
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Blocked isocyanates: from analytical and experimental considerations to non-polyurethane applications
Marianne S.
Rolph
a,
Anna L. J.
Markowska
b,
Colin N.
Warriner
b and
Rachel K.
O'Reilly
*a
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: r.k.o-reilly@warwick.ac.uk
bAWE, Aldermaston, Reading RG7 4PR, UK
First published on 8th November 2016
Abstract
In this review we aim to provide an introductory guide to blocked isocyanates, through discussion on different examples of blocking groups, along with experimental considerations for their application. The review brings together updated examples of functionalities used and places specific emphasis on the analytical techniques used for the determination of deblocking temperature, updating previous reviews with the newly applied technique of X-ray photoelectron spectroscopy (XPS). Additionally, we put forward a brief update on the use of blocked isocyanates for other non-traditional polyurethane based polymer applications. We also highlight the importance of matching the analytical technique to the application of the blocked isocyanate, with differences in material state and experimental parameters having a great impact on the deblocking temperature reported.
Introduction
Since the discovery of polyurethane and its related chemistries by Otto Bayer in 1937, significant developments have been made in the production of polyurethane based materials.1,2 The range of potential properties available results in a wide array of applications, including packaging,3–6 adhesives,7,8 insulation,9–11 coatings,12,13 and fire retardants.14,15 The basic synthesis of polyurethanes involves the reaction of an isocyanate with a hydroxyl-containing compound, such as an alcohol or water. The breadth in combinations of alcohols and hydroxyl compounds alongside a vast catalogue of isocyanate compounds produces an almost limitless collection of structurally different materials (Fig. 1).16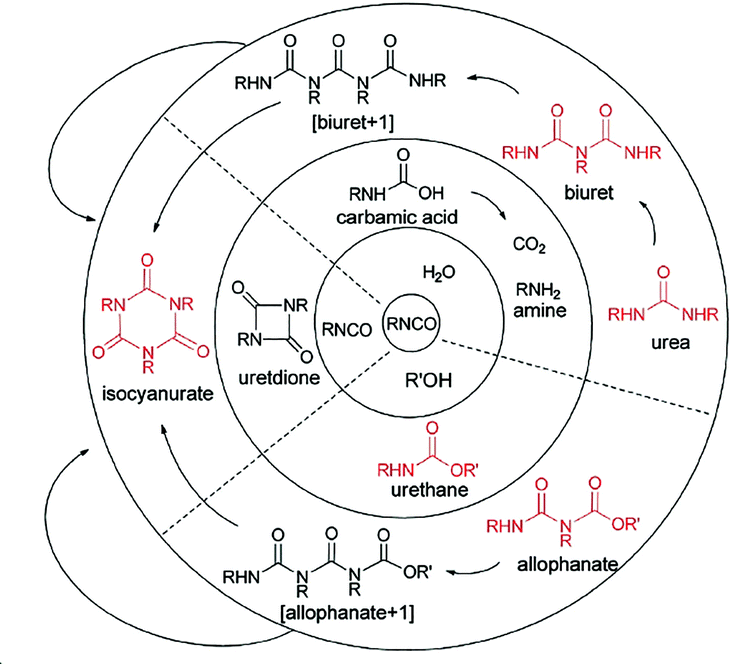 | ||
| Fig. 1 Examples of competing reactions in the synthesis of polyurethane foams. Reproduced from ref. 16 with permission from the Royal Society of Chemistry. | ||
Despite significant developments in the synthesis of polyurethane since the original patent by Bayer et al.,1 a major industrial problem remains: the sensitivity of isocyanates towards moisture. Indeed, the reaction between an isocyanate and a hydroxyl containing compound occurs readily at room temperature regardless of the presence of additives such as catalysts however, industrially, this high sensitivity towards moisture makes prolonged storage of these compounds unfeasible; i.e. careful storage of the isocyanate component is required to prevent reaction and subsequent production of an unusable material.17 In order to overcome this problem, work has focused on the development of isocyanate-free polyurethane formulations, eliminating the isocyanate moisture sensitivity problem,18,19 or through the use of “blocked”, or “masked” isocyanates, i.e. an isocyanate that contains a protected isocyanate. The isocyanate functional group is masked through the use of a blocking agent producing a compound that is seemingly inert at room temperature yet yields the reactive isocyanate functionality at elevated temperatures.20 For the majority of blocked isocyanates, the blocking occurs through a reaction with a compound containing an active hydrogen (Scheme 1). The blocking/deblocking equilibrium, in addition to the weak hydrogen bond on the nitrogen, leads to the regeneration of the blocking agent at raised temperatures and liberation of the reactive isocyanate functionality. Further to this, the relative inertness of the blocked isocyanates towards moisture and other nucleophiles, as well as other free or blocked isocyanates, dramatically increases the shelf life, and advantageously were found to have lower toxicities than free isocyanates.21,22 Furthermore, the implementation of blocked isocyanates has paved the way for a more environmentally friendly and safer synthesis of urethanes, allowed for one-pot (1K) formulations (e.g. for the development of polyurethane based coatings), as well as allowing for a more controlled release of free isocyanates thus introducing another level of control to polyurethane synthesis through minimizing possible side reactions. Moreover, the high industrial applicability of these materials is evidenced by the dominance of blocked isocyanate patents in the related literature on the topic (Fig. 2).23–26
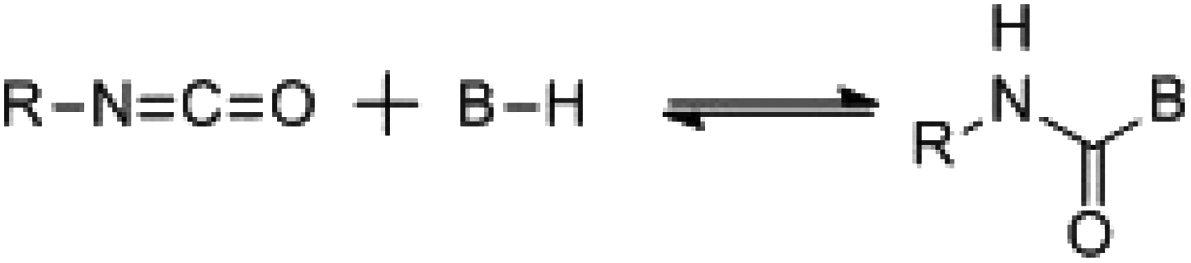 | ||
| Scheme 1 Generalized reaction scheme for the formation of a blocked isocyanate, where B–H is the active hydrogen containing blocking agent. | ||
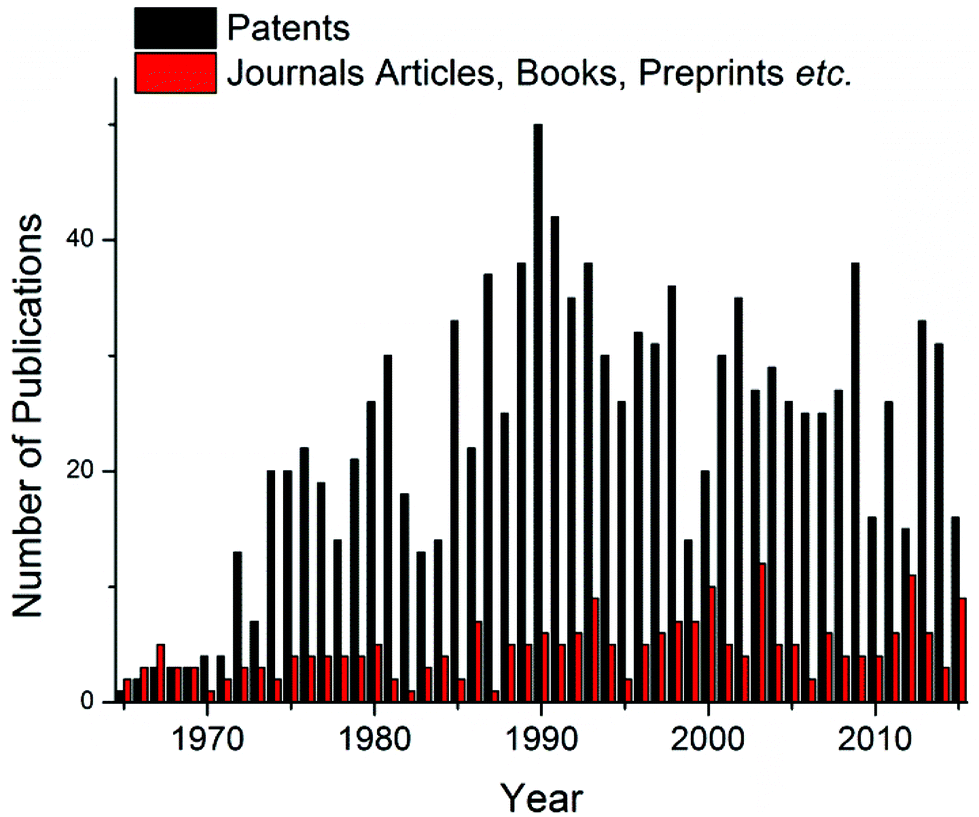 | ||
| Fig. 2 Comparison of the literature published, generated from a SciFinder search on “Blocked Isocyanates” April 2016. | ||
Measuring the deblocking temperature
The temperature at which the isocyanate deblocks is highly dependent on the application of the system, for example heat-cure organic powder coatings tend to require a higher deblocking temperature than those used in other applications including rigid and flexible polyurethane foam.27 There is a wealth of information in the literature on methods used to measure and determine the deblocking temperature, including both isothermal and non-isothermal methods, a summary of which can be found in Table 1. It is of great importance at this point to highlight the use of terminology when discussing blocked isocyanates. There is a broad use of the term “deblocking temperature” in the literature but it should be stressed that this term is not quite accurate. The correct term, as highlighted by Delebecq, is “initial deblocking temperature”, which describes the temperature at which a feature of deblocking (e.g. detection of a free blocking group) can be observed.28 The exact temperature at which scission of the blocking group and isocyanate occurs would involve the determination of reaction rates and extrapolation back through the Arrhenius equation in order to generate an exact deblocking temperature.| Technique | Advantages | Disadvantages | Ref. |
|---|---|---|---|
| IR, FTIR spectroscopy | Immediate result, easy to determine 100% completion of blocking/deblocking. High resolution instruments allow for ppm detection. | Strongly influenced by heating rate. Sample preparation and evaporation can cause discrepancies in results. Results need to be normalized for analysis. | 25, 29–37 and 47 |
| DSC analysis | Relatively high temperature capability (up to 700 °C). No additional sample preparation. | High temperatures may influence the baseline and signal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :noise ratio. :noise ratio. |
38–40 and 42 |
| TGA analysis | High temperature capability (up to 1500 °C). Samples require no additional preparation. μg sensitivity. | High temperatures may influence the baseline and signal:noise ratio. Boiling point of the blocking group must fall within the probed temperature range. |
38–40 and 42 |
| UV-Vis spectroscopy | Determines deblocking temperature within a narrow range. Provides additional information other techniques are not able to provide. | Labour intensive, calculation of multiple molar extinction coefficients required. | 43–45 |
| NMR spectroscopy | Highly accurate measurements. Enables all equilibrium species, and therefore kinetics, to be monitored. Small amount of sample (μg) required. | Requires high boiling point deuterated solvents. Limited to the temperature range of the instrument probe (typical limit = 160 °C). Significantly lower sensitivity vs. other techniques (e.g. XPS). | 28, 33, 46 and 48 |
| MS, GC, GC-MS analysis | Confirmation of starting materials, deblocked product and blocking agent. GC-MS enables the degree of deblocking to be calculated. Highly sensitive (MS limit = ppm, GC limit = ppb) | For degree of deblocking, GC-MS requires calibration. Limited by the maximum oven operating temperature (typically 300 °C). Samples must be volatilized for analysis. | 32, 49 and 50 |
| Titration | Only technique to allow for urethane type | Labour intensive, requires additional work-up prior to titration. | 51–56 |
| CO2 evolution | Immediate result. Produces a single temperature of deblocking. | Requires specialist glassware. Qualitative technique. | 57–59 |
| Physical change | Rheological studies able to produce fairly narrow deblocking temperature. | Highly inaccurate: monitoring of physical change by eye leading to a large source of error. | 60 and 61 |
| XPS | Highly sensitive technique (ppm detection). Provides elemental surface composition. | Samples must be annealed outside of the instrument. Lengthy fitting process. Analysis through comparison to a database. | 62 and 63 |
