); 13C-NMR (75 MHz, CD2Cl2, δ): 176.52 (–COOCH2–), 66.15 (–COOCH2–), 43.46 (–CH2–CH–), 37.00 (–CH2–CH–), 32.33 (–CH2–CH2–CH2–CH3), 21.09 (–CH2–CH2–CH2–CH3), 15.37 (–CH2–CH2–CH2–CH3) ppm.
Under these conditions, only signals corresponding to the PnBA segment of the block copolymer can be detected. Hence, we also conducted NMR studies in deuterated TFA:
1 H-NMR (300 MHz, d-TFA, δ): 4.15 (–COOCH2–), 3.95 (–COOCH3), 3.4–2.4 (overlaying NH3+ and PAMA-backbone), 2.39 (–CH2–CH–), 1.98 (–CH2–CH–), 1.85 (–COO–CH2–CH2–), 1.45 (–CH2–CH3), 1.10 (–CH2–CH3); 13C-NMR (75 MHz, d-TFA, δ): 180.52 (–COOCH2–), 180.34 (–COO–), 66.15 (–COOCH2–, –CH2–C–, –CH2–CH–), 65.00 (–COOCH3), 43.46 (–CH2–CH–), 37.00 (–CH2–C), 32.33 (–CH2–CH2–CH2–CH3), 21.09 (–CH2–CH2–CH2–CH3), 15.37 (–CH2–CH2–CH2–CH3) ppm.
Synthesis of PnBA25-b-(PtBAA20-co-PDha30)
PnBA40-b-PtBAMA50 (300 mg) was dissolved in DMSO (5 mL). Afterwards, LiOH·H2O (1.32 g, 21 eq. per monomer unit) was suspended in the solution and the reaction mixture was heated to 80 °C for 3 h. After cooling down to RT the precipitated block copolymer was mixed with water (10 mL), and the dispersion was neutralized with HClaq. After dialysis against water, the block copolymer was dried in vacuo (159 mg).1 H-NMR (300 MHz, D 2 O, δ): 4.0 (–COO–CH2–), 2.40 (overlapping, –CH2–CH–, –CH2–C–, –CH2–CH–)), 1.40 (–NH–COO(CH3)3, –COO–CH2–CH2–CH2–CH3, –COO–CH2–CH2–CH2–CH3), 0.95 (–CH2–CH3); 13C–NMR (75 MHz, D2O, δ): 176.52 (–CH–COO–), 174.14 (–C–COOH), 153.59 (–NH–COO(CH3)3), 81.20 (–C(CH3)), 66.10 (–COO–CH2, CH2–CH–), 60.21 (–CH2–C–), 41.2 (–CH2–C–), 38.00 (–CH2–CH–), 32 (–COO–CH2–CH2–), 28.00 (–NH–COO(CH3)3), 19.00 (–CH2–CH3), 12.00 (–CH2–CH3) ppm.
Under these conditions, mainly signals corresponding to the PnBA segment of the block copolymer can be detected. Hence, we also conducted NMR studies in deuterated TFA:
1 H-NMR (300 MHz, d-TFA, δ): 4.50 (–COO–CH2–), 3.60–2.60 (overlapping NH3+, –CH2–C–), 2.45 (–CH2–CH–), 2.20 (–CH2–CH–), 1.98 (–COO–CH2–CH2–), 1.85 (–CH2–CH3), 1.65 (–NH–COO(CH3)3), 1.2 (–CH2–CH3); 13C-NMR (75 MHz, d-TFA, δ): 179.24 (–CH–COO–), 179.14 (–C–COOH), 161.00 (overlapping with solvent signal, –NH–COO(CH3)3), 92.00 (–C(CH3)), 70.10 (–COO–CH2), 65.21 (–CH2–C–, –CH2–CH–), 42.00 (–CH2–CH–), 40.00 (–CH2–C–), 37.00 (–NH–COO(CH3)3), 28.7 (–CH2–CH2–CH3), 19 (–CH2–CH3), 15.00 (–CH2–CH3) ppm.
Synthesis of PAA25-b-PDha50
PnBA25-b-(PtBAA20-co-PDha30) (100 mg) was dissolved in water (10 mL) and afterwards trifluoroacetic acid (1 mL, 0.0129 mol, 26 eq. per monomer unit) was added. The reaction mixture was stirred for 96 h at RT, and then neutralized using a sodium hydroxide solution (0.2 M). Afterwards, the dissolved block copolymer was further purified by dialysis against water and dried in vacuo (112 mg).1 H-NMR (300 MHz, D 2 O, δ): 3.2–1.2 (–CH2–CH–, –CH2–CH–, –CH2–C–), 13C-NMR (75 MHz, D2O, δ): 184.01 (–C–COOH), 179.42 (–CH–COOH), 61.50 (–CH2–C–), 59.40 (–CH2–CH–), 45.06 (–CH2–C–), 43.64 (–CH2–CH–) ppm.