One of the most common techniques employed in the literature is the use of Infrared (IR) spectroscopy, and Fourier Transform Infrared (FTIR) spectroscopy, with the latter occasionally coupled with Dynamic Mechanical Analysis (DMA).29 The IR spectra exhibit strong bands between ν = 2230 cm−1 and ν = 2270 cm−1 (attributed to free N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO), the appearance of which can be compared to the disappearance during deblocking of the bands associated with the blocked isocyanate (CO between ν = 1640 cm−1 to ν = 1720 cm−1 and N–H band at approximately ν = 1535 cm−1).30 Whilst being an established method for determination of deblocking temperature, with relatively facile sample preparation, the technique is not without disadvantages. Indeed, for quantification of the degree of deblocking the isocyanate or blocking agent band absorbance must be normalized, typically to an unchanging C–H stretch.31 This normalization, unless carried out by the spectrometer software, can often be time consuming. Additionally, care needs to be taken with regards to the sample preparation and the running conditions. Samples prepared for IR/FTIR spectroscopic analysis can be either in the solid or liquid phase, and either prepared in pellet form or between two potassium bromide plates, with variation in sample preparation found to cause discrepancies: Gedan-Smolka and co-workers noted the difference in the deblocking temperature obtained by Differential Scanning Calorimetry (DSC) and FTIR for low molecular weight isocyanates, attributing the difference to secondary reactions in the KBr pellet.32 Moreover, analysis involved evacuating the spectrometer cell to remove the effects of gasses and moisture present, likely favoring the deblocking reaction, especially for more volatile blocking agents. Indeed, Wicks and co-workers noted that, whilst analyzing methyl ethyl ketoxime (MEKO) blocked isocyanates with an attenuated total reflectance (ATR) fitted FTIR and using a cover slide, the deblocking had a slower rate than the same experiment using an uncovered cell (Fig. 3).25 In their study, it was proposed that the evaporation occurring in the uncovered cell shifts the equilibrium of blocking/deblocking to favor the free isocyanate formation. Additionally, owing to the dynamic nature of the process, the rate of heating is found to have an effect on the reported deblocking temperature. Despite these problems, FTIR analysis proves to be one of the most used techniques in the literature. Further to this, the addition of a hot stage allows for kinetic analysis, for both the blocking and deblocking reactions.33–36 The combination of FTIR with DMA allows for probing of the effect of the blocking agent on material properties.29,37
CO), the appearance of which can be compared to the disappearance during deblocking of the bands associated with the blocked isocyanate (CO between ν = 1640 cm−1 to ν = 1720 cm−1 and N–H band at approximately ν = 1535 cm−1).30 Whilst being an established method for determination of deblocking temperature, with relatively facile sample preparation, the technique is not without disadvantages. Indeed, for quantification of the degree of deblocking the isocyanate or blocking agent band absorbance must be normalized, typically to an unchanging C–H stretch.31 This normalization, unless carried out by the spectrometer software, can often be time consuming. Additionally, care needs to be taken with regards to the sample preparation and the running conditions. Samples prepared for IR/FTIR spectroscopic analysis can be either in the solid or liquid phase, and either prepared in pellet form or between two potassium bromide plates, with variation in sample preparation found to cause discrepancies: Gedan-Smolka and co-workers noted the difference in the deblocking temperature obtained by Differential Scanning Calorimetry (DSC) and FTIR for low molecular weight isocyanates, attributing the difference to secondary reactions in the KBr pellet.32 Moreover, analysis involved evacuating the spectrometer cell to remove the effects of gasses and moisture present, likely favoring the deblocking reaction, especially for more volatile blocking agents. Indeed, Wicks and co-workers noted that, whilst analyzing methyl ethyl ketoxime (MEKO) blocked isocyanates with an attenuated total reflectance (ATR) fitted FTIR and using a cover slide, the deblocking had a slower rate than the same experiment using an uncovered cell (Fig. 3).25 In their study, it was proposed that the evaporation occurring in the uncovered cell shifts the equilibrium of blocking/deblocking to favor the free isocyanate formation. Additionally, owing to the dynamic nature of the process, the rate of heating is found to have an effect on the reported deblocking temperature. Despite these problems, FTIR analysis proves to be one of the most used techniques in the literature. Further to this, the addition of a hot stage allows for kinetic analysis, for both the blocking and deblocking reactions.33–36 The combination of FTIR with DMA allows for probing of the effect of the blocking agent on material properties.29,37
 | ||
| Fig. 3 ATR spectra of deblocking reaction of methyl ethyl ketoxime blocked HDI isocyanurate as a function of time at 140 °C (a) without a cover slide and (b) with a cover removed after 40 min as indicated by the arrow. Reprinted from ref. 25, with permission from Elsevier. | ||
Highly prevalent in the literature, and frequently used in conjunction with FTIR studies, DSC and thermogravimetric analysis (TGA) are two of the most commonly applied techniques to determine isocyanate deblocking. DSC monitors the deblocking reaction by detecting the endothermic transition resulting from urethane bond scission. In contrast, TGA measures the weight loss associated with the deblocking of the blocked isocyanate. Both of these techniques have the advantage of the high temperature range of the instrument, with DSC able to monitor deblocking typically up to 700 °C and TGA able to measure up to 1500 °C, enabling analysis of compounds with very high deblocking temperatures. DSC and TGA are regularly applied as complimentary techniques; Lee et al. used both techniques to probe a series of ε-caprolactam and benzotriazole blocked diisocyanates, with TGA curves being measured up to 500 °C and DSC thermograms measured up to 225 °C. Whilst DSC analysis was able to provide the deblocking temperature range for a diisocyanate blocked with a mixture of ε-caprolactam and benzotriazole, the wide exotherm was not able to distinguish between the different deblocking temperatures of the two blocking agents. However, TGA analysis was able to resolve both deblocking temperatures, exhibiting two decomposition points in the curve. Moreover, Kothandaraman and Thangavel measured TGA curves up to 750 °C, allowing for analysis of the deblocking reaction at temperatures significantly higher than most of the other techniques available.38,39 It is important to note that, for TGA analysis to be successful, the blocking agent must evaporate within the temperature range set on the TGA instrument.40 Whilst the high temperatures are particularly useful for probing compounds for curing applications, the increased temperature is not without a downside. For DSC, high temperature can result in altering of the baseline, with increased heating rates resulting in broader endotherms and therefore a loss in sensitivity. Moreover, vaporization of the sample during DSC analysis could potentially affect the results through altering of the signal to noise ratio, with Tabaddor et al. demonstrating that evaporation of cure products during analysis of a thermoset material resulted in significant noise, rendering the process more difficult to monitor.41 Moreover, the evaporation of a blocking agent has been shown to have an influence on the blocking/deblocking equilibrium (favoring the production of free isocyanate), therefore generating inconsistencies in results and potentially limiting the applicability of both DSC and TGA.42 This problem of evaporation can partly be solved, however, through the use of a sealed pan, though this may itself pose a problem in the form of the pan breaking apart in the cell of the instrument.
The use of ultraviolet (UV) spectroscopy is far less popular than both IR spectroscopy and DSC/TGA, likely as a consequence of a significantly more protracted preparation, but nevertheless enables production of one of the most accurate measurements of blocking/deblocking temperature. Tarasov et al. studied the n–π* transition at λ = 282 nm of the carbonyl functionality in the blocked isocyanate, to produce a conversion ratio of blocked to free isocyanates throughout the deblocking process.43 This technique is one of the more labor-intensive measurements, requiring the determination of molar extinction coefficients (ε) for multiple compounds in the system and thus the technique is not as immediate as alternatives found in the literature. Mao and co-workers utilized the technique to investigate dye-bonded waterborne polyurethanes, in which the shift in the dye absorption indicated the deblocking of the isocyanates, with the temperature compared to that generated through TGA analysis.44 Whilst the high level of preparation required was alluded to by Kiguchi and co-workers, it was noted however that the technique provides additional information not easily obtained by other methods. In their study investigating the blocking of pendent isocyanate functionalities, UV-vis spectroscopy not only provided the kinetics of the blocking reaction, it also enabled determination of the molecular weight of the polymers as well as the viscosity of the solution.45 Despite the wealth of information that can be garnered, the aforementioned problems with experimental set-up have rendered this technique rarely used.
Another potentially underexploited technique, described as producing the most accurate deblocking temperature, nuclear magnetic resonance (NMR) spectroscopy has the advantage of allowing all the species in the system to be monitored simultaneously.28 Whilst both hydrogen (1H) and carbon (13C) NMR spectroscopy are regularly used to show the presence of the blocking group, for example to prove successful blocking, the technique is not readily applied to monitoring of kinetics. Cholli et al. have demonstrated the use of solid-state 13C NMR spectroscopy to prove the generation of free isocyanate species when investigating the deblocking of an oxime blocked isocyanate. Solid state 13C NMR spectroscopy indicated successful deblocking through an increase in peak intensity of the carbons of the oxime blocking group, as well as a decrease in the intensity of carbons arising from the blocked methylenebis (phenyl isocyanate) (MDI).46 However, preparation involved external heating of the sample prior to analysis which, owing to the equilibrium for the deblocking/blocking reaction, may result in an inaccurate determination of the degree of deblocking when attempting to use integrals to quantify the degree of blocking/deblocking. A review of the literature was unable to produce many publications using NMR spectroscopy to follow kinetics or determine the deblocking temperature. One study by Sankar and co-workers did successfully analyze the deblocking using variable temperature 1H NMR spectroscopy to confirm deblocking. Whilst investigating N-methylaniline-blocked polyisocyanates, it was proposed that deblocking occurred via a four-centered transition state involving hydrogen abstraction from the urea linkage by the nitrogen of the blocking group. Owing to the thermal sensitivity of hydrogen bonding, variable temperature proton NMR spectroscopic analysis was able to confirm the structure of the transition state through shifts in the protons associated with the urea linkage (blocked isocyanate) and the hydrogen bonded urethane.33 Ying and co-workers used in situ NMR experiments to monitor the interchange of different blocking groups on isocyanatoethyl methacrylate, confirming the presence of the equilibrium between the blocked isocyanate and the released free blocking agents and isocyanate (Fig. 4).48 Similar to IR spectroscopy, sample preparation is of great importance as it will affect the accuracy of the measurement. The majority of blocking agents deblock at relatively high temperatures requiring the use of high boiling point deuterated solvents such as dimethyl sulfoxide (DMSO-d6) and dimethyl formamide (DMF-d7), the former of which is notoriously difficult to remove water from. This may potentially cause further problems arising from the reaction of this water with the liberated isocyanate artificially decreasing the appearance of the signal associated with the free isocyanate. Moreover, this reaction can further result in inaccurate results: the presence of nucleophiles has been shown by Delebecq and co-workers, through the use of NMR spectroscopy, to reduce the deblocking temperature, likely through a shift in the equilibrium towards the formation of the free isocyanate and blocking agent.28 Moreover, such a technique is limited to the temperature range of the probe in the instrument, adding a further complication when higher temperatures (>150 °C) are required to deblock compounds.
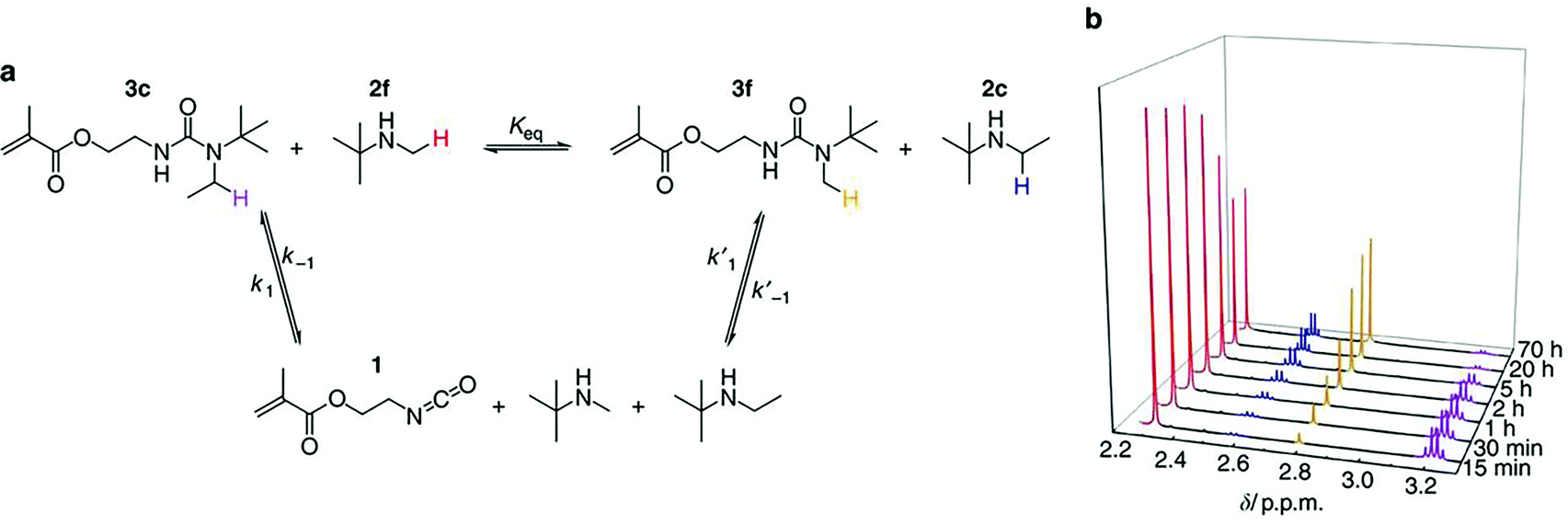 | ||
| Fig. 4 (a) Exchange reaction between 3c and 2f produces 3f and 2c with isocyanate 1 as the intermediate. (b) 1H NMR spectra after mixing 3c and 2f for different durations at 37 °C. The proton peak intensities of 3c and 2f gradually decreased over time, along with the gradual increase of proton peak intensities of 3f and 2c until an equilibrium of the exchange reaction was reached at ∼20 h. The protons subject to analysis from these four compounds were labelled with different colours in a, which correspond to the colours of peaks in b. Reprinted by permission from Macmillan Publishers Ltd: Nature Communications, ref. 48, copyright 2014. | ||
The application of mass spectrometry (MS), gas chromatography (GC) or a combination of the two techniques (GC-MS) sparingly appears in the literature, but has the potential to provide a wealth of information. Mass spectrometry enables confirmation of the starting blocked materials as well as the final deblocked isocyanates. Nasar et al. showed that the degree of deblocking could be monitored by GC analysis with each component (in this instance N-methylaniline, toluene diisocyanate (TDI) and the adduct) eluting separately.49 Mühlebach similarly used GC to separate the products of a deblocking reaction of pyrazole-blocked isocyanates, with subsequent MS analysis confirming the presence of the free isocyanate and blocking group.61 Mass spectral analysis has also been coupled with other techniques, for example in the employment of quadrupole mass spectrometry coupled to a simultaneous thermal analyzer to measure the deblocking temperature of alcohol blocked isocyanates.32 More recently, Nagy and co-workers have carried out an in depth study into the blocking kinetics of a series of blocking agents and MDI and TDI.50 The work used high-performance liquid chromatography (HPLC) in conjunction with electrospray ionization mass spectrometry (ESI-MS) to draw conclusions on the reactivity of different isocyanate groups within diisocyanates.
The most widespread technique in the early literature on blocked isocyanates, though still commonly used, is titration for the detection of isocyanate. Muramatsu et al. describe the process in great detail, in which the reaction mixture is heated and aliquots are removed, undergo a brief work-up and are titrated with di-n-butylamine.51 This work-up does however involve the addition of di-n-butylamine and usually a colored indicator, rendering the technique less immediate than others. The titration method is frequently used to calculate the percentage of free isocyanates during a blocking reaction,52,53 as demonstrated by Pilch-Pitera for the monitoring of the blocking reaction of various isocyanates with both fluorinated and non-fluorinated alcohols.54 The content of free NCO, as determined by titration, was compared with the theoretical free NCO content to ensure reaction completion. Despite the additional work-up required, its continued prevalence in the literature is likely attributed to it having one main advantage over most of the other techniques: the ability to calculate the percentage of isocyanate groups present in either the allophanate or the biuret form. Allophanate and biuret groups (Fig. 1) react differently with the added amine; Furukawa and co-workers demonstrated the selective degradation of these two forms during polyurethane synthesis, whilst showing that the urea formed remained unaffected.55 Titration was also reported by Davis, applying a method developed by Tremblay and Boivin, to allow for the determination of monomeric TDI during the synthesis of a TDI based uretdione, through the use of a weak base dicyclohexylamine which was able to only detect single TDI molecules without any interactions with the dimer ring.56
A less commonly applied technique, developed by Griffin et al. in the early 1960s involves the measuring of carbon dioxide gas evolved during the deblocking reaction.57 This technique, mainly unexploited as a consequence of the necessity of using specialist glassware, involves the blocked isocyanate being heated in a moisture saturated atmosphere until the evolved carbon dioxide (formed during the reaction between the unmasked isocyanate and the atmospheric moisture) is detected with a barium hydroxide solution (both visually, and more accurately through turbidimetry). The technique is frequently used for comparison with other techniques, such as DSC as an example.58 It was noted by Kothandaraman and Nasar that the method produced a lower deblocking temperature than that obtained by IR analysis, likely an effect of the solvent, highlighting a major flaw in the method.59 Furthermore, without additional equipment (e.g. for the measurement of turbidity) it is unable to provide an accurate degree of deblocking, producing only a single temperature, in the same way that other techniques are able to provide a range.