Results and discussion
We herein report the synthesis and solution behavior of block copolymers containing a polydehydroalanine (PDha) segment and (at that stage) either a hydrophobic PS or a hydrophilic PAA block. We are interested in such materials as, depending on the co-block, either amphiphilic or double hydrophilic block copolymers with pH-dependent charge characteristics are obtained and these could serve as interesting building blocks in interpolyelectrolyte complexes (IPECs). The materials are synthesized using sequential atom transfer radical polymerization (ATRP) of styrene/n-butyl acrylate, followed by tBAMA, as reported recently.28 Subsequently, deprotection of either the carboxyl moiety or the amino functionality is carried out, followed by conversion to polyampholytic PDha-containing block copolymers (Schemes 1 and 2). First, macroinitiators of PS30-Br and PnBA25-Br were synthesized (the subscripts denote the corresponding degree of polymerization).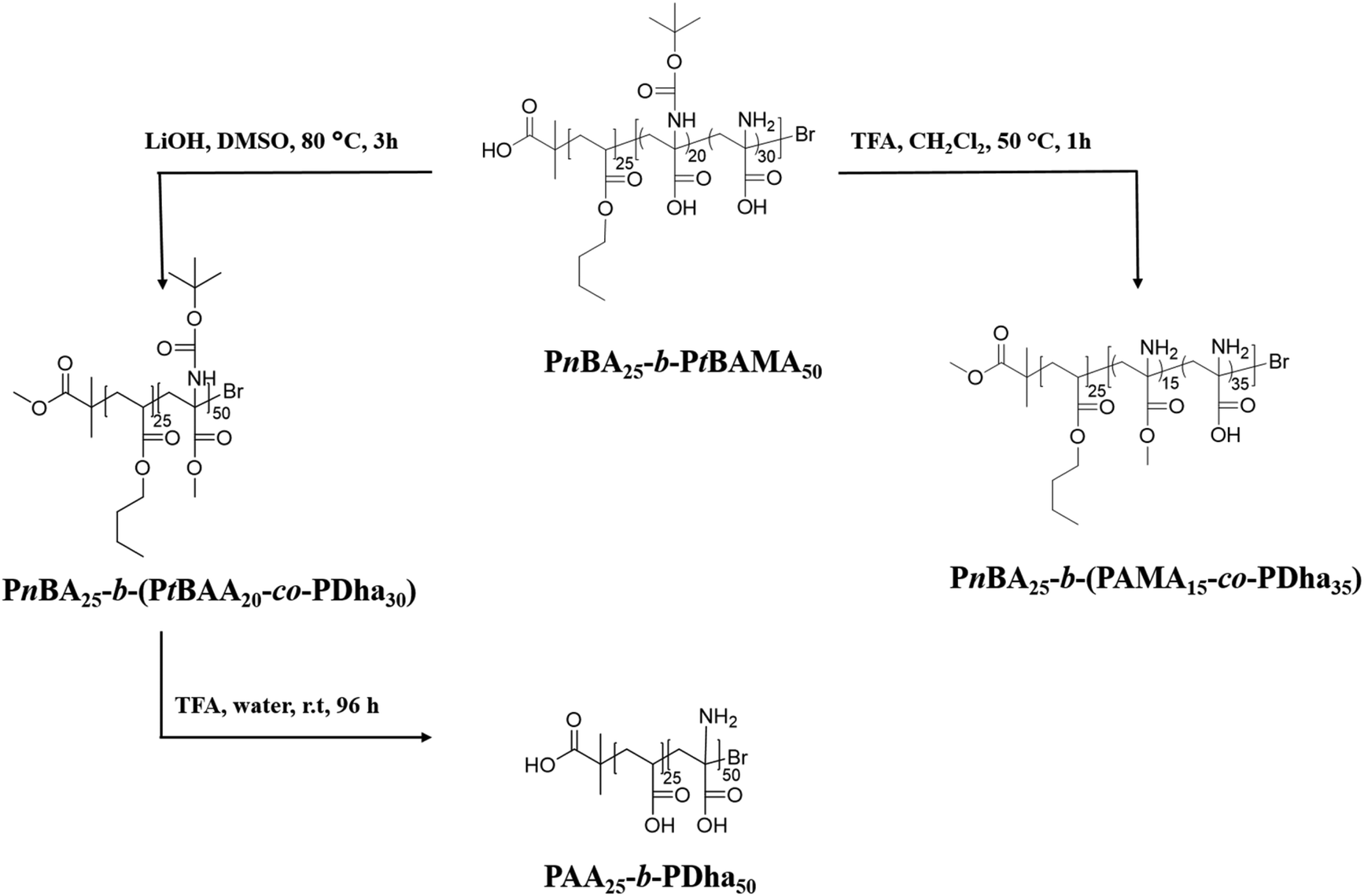 | ||
| Scheme 1 Structure of PnBA25-b-PtBAMA50 and the resulting block copolymers after the respective deprotection steps. | ||
 | ||
| Scheme 2 Structure of PS30-b-PtBAMA40 and the corresponding block copolymers after different deprotection steps. | ||
Subsequent block extension in anisole at 50 °C yielded PS30-b-PtBAMA40 and PnBA25-b-PtBAMA50 block copolymers with narrow dispersity (Table 1). The composition was determined using 1H-NMR spectroscopy. Thereby, hydrolysis of PS30-b-PtBAMA40 will lead to amphiphilic block copolymers, whereas in the case of PnBA25-b-PtBAMA50 double hydrophilic PAA25-b-PDha40 will be obtained (Scheme 1).
| Entry | (Block co-) polymer | M n [g mol−1] | M n, theo [g mol−1] | Đ |
|---|---|---|---|---|
| a Determined by SEC (CHCl3/TEA/i-PrOH: 94/4/2). PS-calibration. b Determined by SEC 0.1 M Na2HPO4 pH 9. Pullulan calibration. | ||||
| 1 | PnBA25-Br | 3600a | 3200a | 1.13a |
| 2 | PnBA25-b-PtBAMA50 | 8100a | 23600a |
1.18a |
| 3 | PnBA25-b-(PAMA15-co-PDha35) | — | 18500 |
— |
| 4 | PnBA25-b-(PtBAA20-co-PDha30) | — | 20000 |
— |
| 5 | PAA25-b-PDha50 | 21800b |
14900b |
1.35b |
| 6 | PS30-Br | 3400a | 3100a | 1.10a |
| 7 | PS30-b-PtBAMA40 | 7900a | 23400a |
1.38a |
| 8 | PS30-b-(PAMA10-co-PDha30) | — | 19400 |
— |
| 9 | PS30-b-(PtBAMA3-co-PDha37) | — | 22800 |
— |
| 10 | PS30-b-PDha40 | — | 18800 |
— |
Synthesis of PAA25-b-PDha50
We have earlier reported on the (selective) deprotection of either the Boc protective group using TFA at 50 °C or the hydrolysis of the methyl ester of PtBAMA homopolymers at 100 °C using LiOH·H2O.29,30 However, during the course of these studies we observed that both for PnBA25-b-PtBAMA50 and PS30-b-PtBAMA40 also significant deprotection of the other moiety occurred, e.g. treatment with TFA at RT removed both Boc group and the methyl ester. We therefore varied the conditions (50 °C and 1 h reaction time) under which these reactions have been carried out, although without obtaining a truly orthogonal procedure so far. Nevertheless, in our opinion the final amphiphilic and double hydrophilic block copolymers featuring PDha segments represent the most interesting materials and for selective deprotection of either –COOH or –NH2 presumably another combination of protective groups might be favorable, at least in the case of block copolymers. Below we describe the so far most selective reaction conditions whereas more detailed descriptions including 1H-NMR and 13C-NMR spectroscopy are discussed in the ESI (Fig. S1 and S2†).We first start by partial hydrolysis of PnBA25-b-PtBAMA50. Starting from PnBA25-b-PtBAMA50, the Boc protective group was cleaved by adding TFA (10 eq. per monomer unit) in dichloromethane at 50 °C for 1 h.36 After precipitation in hexane, the block copolymer was characterized by NMR (Fig. 1). As can be seen, the signal of the Boc group at 1.5 ppm disappears (∼100%), indicating complete cleavage. A new broad signal ranging from 2.4 to 3.4 ppm can be found, indicating the presence of a protonated amine functionality. Further, the alkyl chain of the nBA ester (4.15, 1.85, 1.45 and 1.1 ppm) and the corresponding backbone signals (2.39 and 1.98 ppm) have not been affected, whereas the signal for the methyl ester of the former PtBAMA segment (3.95 ppm) decreased to approximately 30% intensity (resulting in PnBA25-b-(PAMA15-co-PDha35)). Obviously, the chosen conditions are too harsh for the methyl ester, leading to partial cleavage. Compared to earlier studies, here the second block can be used as internal reference, thereby improving the accuracy of NMR characterization. The cleavage of the methyl ester (and the butyl ester of PnBA) was carried out at 80 °C for 3 h and using 21 eq. LiOH·H2O.29,37 After neutralization using HClaq and subsequent dialysis against water, 1H-NMR in deuterated TFA showed typical signals for PnBA (4.5, 2.45, 2.20, 1.98, 1.85 and 1.2 ppm) and PtBAA (3 ppm and 1.65 ppm). The intensity of the Boc group at 1.5 ppm decreased to 30%, again indicating unselective cleavage (PnBA25-b-(PtBAA20-co-PDha30)). The removal of the Boc protective group during treatment with LiOH·H2O was already observed. In further studies, simultaneous cleavage of the Boc and methyl ester in N-Boc alanine methyl ester was justified by the possibility of water to act as dual acid/base catalyst.38,39