The earliest studies into blocked isocyanates were carried out from an industrial perspective for the development of coatings for applications in e.g. the automotive industry. Subsequently the main detection method, prevalent in the early patents, is based on monitoring of a physical change. Frequently the physical change monitored is gel time, with Katsamberis and co-workers defining the gel time as the time taken until no solution flow was observed.60 However the method affords a 5% error, significantly greater than the error generated with DSC and/or TGA instrumentation. Since the initial studies monitoring a physical change, significant developments have been made in the last 30 years, with measurements becoming increasingly more precise, in particular with the introduction of rheological methods. Mühlebach successfully demonstrated the applicability of rheology to determine the effect of Jeffamine®, produced by Huntsman International LLC, on the curing reaction of a pyrazole blocked hexamethylene diisocyanate (HDI).61
Most recently X-ray photoelectron spectroscopy (XPS) has been employed to probe the deblocking. A method of surface chemical analysis, the technique yields information about the surface elemental composition and chemical state thus enabling identification of the free and the blocked isocyanates. Zhang and co-workers used the technique whilst investigating the mechanism of thermal deblocking of sodium bisulfite blocked polymeric MDI (pMDI), with the spectra clearly demonstrating the appearance of the urethane bond (from the reaction of the free isocyanate with polyurethane).62 Not only was the technique able to indicate the temperature of deblocking, the equilibrium in the process was highlighted through a changing peak area (Fig. 5) observed as a consequence of a change in the local bonding environment of both the carbons and nitrogens when undergoing blocking/deblocking. Integration of peak areas additionally affords the potential to calculate degrees of deblocking. Zuo et al. demonstrated the stability of their system by running an XPS analysis on samples of a freeze-dried blocked isocyanate emulsion.63 Whilst the technique can be applied to both solid and liquid materials (with the latter being absorbed onto a base material up to the point where the base material is no longer detected by XPS), the lengthy fitting of the data, and analysis by comparison to databases may explain why the technique is not greatly employed. Moreover, most samples have to be annealed/heated externally from the instrument owing to a large number of instruments being unable to heat accurately at lower temperatures (<50 °C), thus preventing in situ analysis.
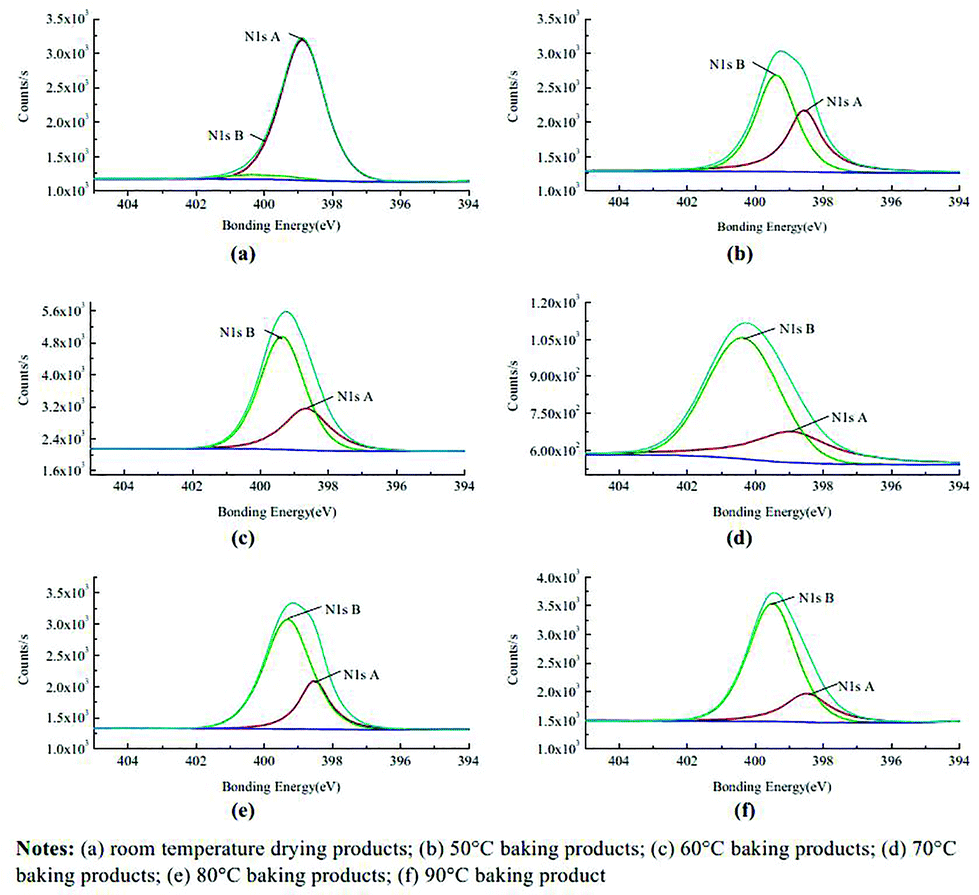 | ||
| Fig. 5 XPS spectra of different-temperature baking products for sodium bisulphite blocked isocyanates. Reproduced from ref. 62 © Emerald Group Publishing Limited, all rights reserved. (a) Room temperature drying products; (b) 50 °C baking products; (c) 60 °C baking products; (d) 70 °C baking products; (e) 80 °C baking products; (f) 90 °C baking products. | ||
As mentioned earlier, the use of the term “deblocking temperature” is frequently actually the initial temperature at which deblocking is observed. As demonstrated in the preceding section, the deblocking temperature can be measured in multiple different ways, and it has been found that different methods produce great deviations in the resulting deblocking temperatures or rates. Whilst studies have been known to utilize multiple techniques to confirm the validity of the results, the outcome generated can be mixed. Although Muramatsu et al. and Subramani et al. recorded approximately the same results for the deblocking temperature when using DSC and DTA-TGA,40,51 Lee and co-workers found differences in the deblocking temperatures generated by DSC and TGA. It was reported that DSC afforded a deblocking temperature of 215–240 °C for benzotriazole blocked MDI, yet TGA provided a deblocking temperature of 190–220 °C, a difference of 20 °C.38 Sankar and Yan further noted differences in deblocking temperature when using thermal analysis to evaluate deblocking. When investigating imidazole blocked poly(MDI) FTIR analysis produced a deblocking temperature of 170 °C, considerably lower than that produced by TGA (216 °C) and 120 °C lower than that produced by DSC (291 °C).64 Furthermore, Gedan-Smolka et al. reported differences in deblocking temperature when probing hydroxybenzoic acid ethyl ester blocked HDI. Analysis using FTIR afforded a deblocking temperature of 118 °C, yet analysis using simultaneous thermal analysis coupled with a quadrupole mass spectrometer (STA-QMS) revealed three deblocking temperatures (DSC/TGA = 150 °C, DSC = 170 °C and MS = 155 °C), all of which were significantly higher than that produced by FTIR.32 Discrepancies are theorized to be related to the equilibrium established for the addition and dissociation of the blocking agents. As already mentioned, elimination of the blocking agent, through evaporation in thermogravimetric analysis for example, forces the equilibrium in favor of the free isocyanates. Further to this, experimental conditions such as nucleophilicity of solvents can also potentially affect the deblocking reaction, and technique parameters such as the temperature ramping rate further influence the equilibrium, therefore the temperature determined is unlikely to be the true deblocking temperature, and so it is imperative that the quoted temperature is used only as a guide as to the range in which the isocyanate deblocks.
Blocking agents
The classes of functional group that have been investigated as blocking agents for isocyanates, with specific examples of blocking agents and the corresponding deblocking temperatures, are presented in Table 2. The list includes both external agents (i.e. when an additional compound has been used to block the isocyanate) and internal blocking agents (when the isocyanate is used to block itself). It should be highlighted, especially given the preceding section, that the deblocking temperatures quoted must be taken as a range for which these functionalities deblock. The temperatures quoted result from a wide range of analytical techniques, and as previously described these techniques can provide very different deblocking temperatures even for the same compounds. Indeed, blocking agents are only able to be directly compared if analyzed using the same technique and under identical conditions, as evidenced in the comprehensive study by Regulski et al. in which the crosslinking temperatures of various blocked isocyanatoethyl methacrylates were investigated.65 Despite this, it is evident that there is a vast array of blocking agents available for use encompassing a wide range of deblocking temperatures, with further temperatures able to be targeted through altering substituents on the blocking group, as well as changing the experimental conditions and the isocyanate to be blocked.| Blocking functional group | Specific examples | Additional information | Deblocking Temp.a,b (°C) |
|---|---|---|---|
| a The deblocking temperatures are those quoted in the literature and as such are determined by varying methods. Different methods can yield different deblocking temperatures for the same blocking group and so these temperatures should only be taken as an approximate guide of the deblocking temperature range. Moreover, these can readily be influenced by the presence of catalysts and other additives. b Blocking agents without deblocking temperatures have been found in literature in which the deblocking occurs during either curing studies, kinetic studies at a constant temperature known to be greater than the deblocking temperature, or they are synthesized as a pre-polymer and so the deblocking temperature is not quoted. | |||
| Alcohol | Butanol24,67 | Generally found to have high deblocking temperatures. The presence of halogens found to significantly decrease the deblocking temperature.25 | 95–200 |
| Ethanol24 | |||
| Isopropanol32,68,69 | |||
| Phenol | Phenol58,70,71 | Easy to demonstrate the effect of substituent type and position on the deblocking temperature.59 | 60–180 |
| o-Cresol59 | |||
| p-Chlorophenol72–74 | |||
| Pyridinol | 2-Pyridinol25 | Deblocking at a lower temperature than phenol due to the hydrogen bond formation between the amine and the urethane bond.25 | 110 |
| 2-Chloro-3-pyridinol25 | |||
| Oxime | MEKO51,75–77 | MEKO found to be most common blocking agent in the literature, with the blocking reaction requiring no catalyst. Strong substituent effect on deblocking temperature.78 | 85–260 |
| Benzophenone Oxime71,78,79 | |||
| Thiophenol | Thiophenol80 | Thiophenol found to have faster deblocking rates than phenol.73 | 130–170 |
| Pentafluorothiophenol37 | |||
| Mercaptan | 1-Dodecanethiol57 | Restricted applicability due to odours produced in production and deblocking.25 | 75–115 |
| Amide | Acetanilide51 | Found to have lower deblocking temperatures than MEKO blocked isocyanates.51 | 100–130 |
| Methylacetamide51 | |||
| Cyclic amide | Pyrrolidinone81 | ε-Caprolactam does not volatilize after deblocking and is able to act as a plasticizer.84 | 70–170 |
| ε-Caprolactam40,82,83 | |||
| Imide | Succinimide51 | Along with amines, the deblocking temperature is heavily influenced by the polarization of the NH bond.51 | 110–145 |
| N-Hydroxyphthalimide85 | |||
| Imidazole/imidazoline | Imidazole64,86 | Basicity of imidazole able to accelerate the blocking reaction without additional catalyst.22 Large substituent effect on deblocking temperature.87 | 120–290 |
| 2-Methylimidazole53,86,87 | |||
| 2-Phenylimidazole88 | |||
| Pyrazole | Dimethylpyrazole61,89,90 | When deblocked in the presence of amines, deblock via a cyclic transition state releasing the isocyanate, in contrast to phenol blocked isocyanates in which the amine attacks the urethane bond.61 Deblocking temperature is lowered when the basicity of the pyrazole is increased.25 | 85–200 |
| 2-Methyl-4-ethyl-5-methylpyrazole61 | |||
| Triazole | Benzotriazole38,91 | Along with pyrazoles, triazoles produce less yellowing than oximes.25 | 120–250 |
| Triazole31,92,93 | |||
| Amidine | Bicyclic amidine94,95 | Radical intermediates formed during cleavage.95 | 70–175 |
| Hydroxamic acid ester | Benzylmethacrylo-hydroxamate96 | Deblocking proceeds via a six membered transition state.96 | 50 |
| Intra-molecular | Uretdione97–99 | Uretdione is a self-condensation product, and can be further transformed into a trimeric species (isocyanurates).101 | 150–200 |
| 2-Oxo-1,3-diazepane-1-carboxylate100 | |||
| Other | Sodium bisulphite102–104 | Sodium bisulphite is frequently used in waterborne coatings as the blocked product is water soluble,25 as well as being relatively cheap with no pollution.104 | 50–160 |
| N-Methylaniline64,105,106 | N-Methylaniline deblocks via a four membered transition state.106 | — | |
It is worth mentioning that the use of external blocking agents, whilst providing access to a greater temperature range of deblocking, is not without disadvantage. One particular problem is that once deblocked, unless the temperature range is similar to the boiling point of the blocking agent, with evaporation of alcohol based blocking agents affording a facile method of blocking agent removal from the product material, the free blocking agent remains in the product material. Meier-Westhues reported that, especially in a one-component system, the blocked isocyanate was responsible for increased thermal yellowing, though this could be mitigated through the use of correct stabilizers. Moreover it was noted that residual blocking agents in a paint film affected film quality resulting in lower etch resistance.66 Though the residual blocking agent may have a negative impact on the application, it is not necessarily detrimental to the final product. Carter and co-workers found that ethyl acetoacetate oxime, upon deblocking, underwent intramolecular cyclization to produce an unreactive oxazolone product which acted as a plasticizer.107 Even if the blocking agent does evaporate, again this is not without problems and can result in the formation of bubbles in the product material, which is especially harmful for coating applications.