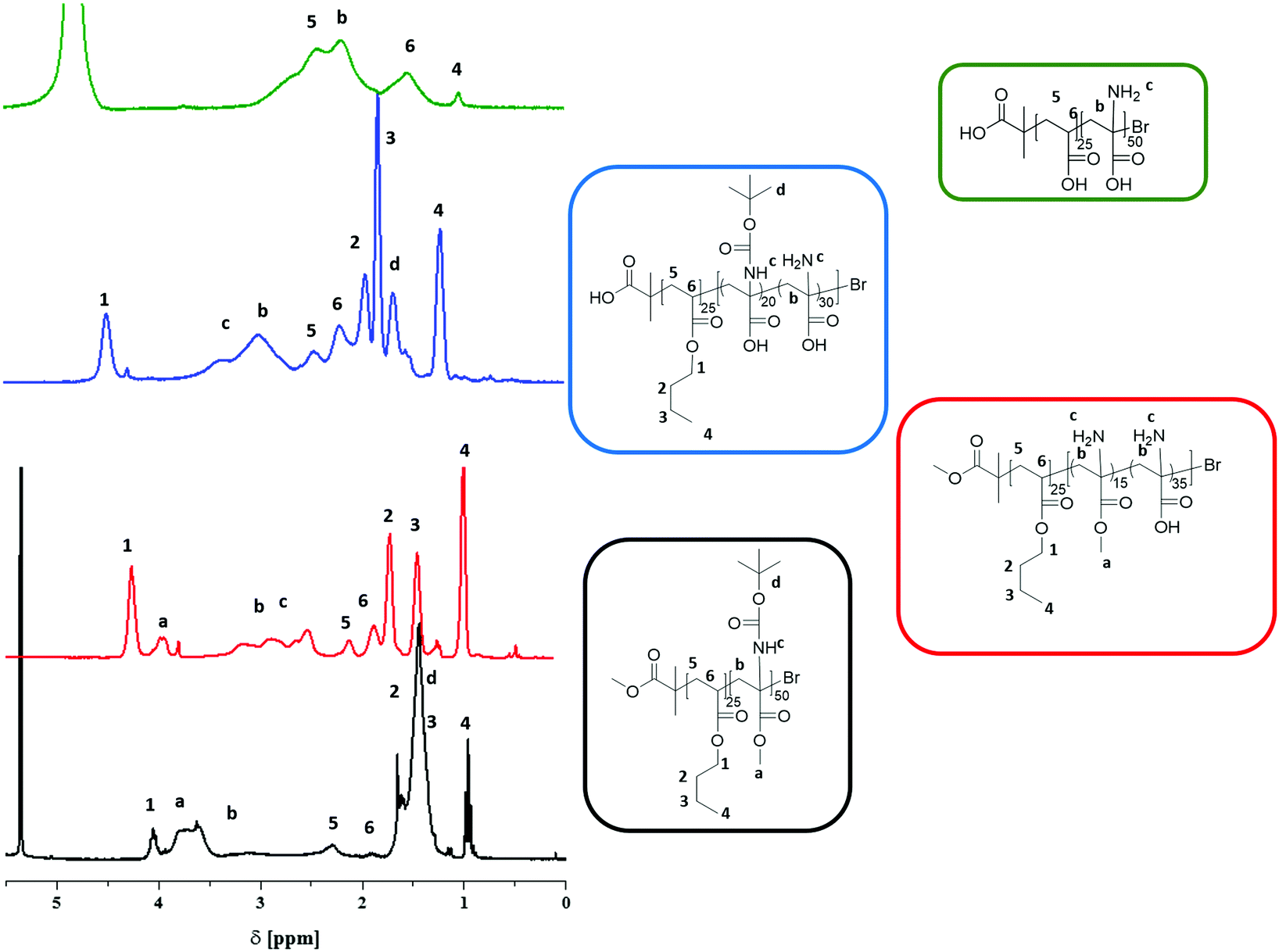 | ||
| Fig. 1 1H-NMR of PnBA25-b-PtBAMA50 (black line, CDCl3), PnBA25-b-(PAMA15-co-PDha35) (red line, d-TFA), PnBA25-b-(PtBAA20-co-PDha30) (blue line, D2O), PAA25-b-PDha50 (green line, D2O), numbers indicate signals which have been allocated to PnBA/PAA, letters indicate signals related to PtBAMA, PAMA, PtBAA and PDha. | ||
Subsequent full deprotection was achieved by treatment of PnBA25-b-(PtBAA20-co-PDha30) with TFA at RT in water for 96 h. The resulting PAA25-b-PDha50 block copolymer is well soluble in water and 1H-NMR in D2O shows only signals for the polymer backbone (3.2–1.2 ppm), with the exception that a small fraction of remaining PnBA can still be observed (below 10%).
The results obtained by 1H-NMR were confirmed by 13C-NMR (Fig. 2). For PnBA-b-PtBAMA, the signals of the backbone (61 and 41 ppm), the carbonyl group of PnBA (172 ppm), the methyl ester of PtBAMA (172 ppm) and the Boc protective group (153 ppm, 79 ppm, 28 ppm) can be observed. The signals of the butyl group of PnBA can be detected at 64, 30, 19 and 13 ppm and the methyl ester at 52 ppm. For PnBA25-b-(PAMA15-co-PDha35, measured in d-TFA) the Boc group (153, 79, 28 ppm) is absent and the methyl ester (65 ppm) can still be detected. For PnBA25-b-(PtBAA20-co-PDha30), the signals for the carbonyl moieties could be detected (179.24 ppm and 179.14 ppm) and even the methyl groups of the Boc group (37 ppm) are visible. Also, both PnBA (70, 65, 42, 28, 19 and 15 ppm) and PtBAA (161, 92, 70 and 42 ppm) can be clearly identified. For PAA-b-PDha, mainly carbon atoms of the backbone (61, 59, 45 and 43 ppm) as well as the carbonyl groups (184 and 179 ppm) of the acid functions are observed (NMR in D2O). The end group of PnBA-b-PtBAMA (and also for PS-b-PtBAMA) could not be detected in NMR spectroscopy. However, almost complete re-initiation of PtBAMA prepared via ATRP was possible, hinting to a good endgroup fidelity.28 During the deprotection steps, we expect to hydrolyze both the terminal bromine as well as the ester moiety of the ATRP initiator.
 | ||
| Fig. 2 13C-NMR of PnBA25-b-PtBAMA50 (black line, CDCl3), PnBA25-b-(PAMA15-co-PDha35) (red line, d-TFA), PnBA25-b-(PtBAA20-co-PDha30) (blue line, D2O), PAA25-b-PDha50 (green line, D2O), numbers indicate signals of PnBA/PAA, letters indicate signals of PtBAMA, PAMA, PtBAA and PDha. | ||
Differences in solubility of the purified intermediate stages are discussed in the ESI.† It was possible to measure NMR of PnBA25-b-(PAMA15-co-PDha35) in DCM and TFA as well as PnBA25-b-(PtBAA20-co-PDha30) in TFA and in D2O (Fig. S3–S6†). After complete deprotection, SEC (0.1 M Na2HPO4/NaN3 pH 9, pullulan calibration) of PAA25-b-PDha50 showed a monomodal distribution, indicating an intact polymer backbone after the different reaction steps (Table 1, Fig. 3A).
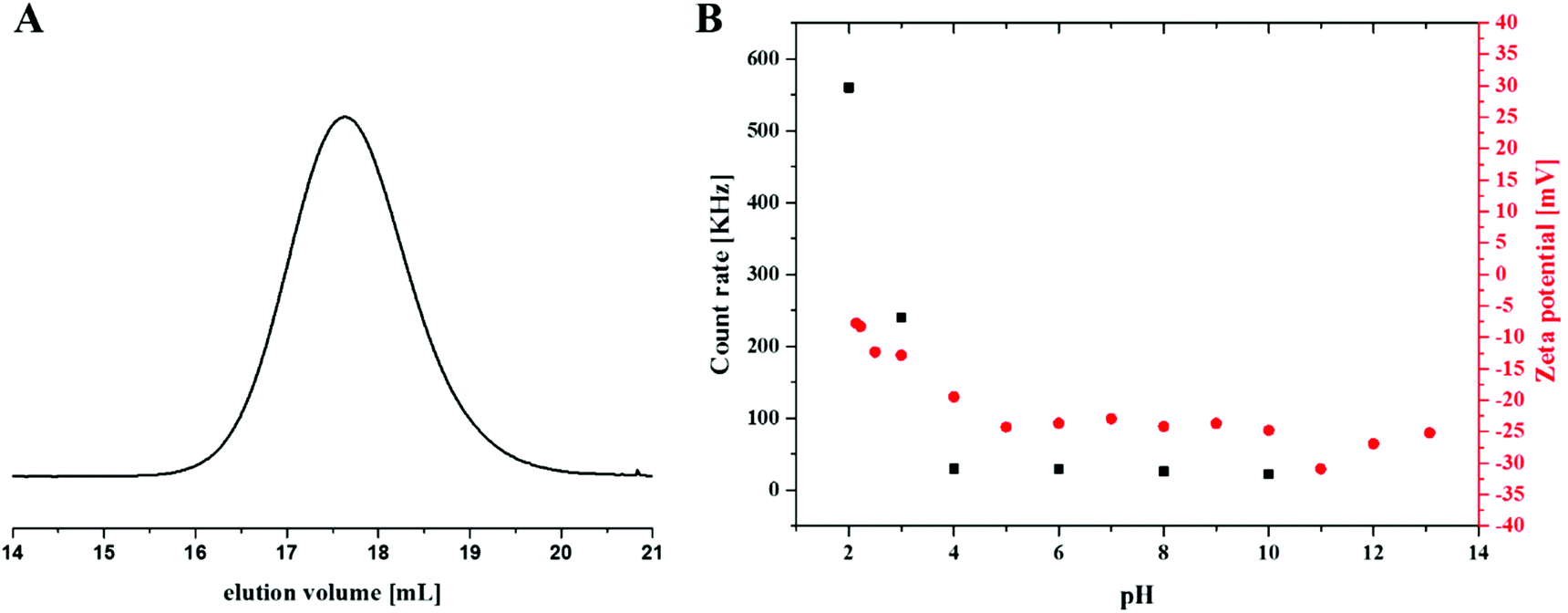 | ||
| Fig. 3 (A) SEC elution trace of PAA25-b-PDha50 in 0.1 M Na2HPO4 at pH 9 (Pullulan calibration); (B) zeta potential measurements of PAA25-b-PDha50 at different pH values (red squares), and the corresponding average count rates of PAA25-b-PDha50 in aqueous media at different pH value as obtained using DLS experiments (black squares). | ||
Synthesis of PS30-b-PDha40
PS30-b-PtBAMA40 exhibits signals for PS (6.5–7.5 ppm, and 2–1.5 ppm) and PtBAMA (5.7–5.2 ppm –NH, 3.45–3.89 ppm, –OCH3, 3.3–2 ppm –C–CH2–, 1.6–1.3 ppm –(C–(CH3)3), Fig. S5†). In 13C-NMR, signals for PS (147, 129, 127, 61 and 46 pm) as well as for PtBAMA (174, 156, 81, 61, 55, 42 and 30 ppm) can be observed (Fig. S6†). Deprotection of the Boc group was carried out using 10 eq. TFA in dichloromethane at RT. After 19 h, the reaction mixture was neutralized with a sodium hydroxide solution and the block copolymer was precipitated in hexane. According to 1H-NMR in d-TFA, a deprotection of 93% could be confirmed through the decrease of the signal at 1.5 ppm. Besides, the corresponding signal of the amine function changed from 5.5 ppm in CDCl3 to 3.0 ppm in d-TFA, similar to earlier observations for PnBA-b-PAMA. Also, here this treatment leads to a simultaneous removal of the methyl ester (4.2 ppm, Fig. S5†) of approximately 75% (PS30-b-(PAMA10-co-PDha30)). In 13C-NMR (d-TFA), the backbone carbon signals (59 and 39 ppm) and the signal for the methyl ester (50 ppm) could be detected (Fig. S8†).Removal of the methyl ester was performed as described above for PnBA25-b-PtBAMA50 using 21 eq. LiOH·H2O for 24 h at 100 °C. After neutralization and precipitation, the resulting block copolymer was characterized by NMR in DMSO-d6. Here, only signals for the PS segment (7.3–6.3 ppm) and the block copolymer backbone (2.4–1.32 ppm) were observed, and the intensity of the Boc group decreased to 7% (1.25 ppm). However, almost complete cleavage of the methyl ester was achieved (>90%, PS30-b-(PtBAMA3-co-PDha37)). In this particular case, solid state NMR was carried out due to low solubility of the material in common solvents (1H-NMR: Fig. S7,†13C-NMR: Fig. S8†). Here, the carbons of the aromatic ring (146, 129 ppm) can be detected (Fig. S7†), as well as the carbonyl moiety of the ester (179 ppm) and the Boc group (157 ppm). Further, a small signal for the Boc group was detected at 30 ppm, again confirming the partial cleavage, and additionally the backbone signals (62 and 46 ppm) were visible.