Mechanisms and kinetics
Whilst there is a great deal known about the mechanism of the reaction between a free isocyanate and a nucleophilic species, such as that witnessed in the synthesis of polyurethane, there is still much debate over the mechanism of deblocking. With regards to the reaction between a blocked isocyanate and a nucleophile (e.g. a hydroxyl group), there are two generally accepted mechanisms proposed: an elimination-addition mechanism (Scheme 2a) and an addition-elimination mechanism (Scheme 2b), both of which are supported in the literature.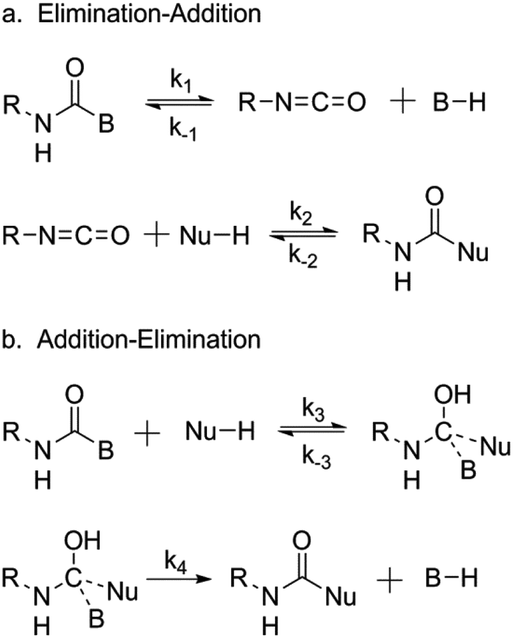 | ||
| Scheme 2 The proposed mechanism for isocyanate deblocking in the presence of a nucleophilic species. (a) elimination-addition mechanism, (b) addition-elimination mechanism, where B = blocking agent, Nu = nucleophilic species, and R = additional functionality. Reprinted from ref. 25, with permission from Elsevier. | ||
Evidence in support of the elimination-addition mechanism, in which the blocked isocyanate deblocks and subsequently reacts, was provided by Wicks and co-workers who demonstrated the homolytic cleavage of the blocked isocyanate through detection of the free isocyanate stretch in the IR spectral analysis.25 Tassel et al. used the same techniques to evaluate the kinetics of the deblocking of formyloxyethyl methacrylate blocked isocyanate and concluded that the appearance of the isocyanate stretch must indicate homolytic cleavage and release of the isocyanate.92 Interestingly, Mühlebach proposed that both mechanisms could occur, and that the mechanism depended on the blocking agent itself.61 It was proposed that a pyrazole-blocked isocyanate underwent cleavage of the blocking agent yielding the free isocyanate and blocking group (consistent with the elimination-addition mechanism), whilst a phenol blocked isocyanate would be directly attacked by a nucleophile (the addition-elimination mechanism). Essenfeld and Wu provided further evidence for this mechanism during their work using tris(alkoxycarbonylamino)triazine (TACT), analogous to an alcohol blocked isocyanate. The reaction of TACT with primary amines was found not to form urea linkages. If the elimination-addition mechanism was applied, elimination of the alcohol would generate a free isocyanate and thus urea linkages.108 Similar to the proposal by Mühlebach in relation to the deblocking mechanism, experimental conditions further influence the observed mechanism. Solvent assisted deblocking has been demonstrated to increase the rate of deblocking based on an elimination-addition pathway,105 and an increased temperature has been demonstrated to favor the elimination-addition pathway in accordance to the Arrhenius parameters.109
The kinetics of both proposed mechanisms are further complicated by the reversible nature of the reaction. Indeed, if the deblocking was brought about through only heating the blocked isocyanate, there is always a possibility of producing dimers or trimers. Further to this, the isocyanates could thermally decompose or react immediately with any nucleophile, rendering detection more difficult.
Experimental considerations
Isocyanate structure
It is widely accepted that an aromatic blocked isocyanate undergoes deblocking at lower temperatures compared to an aliphatic blocked isocyanate, as a consequence of both steric and electronic effects. The aromatic ring, in contrast to the alkyl functionality in aliphatic isocyanates, lowers the deblocking temperature through the conjugation of the π electrons of the ring and the NCO bond.109 Moreover, substitution of the ring with electron-withdrawing groups amplifies this effect and further lowers the deblocking temperature. Nesterov reported that substitution on the aromatic ring resulted in increasing the blocking reaction rate with active hydrogen compounds, evidenced in the changing Hammet constants.110 The Hammet constants, relating to the rate of a reaction when investigating a series of substituents, were found to be positive, indicating an acceleration of the reaction rate between the alcohol and isocyanate with the presence of electron-withdrawing groups. Conversely, the addition of electron-donating groups decreases the blocking rates. Indeed, in their study involving the reaction of different substituted phenyl isocyanates with alcohols, Bailey and co-workers noted that both nitrile and chlorine substituted phenyl isocyanate had much greater rates of reaction than that of the methyl groups which exhibited an inhibiting effect on the reaction.111 Lee et al. further elaborated whilst reporting the deblocking of H12-MDI, TDI and MDI: H12-MDI was found to have the highest deblocking temperature owing to having no aromatic ring, followed by MDI and then TDI.38 The π electrons of the ring attract the lone pair of the nitrogen resulting in a slightly positive character on the nitrogen and thus increasing repulsion between the nitrogen and the hydrogen attached. This has the net result of making the hydrogen more labile and therefore decreasing the deblocking temperature. TDI was also explained as having the lowest deblocking temperature owing to steric effects with the asymmetry of the diisocyanate, thus lowering the steric hindrance.
The combination of electronic and steric effects was further discussed by Muramatsu et al., who demonstrated the MEKO blocked isophorone diisocyanate (IPDI) deblocked at lower temperatures than hexamethylene diisocyanate (HDI).51 The lower deblocking temperature was attributed to the bulky nature of the cyclohexyl ring on the IPDI resulting in a more crowded isocyanate, highlighted with the more sterically crowded tetramethylxylenediisocyanate (TMXDI) having an even lower deblocking temperature. Interestingly, it was observed by Tassel et al. that the isocyanates on IPDI did not block at the same time, with the cyclic isocyanate blocking before the aliphatic isocyanate.92 Indeed, this effect of isocyanate inequivalence was observed by Bailey and co-workers in the mid-1950s: whilst phenylene diisocyanate was found to undergo a fast blocking reaction when reacted with alcohols, an additional isocyanate in a meta or para position relative to the first further increased reactivity (with meta slightly more active than para).111 This substituent effect is further noted in the synthesis of uretdiones; Otto noted a significantly faster rate of synthesis for 4-nitrophenyl isocyanate based uretdiones than non-ring substituted.112
Blocking agent structure and the substituent effects
Primarily, the blocking/deblocking temperature is determined by the blocking agent. As demonstrated in Table 2, it is clear that different blocking agents have different deblocking temperatures, and as a result it is possible, to a certain degree, to tailor the deblocking temperature simply by selecting the correct blocking group. However, owing to the problems associated with the high variance in deblocking temperature with detection method selected, it is important to have an alternative method to tailor the deblocking temperature. This is achieved through altering functional groups bound to the blocking agent. As previously mentioned, aromatic systems, for example phenolic blocked isocyanates, can easily be tailored through varying the ring substituents. Caution must be taken however in choosing the correct base agent: addition of the same functional groups to different aromatic systems has been demonstrated to have different effects. Mühlebach et al. demonstrated that varying the substituents present on the pyrazole ring on the blocking agent, from electron withdrawing groups (decreased the curing rate), to electron donating groups (increased the curing rate) was the opposite to the addition of these groups to phenolic blocking agents, demonstrated by Rand and co-workers investigating the effect of ortho and para substituents on aryl carbanilates.61,113Work by Kothandaraman and Nasar further elaborated the effect that substituents had to play on deblocking temperature: the urethane carbonyl formed during the blocking reaction has a partial positive charge on the carbon and the addition of electron donating substituents enhance the negative charge density on the blocking group and this intensification of charge difference between the urethane carbonyl and the blocking agent increases the bond strength and therefore increases the deblocking temperature.59 It is not, however, just electronic effects of substituents that have an impact on deblocking temperature. In the same study as the investigations into electronic effects, Kothandaraman and Nasar concluded that sterics also had a significant impact, yet the electronic effects predominate over the steric effects.