The preparation of PS30-b-PDha40 was carried out from PS30-b-(PtBAMA3-co-PDha37) by hydrolysis with TFA as described above. After neutralization and dialysis against water, the block copolymer was characterized by 1H-NMR in DMSO. Signals for the PS segment at 7.2–6.4 ppm and the block copolymer backbone (2.2–1.5 ppm) were observed, as well as a small amount of remaining Boc group (7%, 1.5 ppm, Fig. S7†). Solid state 13C-NMR of PS40-b-PDha30 revealed signals at 180, 146, 62 and 46 and 42 ppm (Fig. S8†).
Solution behavior of PAA25-b-PDha50 and PS30-b-PDha40
We were interested in the solution behavior of the above described amphiphilic and double hydrophilic block copolymers. Starting with PAA25-b-PDha50, we investigated the pH-dependent solubility and net charge using a combination of zeta-potential measurements and dynamic light scattering (Fig. 3B). Starting under basic conditions (pH 14), the block copolymer shows a negative zeta potential (approximately −25 mV) and this value remains more or less constant until pH 5, presumably due to the presence of deprotonated –COO− moieties. Upon acidification, at pH 4 the zeta potential starts to increase up to −5 mV at pH 2. This hints towards both the protonation of the carboxylates as well as the amino moiety. We explain the fact that the zeta potential remains slightly negative mainly by partial aggregation of the block copolymer at lower pH values.These findings were supported by DLS experiments at different pH values in aqueous solution at a concentration of 1 g L−1 (Fig. 3B and 4). As can be seen, below pH values of 4, a significant increase in apparent count rate occurs, hinting towards aggregation of PAA25-b-PDha50. This process is accompanied by increasing turbidity. DLS CONTIN plots of PAA25-b-PDha50 at different pH values show mainly unimers at pH 10, as well as at pH 6 and 4 (Fig. 4, hydrodynamic radii of 1.5 to 3 nm are detected, hinting towards the presence of mainly block copolymer unimers). Under acidic conditions (pH 3), distinct differences were observed as the degree of protonation for both the carboxylic acid groups of PAA and PDha increases. This leads to a decreased solubility, followed by aggregation and the respective DLS experiments revealed an apparent hydrodynamic radius of 220 nm, pointing towards the presence of loosely defined block copolymer aggregates (Fig. 4).
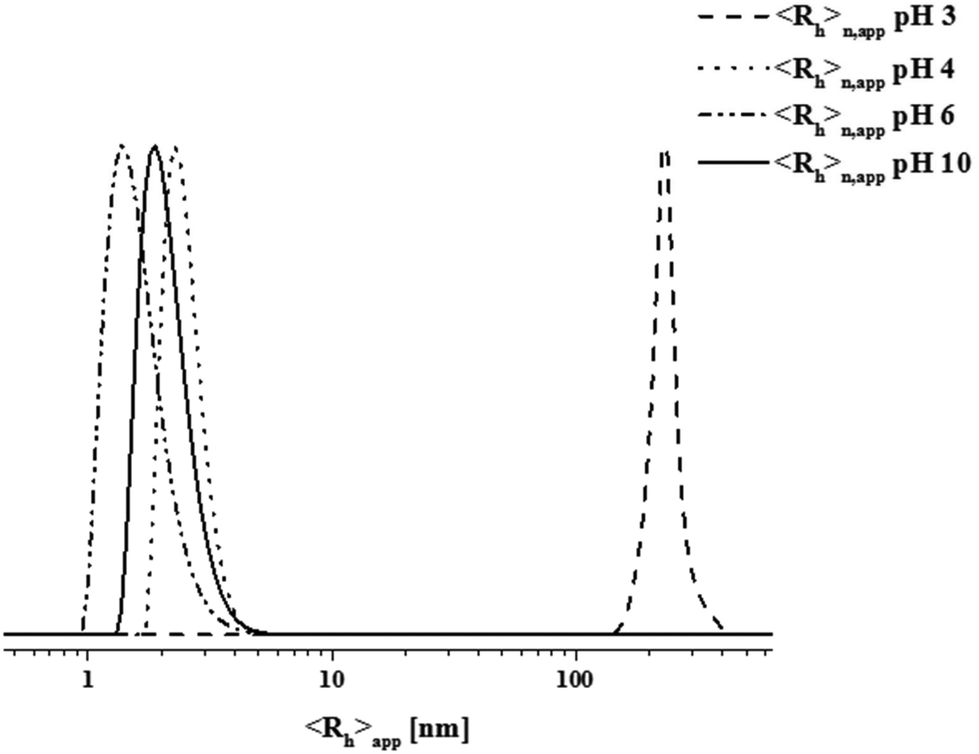 | ||
| Fig. 4 DLS CONTIN plots of PAA25-b-PDha50 at pH 10 (solid black line, <Rh>n, app = 2 nm), pH 6 (dashed–dotted line, <Rh>n, app = 1.5 nm), pH 4 (dotted line, <Rh>n, app = 2.5 nm), and pH 3 (dashed line, <Rh>n, app = 220 nm) – concentration was 1 g L−1 in all cases. | ||
We also carried out potentiometric titrations of PAA25-b-PDha50 at a concentration of 3 g L−1, starting under basic conditions (pH 13, 0.1 M NaOH, double negatively charged block copolymer unimers are present as discussed above) by step-wise addition of HCl (0.1 M, Fig. 5). Here, the first steps (pH 13.0–11.0) mainly correspond to the neutralization of excess NaOH. According to previous studies, the pKB of the –NH2 moiety in PDha is around 9.2, indicating a pH-range of increasing protonation from pH 11–7.29 In that region, the block copolymer consists of a negatively charged segment (PAA) and a zwitterionic block (PDha). Further decrease of the pH value leads to continuous protonation of the amino functionality. For the –COOH groups, typical pKA values are reported around 6–7.40–44 Therefore, at a pH value of 5 the majority of –COOH moieties is protonated, leading to a charge neutral PAA segment and mainly positive charges within the PDha block. This then leads to aggregation at lower pH values (pH < 4), accompanied by increasing turbidity and precipitation at pH 2.3 (see also DLS experiments).
 | ||
| Fig. 5 Potentiometric titration of PAA25-b-PDha50 with 0.1 M HCl starting from 0.1 M NaOH. | ||
We were also interested in the solution properties of amphiphilic PS30-b-PDha40 and therefore dissolved the block copolymer in water (D2O) by short heating to 80 °C at a concentration of c = 0.1 g L−1. After cooling down, a turbid solution was obtained, which we ascribed to the micellization of PS30-b-PDha40 (Fig. 6). The core of these aggregates is formed by the hydrophobic PS block and the corona is formed by zwitterionic PDha under these conditions. Subsequent DLS measurements revealed the presence of micelles with a hydrodynamic radius of 51 nm, as well as some larger aggregates above 200 nm in radius (Fig. 6A). Additional cryo-TEM measurements revealed the presence of aggregates with a diameter of 67 nm ± 10 nm (measurement of 50 particles), being in relatively good agreement with the size regime obtained from DLS experiments as here only the PS core is directly visible (Fig. 6B).
 | ||
| Fig. 6 DLS CONTIN plot of PS30-b-PDha40 in D2O (0.1 g L−1, <Rh>n, app = 51, 224 nm, (A) and cryo-TEM micrograph of a micellar solution of PS30-b-PDha40 (B). | ||
Conclusion
We have demonstrated the transformation of previously synthesized PS-b-PtBAMA and PnBA-b-PtBAMA diblock copolymers into double hydrophilic PAA-b-PDha and amphiphilic PS-b-PDha materials by sequential deprotection steps. Thereby, we discovered that neither the TFA-mediated deprotection of the Boc moiety nor the hydrolysis of the methyl ester under basic conditions using LiOH are truly selective. Instead, in both cases already considerable amounts of PDha are generated directly – as evidenced mainly by in-depth NMR investigations. Although the focus of this work here was mainly put on the final PDha-based block copolymers, future work will also be directed towards different monomer substitution patterns which can be orthogonally addressed. Nevertheless, the resulting block copolymers revealed interesting solution properties in aqueous media, as shown for example by pH-dependent DLS and zeta potential measurements in the case of PAA-b-PDha or first micellization studies of PS-b-PDha. Such materials in our opinion are of interest as building blocks for micellar interpolyelectrolyte complexes (IPECs) or for the preparation of membranes with charge-tunable separation layers.Acknowledgements
The authors thank Kristin Schreyer (FSU Jena) for help with SEC measurements and Philip Biehl for performing cryo-TEM. FHS is grateful to the Thuringian Ministry of Science, Education, and Culture (TMBWK; grants #B515-10065, ChaPoNano and #B514-09051, NanoConSens). We would further like to acknowledge the NMR-platform at the Friedrich-Schiller-University Jena for support in NMR spectroscopy. FHS is also grateful to Ulrich S. Schubert for continuous support.References
- A. V. Dobrynin and M. Rubinstein, Prog. Polym. Sci., 2005, 30, 1049–1118 CrossRef CAS.
- A. Laschewsky, Curr. Opin. Colloid Interface Sci., 2012, 17, 56–63 CrossRef CAS.
- D. V. Pergushov, A. H. E. Müller and F. H. Schacher, Chem. Soc. Rev., 2012, 41, 6888–6901 RSC.
- A. V. Dobrynin, R. H. Colby and M. Rubinstein, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 3513–3538 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Chem. Rev., 2002, 102, 4177–4190 CrossRef CAS PubMed.
- A. Laschewsky, Polymers, 2014, 6, 1544 CrossRef CAS.
- S. Kudaibergenov, W. Jaeger and A. Laschewsky, in Supramolecular Polymers Polymeric Betains Oligomers, Springer Berlin Heidelberg, Berlin, Heidelberg, 2006, pp. 157–224 Search PubMed.
- I. Roy and M. N. Gupta, Chem. Biol., 2003, 10, 1161–1171 CrossRef CAS PubMed.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS PubMed.
- D. A. Mortimer, Polym. Int., 1991, 25, 29–41 CrossRef CAS.
- S. Jiang and Z. Cao, Adv. Mater., 2010, 22, 920–932 CrossRef CAS PubMed.
- F. Xuan and J. Liu, Polym. Int., 2009, 58, 1350–1361 CrossRef CAS.
- P.-S. Liu, Q. Chen, S.-S. Wu, J. Shen and S.-C. Lin, J. Membr. Sci., 2010, 350, 387–394 CrossRef CAS.