Reaction medium
The majority of literature available stresses that the effects of reaction medium are mainly due to the ability of the medium to hydrogen bond with the reactants.105,106,114 Strong hydrogen bond-acceptor solvents have been shown to significantly reduce the rate of recombination of a blocking agent and isocyanate (Scheme 2, k−1), and therefore the rate of deblocking would increase in hydrogen bond accepting solvents in comparison to aprotic solvents. Indeed, this effect was noted by Nasar and co-workers who reported a shorter reaction time when using hexamethyl phosphoric acid triamide and mesitylene when studying the impact of different solvents on the polymerization rate of blocked isocyanate prepolymers.105 It was suggested that the tertiary amine nitrogen in the solvent abstracts the proton from the blocked isocyanate nitrogen and facilitates transfer back to the blocking agent, increasing the rate of evolution of the free isocyanate. Gnanarajan et al. also investigated the effects of both dipolar aprotic solvents and non-polar solvents on the reaction between a N-methylaniline blocked polyurethane prepolymer and an anhydride, again concluding that the basicity and polarity of the solvents impacted the rate of deblocking.106 Conversely, as reported by Baker and Gaunt for the blocking of phenyl isocyanate with alcohols/amines, the reaction proceeds faster in nonpolar solvents as these reduce the rate of deblocking, k1.114The impact of reaction medium is not, however, singularly an effect of the H-bonding ability. Of potentially similar importance is the ability of the solvent to fully dissolve the blocked isocyanate. Noted by both Griffin and Willwerth, and by Mohanty and Krishnamurti, the higher the solubility of the adduct, the lower the observed deblocking temperature, with it being widely acknowledged that liquid blocked isocyanates deblock at lower temperatures than solids.57,115
Catalysts
It should be noted that for the synthesis of blocked isocyanates, no catalyst is necessary in the majority of instances, with Sankar and Yan noting that the aromatic secondary amines present in their system were sufficiently reactive without the addition of a catalyst.116 In spite of this, dibutyltin dilaurate (DBTL) is frequently used to ensure both a successful reaction and that the reaction goes to completion, with catalysis proposed to proceed via a four-membered transition state.117 One system that often does require the use of a catalyst is the production of uretdiones. Multiple patents and papers report the use of phosphine based catalysts for the synthesis of uretdiones and the trimeric isocyanurate species.16,118–120In the context of catalysis during the deblocking process, it is important to differentiate between what the catalyst is doing with regards to the blocked isocyanate. The catalyst is likely to be catalyzing one of four main reactions: the deblocking reaction, the reaction of the free isocyanate with a nucleophile, the addition-elimination reaction, or side reactions in the system. Most prevalent in the literature, DBTL is further used as a catalyst for the deblocking reaction, as well as catalyzing the reaction between a free isocyanate and a nucleophilic species. Indeed, Lomölder and co-workers found DBTL to be the best catalyst for the reaction between isophorone diisocyanate (IPDI) and 1-butanol.121 It was proposed that the metal acts as a Lewis acid and is able to activate the isocyanate by coordination to the carboxyl group. DBTL was further demonstrated by Carlson et al. to catalyze the side reaction producing allophanate.122 It was noted that the rates of the deblocking were no different for the catalyzed and uncatalyzed deblocking, yet that the level of free isocyanate was much lower with the DBTL. This was postulated to result from the DBTL catalyzing the allophanate producing reaction, further evidenced by additional studies.123
Whilst the applications of DBTL are numerous, its use is not without problems, specifically the stability of the catalyst. DBTL is hydrolytically unstable, limiting the use in one-component aqueous polyurethane formulations. Moreover, various other metal catalysts have been found to offer better selectivity or reaction temperatures, important for application.124 Indeed, zirconium based complexes have been demonstrated to produce significantly less carbon dioxide than DBTL as a result of a different mechanism of catalysis, which promotes the isocyanate–alcohol reaction in contrast to the isocyanate–water reaction.125 In a study involving various different metal based catalysts, Edelmann concluded that zinc(II)-acetylacetonate allowed for the lowest curing temperature, yet no catalysis was observed using bismuth(II) citrate.99 Contrastingly, work on MEKO blocked HDI by Blank and co-workers found bismuth tris(2-ethylhexanoate) produced the greatest catalytic response.126 These differing results highlight the importance of catalyst ligands on the selection of suitable catalyst, as well as for tailoring the catalyst to application (e.g. polyurethane coatings vs. rigid foams).
It is widely accepted that, along with organo-tin complexes, tertiary amines are also highly efficient at catalyzing the reaction between isocyanates and nucleophilic species. Most established amongst tertiary amine catalysts, 1,4-diazabicyclo[2.2.2]octane (DABCO) is frequently implemented for a range of blocked isocyanate reactions. The efficiency of a tertiary amine catalyst depends primarily on their chemical structure and basicity,106 with amine based catalysis found to be unsuitable for deblocking mechanisms requiring proton transfer from the nitrogen of the NCO moiety to the blocking agent.79 Katsamberis et al. further highlighted the importance of tertiary amine structure and basicity whilst investigating the catalytic effects of four amines,60 with bicyclic amidine found to be most catalytically active, observed in faster gelling times. The rate was attributed to a combination of the catalysts ability to carry out the deprotonation of the alcohol whilst simultaneously protonating the amine. Quinuclidine, with a similar pKa, is unable to form the correct conformation and thus is unable to carry out this concurrent proton exchange, resulting in lower catalytic activity. The quinuclidine, however, was found to be more reactive than DABCO (similar in sterics but less basic) and triethylamine (less basic and significantly more sterically hindered), with triethylamine the least catalytically active, emphasizing the importance of both steric and electronic effects. Whilst there are few studies, it has been suggested that tertiary amines have an increased catalytic effect for aromatic isocyanates compared to aliphatic isocyanates.127
Blocked isocyanates for non-polyurethane applications
Blocked isocyanates have long been used for the synthesis of hyperbranched/dendritic polyurethane materials.128,129 In addition to the vast range of polyurethane based applications for blocked isocyanates, another potential aspect of use that has recently begun to garner attention is within post-polymerization modifications. Despite post-polymerization modifications of isocyanate containing polymers being carried out since the early 1960/1970s, the development of controlled radical techniques in the 1990s allowed for significant developments in the use of post-polymerization modifications of these polymers.130 Indeed, owing to their significant reactivity with nucleophilic species such as amines, alcohols and thiols, modification of the isocyanate functionality is relatively quick and generally does not require a catalyst. Bode and co-workers used amine and alcohol functionalized anthracene to confirm the loss of the dimethyl pyrazole blocking groups on either end of the polymer chain (through the reaction of the released isocyanate with the nucleophilic anthracene), as well as exploiting this reaction for the synthesis of conjugated polymers with defined end groups.131 Work by Hoff et al. further exploited the use of blocked isocyanates not only through post-polymerization modification with thiols and amines, but further demonstrated the ability to use different blocking groups to allow for sequential polymer modification.93 Based on the different deblocking temperature of dimethylpyrazole and triazole blocked isocyanates, it was demonstrated that, through careful selection of reaction conditions, the triazole blocked isocyanates could be first deblocked and reacted with piperidine, prior to the deblocking of the pyrazole blocked isocyanates and their subsequent reaction with benzyl mercaptan (Fig. 6). Interestingly, other work has involved the in situ generation of isocyanates, without the need to directly block the