- A. B. Ihsan, T. L. Sun, T. Kurokawa, S. N. Karobi, T. Nakajima, T. Nonoyama, C. K. Roy, F. Luo and J. P. Gong, Macromolecules, 2016, 49, 4245–4252 CrossRef.
- J. Zhao, N. A. D. Burke and H. D. H. Stöver, RSC Adv., 2016, 6, 41522–41531 RSC.
- X. Shen, X. Yin, Y. Zhao and L. Chen, Colloid Polym. Sci., 2015, 293, 1205–1213 CAS.
- Y. K. Jhon, S. Arifuzzaman, A. E. Özçam, D. J. Kiserow and J. Genzer, Langmuir, 2012, 28, 872–882 CrossRef CAS PubMed.
- Q. Zhang and R. Hoogenboom, Chem. Commun., 2015, 51, 70–73 RSC.
- R. Wang and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2468–2483 CrossRef CAS.
- M. Kamachi, M. Kurihara and J. K. Stille, Macromolecules, 1972, 5, 161–167 CrossRef CAS.
- E. Giebeler and R. Stadler, Macromol. Chem. Phys., 1997, 198, 3815–3825 CrossRef CAS.
- E. Betthausen, M. Drechsler, M. Förtsch, F. H. Schacher and A. H. E. Müller, Soft Matter, 2011, 7, 8880–8891 RSC.
- F. Schacher, A. Walther and A. H. E. Müller, Langmuir, 2009, 25, 10962–10969 CrossRef CAS PubMed.
- E. Betthausen, M. Drechsler, M. Förtsch, D. V. Pergushov, F. H. Schacher and A. H. E. Müller, Soft Matter, 2012, 8, 10167–10177 RSC.
- A. C. Rinkenauer, A. Schallon, U. Günther, M. Wagner, E. Betthausen, U. S. Schubert and F. H. Schacher, ACS Nano, 2013, 7, 9621–9631 CrossRef CAS PubMed.
- Z. A. Jiménez and R. Yoshida, Macromolecules, 2015, 48, 4599–4606 CrossRef.
- I. Javakhishvili, K. Jankova and S. Hvilsted, Polym. Chem., 2013, 4, 662–668 RSC.
- M. Billing and F. H. Schacher, Macromolecules, 2016, 49, 3696–3705 CrossRef CAS.
- U. Günther, L. V. Sigolaeva, D. V. Pergushov and F. H. Schacher, Macromol. Chem. Phys., 2013, 214, 2202–2212 Search PubMed.
- M. von der Lühe, U. Günther, A. Weidner, C. Gräfe, J. H. Clement, S. Dutz and F. H. Schacher, RSC Adv., 2015, 5, 31920–31929 RSC.
- R. E. Hermes, L. J. Mathias and J. W. Virden, Macromolecules, 1987, 20, 901–903 CrossRef CAS.
- L. J. Mathias and R. E. Hermes, Macromolecules, 1988, 21, 11–13 CrossRef CAS.
- L. J. Mathias and R. E. Hermes, Macromolecules, 1986, 19, 1536–1542 CrossRef CAS.
- C. R. Morcombe and K. W. Zilm, J. Magn. Reson., 2003, 162, 479–486 CrossRef CAS PubMed.
- P. Leroy, C. Tournassat, O. Bernard, N. Devau and M. Azaroual, J. Colloid Interface Sci., 2015, 451, 21–39 CrossRef CAS PubMed.
- A. M. Walji, E. D. Hostetler, H. Selnick, Z. Zeng, P. Miller, I. Bennacef, C. Salinas, B. Connolly, L. Gantert, M. Holahan, S. O'Malley, M. Purcell, K. Riffel, J. Li, J. Balsells, J. A. Obrien, S. Melquist, A. Soriano, X. Zhang, A. Ogawa, S. Xu, E. Joshi, J. Della Rocca, F. J. Hess, J. Schachter, D. Hesk, D. Schenk, A. Struyk, K. Babaoglu, T. G. Lohith, Y. Wang, K. Yang, J. Fu, J. L. Evelhoch and P. J. Coleman, J. Med. Chem., 2016, 59, 4778–4789 CrossRef CAS PubMed.
- T. Zhou, R. C. Hider, P. Jenner, B. Campbell, C. J. Hobbs, S. Rose, M. Jairaj, K. A. Tayarani-Binazir and A. Syme, Eur. J. Med. Chem., 2010, 45, 4035–4042 CrossRef CAS PubMed.
- J. Wang, Y.-L. Liang and J. Qu, Chem. Commun., 2009, 5144–5146 RSC.
- H. Gunaydin and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 15232–15233 CrossRef CAS PubMed.
- B. N. Dickhaus and R. Priefer, Colloids Surf., A, 2016, 488, 15–19 CrossRef CAS.
- W. Guo and N. Hu, Biophys. Chem., 2007, 129, 163–171 CrossRef CAS PubMed.
- M.-K. Chun, C.-S. Cho and H.-K. Choi, J. Controlled Release, 2002, 81, 327–334 CrossRef CAS PubMed.
- M. Guo, Y. Yan, H. Zhang, H. Yan, Y. Cao, K. Liu, S. Wan, J. Huang and W. Yue, J. Mater. Chem., 2008, 18, 5104–5112 RSC.
- R. Dong, M. Lindau and C. K. Ober, Langmuir, 2009, 25, 4774–4779 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py02076c |
| This journal is © The Royal Society of Chemistry 2017 |