isocyanate or polymerize an isocyanate-functionalized monomer. Klinger et al. synthesized 4-benzoyl azide monomers which were polymerized using free radical polymerization, yet upon heating underwent a Curtius rearrangement evolving nitrogen gas and producing a free isocyanate group, which was further reacted with an alcohol to form various carbamates in quantitative yields (Scheme 3).132 Indeed, the Curtius rearrangement was also used by Van Den Berghe and co-workers for the production of poly(lactic acid) with activated chain ends,133 and by Gody et al. who produced an azide functionalized RAFT (reversible addition-fragmentation chain-transfer) agent for the synthesis of α-end functionalized polymer,134 with the same RAFT agent used by Le-Masurier for coupling polymers onto silica particles.135 More recently, Gody et al. used the Curtius rearrangement on an azide containing RAFT agent, and the reaction between an amine and an isocyanate for polymer–polymer coupling.136 Additionally, the Curtius rearrangement has been further utilized in the aforementioned synthesis of dendritic/hyperbranched polymers using blocked isocyanates, with Okaniwa and co-workers producing dendritic poly(amide-urea)s,137 and Sivakumar and Nasar synthesizing amine and hydroxyl terminated hyperbranched polyurethanes.138 Another application was shown by Sagi et al. who used a blocked isocyanate polymer backbone to produce self-immolative polymers able to be used as molecular sensors.139 The emergence of the use of blocked isocyanates in post-polymerization modifications provides a relatively facile method for polymer modification.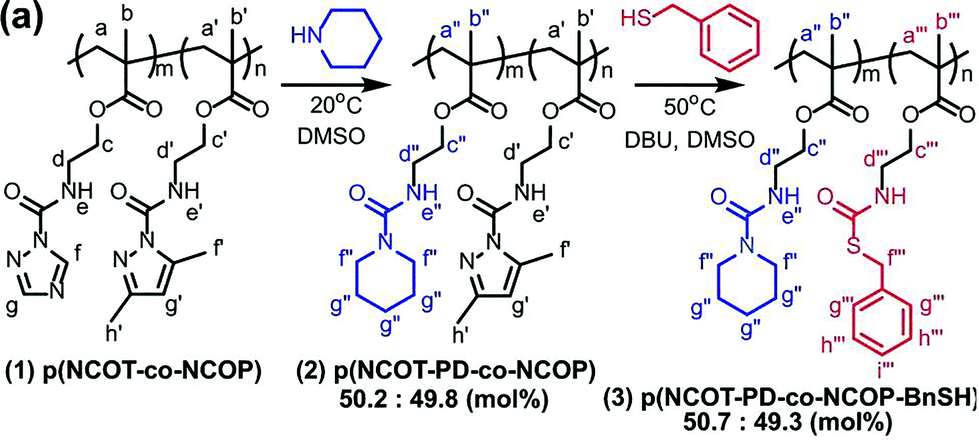 | ||
| Fig. 6 Polymers with pendent blocked isocyanate groups sequentially modified through the exploitation of different deblocking temperatures between the pyrazole and the triazole. Reprinted from ref. 93. Copyright 2016 American Chemical Society. | ||
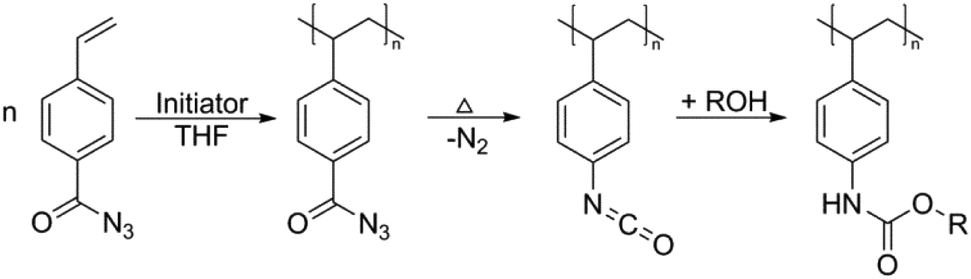 | ||
| Scheme 3 In situ generation of an isocyanate via the Curtius rearrangement, and reaction with an alcohol to yield carbamate functionalized polymers, as reported by Klinger et al. Reproduced from ref. 132 with permission from John Wiley and Sons. | ||
Conclusions
In lieu of the vast array of techniques utilized for the measurement of deblocking temperature of isocyanates, and the significant influence of experimental parameters, it is very challenging to complete an accurate list of blocking agents with their corresponding deblocking temperatures. Indeed, the problems associated with comparisons of deblocking temperatures are highlighted in this review, and caution is hence recommended when implementing a group based on data found in the literature. It is possible to identify a range of deblocking temperatures for a series of isocyanates, based on the literature, but this should only be taken as a rough guide, with further potential for the temperature to be tuned through the use of catalysts, changing experimental conditions or altering blocking group substituents. Most crucially, this review highlights that different techniques generate different deblocking temperatures. As such, the technique chosen for analysis should be carefully selected based on the application of the blocked isocyanate. Indeed, a vast difference is observed when the technique involves solid vs. liquid samples, with liquid samples exhibiting lower deblocking temperatures. For applications such as coatings, where the blocked isocyanate is likely within a liquid formulation, a solid phase technique such as solid state FTIR would produce a deceptively high deblocking temperature impacting the temperature at which the coating would need to be cured at. Moreover, care needs to be taken when analyzing samples in situ, with ramping experiments found to produce lower deblocking temperatures than constant heating at a set temperature, and with different ramping rates further having an impact on the deblocking temperature. Further to this, it should be noted that the deblocking temperature itself, whilst useful, does not provide information on the rate of deblocking, something that less of the literature covers in detail, although it is very important industrially.Acknowledgements
With support and contribution of AWE plc., Aldermaston. The University of Warwick and AWE are thanked for co-funding the PhD studentship to M. S. R. EPSRC are also acknowledged for funding to support R. K. O. (Career Acceleration Fellowship). Dr Guillaume Hedir and Miss Annette Christie are thanked for corrections and suggestions.Notes and references
- O. Bayer, W. Siefhen, H. Rinke, L. Orther and H. Schild, DE Pat, 728981C, 1937 Search PubMed.
- O. Bayer, Angew. Chem., 1947, 59, 257–272 CrossRef.
- P. J. Coppola, US Pat, 4087389A, 1978 Search PubMed.
- R. J. Raynor, US Pat, 4374934A, 1983 Search PubMed.
- M. F. Hnatow, V. A. Bonaddio, C. Y. Chan, D. V. Tursi and J. R. Rucker, US Pat, 6508362B2, 2003 Search PubMed.
- H. L. S. A. Mispreuve, S. Waddington and U. Tribelhorn, US Pat, 5484820A, 1996 Search PubMed.
- K. P. Somani, S. S. Kansara, N. K. Patel and A. K. Rakshit, Int. J. Adhes. Adhes., 2003, 23, 269–275 CrossRef CAS.
- H. Du, Y. Zhao, Q. Li, J. Wang, M. Kang, X. Wang and H. Xiang, J. Appl. Polym. Sci., 2008, 110, 1396–1402 CrossRef CAS.
- C. Schiller, M. Naumann, R. Landers and C. Eilbracht, US Pat, 8946311B2, 2015 Search PubMed.
- R. W. Tilton, US Pat, 4477399A, 1984 Search PubMed.
- A. Demharter, Cryogenics, 1998, 38, 113–117 CrossRef CAS.
- D. K. Chattopadhyay and K. V. S. N. Raju, Prog. Polym. Sci., 2007, 32, 352–418 CrossRef CAS.
- M. Melchiors, M. Sonntag, C. Kobusch and E. Jürgens, Prog. Org. Coat., 2000, 40, 99–109 CrossRef CAS.
- G. L. Statton and J. M. Gaul, US Pat, 4621105A, 1986 Search PubMed.
- G. Wu, J. Li and Y. Luo, Polym. Degrad. Stab., 2016, 123, 36–46 CrossRef CAS.
- J. N. Gibb and J. M. Goodman, Org. Biomol. Chem., 2013, 11, 90–97 CAS.
- S. R. Sompuram, K. Vani, L. Wei, H. Ramanathan, S. Olken and S. A. Bogen, Anal. Biochem., 2004, 326, 55–68 CrossRef CAS PubMed.
- M. S. Kathalewar, P. B. Joshi, A. S. Sabnis and V. C. Malshe, RSC Adv., 2013, 3, 4110–4129 RSC.
- L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Chem. Rev., 2015, 115, 12407–12439 CrossRef CAS PubMed.
- D. Eaves, Handbook of Polymer Foams, Rapra Technology, Shawbury, U.K., 2004 Search PubMed.
- D. Barceló, Emerging Organic Pollutants in Waste Waters and Sludge, Springer, Berlin Heidelberg, 2004 Search PubMed.
- S. Subramani, A. Sultan Nasar, T. Philip Gnanarajan, N. Padmanabha Iyer and G. Radhakrishnan, Polym. Int., 2000, 49, 546–550 CrossRef CAS.
- Z. W. Wicks, Prog. Org. Coat., 1981, 9, 3–28 CrossRef CAS.
- Z. W. Wicks Jr., Prog. Org. Coat., 1975, 3, 73–99 CrossRef.
- D. A. Wicks and Z. W. Wicks, Prog. Org. Coat., 1999, 36, 148–172 CrossRef CAS.
- D. A. Wicks and Z. W. Wicks Jr., Prog. Org. Coat., 2001, 41, 1–83 CrossRef CAS.
- I. A. Mohammed and G. Sankar, High Perform. Polym., 2011, 23, 535–541 CrossRef CAS.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- J. F. Johnson and P. S. Gill, in Analytical Calorimetry: Volume 5, ed. J. F. Johnson and P. S. Gill, Springer US, Boston, MA, 1984, vol. 5, pp. 377–393 Search PubMed.
- J. Gironès, M. T. B. Pimenta, F. Vilaseca, A. J. F. de Carvalho, P. Mutjé and A. A. S. Curvelo, Carbohydr. Polym., 2007, 68, 537–543 CrossRef.
- C. Decker, F. Masson and R. Schwalm, Macromol. Mater. Eng., 2003, 288, 17–28 CrossRef CAS.
- M. Gedan-Smolka, L. Haussler and D. Fischer, Thermochim. Acta, 2000, 351, 95–105 CrossRef CAS.
- G. Sankar and A. S. Nasar, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1557–1570 CrossRef CAS.
- G. Sankar and A. S. Nasar, J. Appl. Polym. Sci., 2008, 109, 1168–1176 CrossRef CAS.
- P. F. Yang, Y. De Han, J. Y. Li and T. D. Li, Int. J. Polym. Anal. Charact., 2011, 16, 251–258 CrossRef CAS.
- Z. Ranjbar, S. Montazeri, M. M. R. Nayini and A. Jannesari, Prog. Org. Coat., 2010, 69, 426–431 CrossRef CAS.
- S. V. Lonikar, N. Rungsimuntakul, R. D. Gilbert and R. E. Fornes, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 759–775 CrossRef CAS.
- J. M. Lee, S. Subramani, Y. S. Lee and J. H. Kim, Macromol. Res., 2005, 13, 427–434 CrossRef CAS.
- H. Kothandaraman and R. Thangavel, J. Appl. Polym. Sci., 1993, 47, 1791–1796 CrossRef CAS.
- S. Subramani, Y.-J. Park, Y.-S. Lee and J.-H. Kim, Prog. Org. Coat., 2003, 48, 71–79 CrossRef CAS.
- P. L. Tabaddor, C. J. Aloisio, H. E. Bair, C. H. Plagianis and C. R. Taylor, J. Therm. Anal. Calorim., 2000, 59, 559–570 CrossRef CAS.
- Q.-W. Lu, T. R. Hoye and C. W. Macosko, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2310–2328 CrossRef CAS.
- D. N. Tarasov, R. P. Tiger, Y. N. Chirkov, S. G. Entelis and J. J. Tondeur, Kinet. Catal., 2000, 41, 355–358 CrossRef CAS.
- H. Mao, F. Yang, C. Wang, Y. Wang, D. Yao and Y. Yin, RSC Adv., 2015, 5, 30631–30639 RSC.
- T. Kiguchi, H. Aota and A. Matsumoto, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 606–615 CrossRef CAS.
- A. Cholli, J. Koenig, T. Sun and H. Zhou, J. Appl. Polym. Sci., 1983, 28, 3497–3502 CrossRef CAS.
- J. F. Johnson and P. S. Gill, Analytical Calorimetry, Springer, US, 2012 Search PubMed.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 Search PubMed.
- A. S. Nasar, S. Subramani and G. Radhakrishnan, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 1815–1821 CrossRef CAS.
- T. Nagy, B. Antal, K. Czifrák, I. Papp, J. Karger-Kocsis, M. Zsuga and S. Kéki, J. Appl. Polym. Sci., 2015, 132, 42127 CrossRef.
- I. Muramatsu, Y. Tanimoto, M. Kase and N. Okoshi, Prog. Org. Coat., 1993, 22, 279–286 CrossRef CAS.
- C. Lou, J. Gu, M. Di, L. Ma, Y. Wang and X. Liu, Iran. Polym. J., 2011, 20, 247–255 CAS.
- A. Li, G. Fan, H. Chen and Q. Zhao, Res. Chem. Intermed., 2012, 39, 3565–3577 CrossRef.
- B. Pilch-Pitera, J. Appl. Polym. Sci., 2012, 124, 3302–3311 CrossRef CAS.
- M. Furukawa and T. Yokoyama, J. Polym. Sci., Part C: Polym. Lett., 1979, 17, 175–180 CrossRef CAS.
- A. Davis, Makromol. Chem., 1963, 66, 196–204 CrossRef CAS.
- G. R. Griffin and L. J. Willwerth, Ind. Eng. Chem. Res., 1962, 1, 265–268 CrossRef CAS.
- H. Kothandaraman, A. S. Nasar and R. K. Lakshmi, J. Appl. Polym. Sci., 1994, 53, 31–38 CrossRef CAS.
- H. Kothandaraman and A. S. Nasar, Polymer, 1993, 34, 610–615 CrossRef CAS.
- D. Katsamberis and S. P. Pappas, J. Appl. Polym. Sci., 1990, 41, 2059–2065 CrossRef CAS.
- A. Mühlebach, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 753–765 CrossRef.
- Y. Zhang, J. Cao, H. Tan and J. Gu, Pigm. Resin Technol., 2014, 43, 194–200 CrossRef CAS.
- Y. Zuo, J. Gu, Y. Zhang, H. Tan, P. Li and M. Di, J. Adhes. Sci. Technol., 2012, 26, 1685–1698 CAS.
- G. Sankar and N. Yan, J. Macromol. Sci., Part A: Pure Appl. Chem., 2015, 52, 47–55 CrossRef CAS.
- T. W. Regulski and M. R. Thomas, ACS Organic Coatings and Applied Polymer Science Proceedings, Seattle, WA, 1983 Search PubMed.
- U. Meier-Westhues, Polyurethanes: Coatings, Adhesives and Sealants, Vincentz Network, Hannover, 2007 Search PubMed.
- W.-H. Chang, R. L. Scriven, J. R. Peffer and S. Porter, Ind. Eng. Chem. Prod. Res. Dev., 1973, 12, 278–288 Search PubMed.
- X. Wen, R. Mi, Y. Huang, J. Cheng, P. Pi and Z. Yang, J. Coat. Technol. Res., 2010, 7, 373–381 CrossRef CAS.
- R. P. Subrayan, S. Zhang, F. N. Jones, V. Swarup and A. I. Yezrielev, J. Appl. Polym. Sci., 2000, 77, 2212–2228 CrossRef CAS.
- J. Gironès, M. T. B. Pimenta, F. Vilaseca, A. J. F. Carvalho, P. Mutjé and A. A. S. Curvelo, Carbohydr. Polym., 2008, 74, 106–113 CrossRef.
- T. Sadoun, G. Clouet and J. Brossas, Makromol. Chem., 1987, 188, 1367–1373 CrossRef CAS.
- K. S. Ho and L. W. Chen, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 1703–1710 CrossRef CAS.
- P. Kamath and M. Srinivasan, Polym. Int., 1993, 32, 33–38 CrossRef CAS.
- G. Y. Lei, S. R. Angeli, D. S. Kristol and W. H. Snyder, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 607–615 CrossRef CAS.
- P. E. Engonga, V. Marchetti, P. Gérardin, P. Tekely and B. Loubinoux, J. Fluorine Chem., 2000, 101, 19–25 CrossRef CAS.
- S. Subramani, J. M. Lee, J. H. Kim and I. W. Cheong, Macromol. Res., 2005, 13, 418–426 CrossRef CAS.
- S. Subramani, Y.-J. Park, I.-W. Cheong and J.-H. Kim, Polym. Int., 2004, 53, 1145–1152 CrossRef CAS.
- H. Kothandaraman and R. Thangavel, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2653–2657 CrossRef CAS.
- A. W. Levine and J. Fech, J. Org. Chem., 1972, 37, 1500–1503 CrossRef CAS.
- D. C. Phillips, J. D. B. Smith, J. F. Meier and T. D. Kaczmarek, Microchem. J., 1978, 23, 165–177 CrossRef CAS.
- H. G. Fravel, T. W. Reguiski and M. R. Thomas, Ind. Eng. Chem. Res., 1984, 23, 586–590 CrossRef CAS.
- Y. Camberlin, P. Michaud, C. Pesando and J. P. Pascault, Makromol. Chem., Macromol. Symp., 1989, 25, 91–99 CrossRef CAS.
- S. Premkumar, C. Karikal Chozhan and M. Alagar, Polym. Eng. Sci., 2009, 49, 747–755 CAS.
- D. A. Dillard and A. V. Pocius, Adhesion Science and Engineering: Surfaces, Chemistry and Applications, Elsevier Science, Amsterdam, 2002 Search PubMed.
- W.-J. Wei, Z.-R. Guo, Y.-F. Zhang and E.-L. Pan, J. Appl. Polym. Sci., 2002, 84, 1346–1352 CrossRef CAS.
- A. S. Nasar, S. Subramani and G. Radhakrishnan, Polym. Int., 1999, 48, 614–620 CrossRef CAS.
- A. S. Nasar, S. N. Jaisankar, S. Subramani and G. Radhakrishnan, J. Macromol. Sci., Part A: Pure Appl. Chem., 1997, 34, 1237–1247 CrossRef.
- E. Wolf, J. Obendorf and R. Gras, US Pat, 4255551, 1981 Search PubMed.
- T. Wang, S. Qi, B. Ren and Z. Tong, Prog. Org. Coat., 2007, 60, 132–139 CrossRef CAS.
- R. Gertzmann and C. Gürtler, Tetrahedron Lett., 2005, 46, 6659–6662 CrossRef CAS.
- S. W. Wong, A. Damusis, K. C. Frisch, R. L. Jacobs and J. W. Long, J. Elastomers Plast, 1979, 11, 15–36 CAS.
- X. Tassel, D. Barbry and L. Tighzert, Eur. Polym. J., 2000, 36, 1745–1751 CrossRef CAS.
- E. A. Hoff, B. A. Abel, C. A. Tretbar, C. L. McCormick and D. L. Patton, Macromolecules, 2016, 49, 554–563 CrossRef CAS.
- R.-V. Meyer, H. J. Kreuder and E. de Cleur, US Pat, 4424353, 1984 Search PubMed.
- I. Polenz, A. Laue, T. Uhrin, T. Ruffer, H. Lang, F. G. Schmidt and S. Spange, Polym. Chem., 2014, 5, 6678–6686 RSC.
- E. Querat, L. Tighzert and J.-P. Pascault, Angew. Makromol. Chem., 1994, 219, 185–203 CrossRef CAS.
- R. E. Buckles and L. A. McGrew, J. Am. Chem. Soc., 1966, 88, 3582–3586 CrossRef CAS.
- E. Spyrou, H. J. Metternich and R. Franke, Prog. Org. Coat., 2003, 48, 201–206 CrossRef CAS.
- M. Edelmann, M. Gedan-Smolka and D. Lehmann, Prog. Org. Coat., 2006, 57, 251–258 CrossRef CAS.
- L. Ubaghs, H. Keul and H. Höcker, Polymer, 2005, 46, 1459–1465 CrossRef CAS.
- D. Heift, Z. Benko, H. Grutzmacher, A. R. Jupp and J. M. Goicoechea, Chem. Sci., 2015, 6, 4017–4024 RSC.
- M. Viganò, R. Suriano, M. Levi, S. Turri, M. Chiari and F. Damin, Surf. Sci., 2007, 601, 1365–1370 CrossRef.
- K.-D. Vorlop, A. Muscat and J. Beyersdorf, Biotechnol. Tech., 1992, 6, 483–488 CrossRef CAS.
- Y. Zhang, J. Gu, X. Jiang, L. Zhu and H. Tan, Pigm. Resin Technol., 2011, 40, 379–385 CrossRef CAS.
- A. S. Nasar, A. Raghavan and V. S. Kumar, J. Macromol. Sci., Part A: Pure Appl. Chem., 2005, 42, 309–319 CrossRef.
- T. P. Gnanarajan, A. S. Nasar, N. P. Iyer and G. Radhakrishnan, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4032–4037 CrossRef CAS.
- J. W. Carter and S. P. Pappas, J. Coat. Technol. Res., 1992, 64, 29 CAS.
- A. Essenfeld and K.-J. Wu, Polym. Mater. Sci. Eng., 1997, 77, 385 CAS.
- Z. W. Wicks, F. N. Jones, S. P. Pappas and D. A. Wicks, Organic Coatings: Science and Technology, Wiley-VCH Verlag GmbH & Co. KGaA, New Jersey, 2006 Search PubMed.
- S. G. Entelis and O. V. Nesterov, Russ. Chem. Rev., 1966, 35, 917–930 CrossRef.
- M. E. Bailey, V. Kirss and R. G. Spaunburgh, Ind. Eng. Chem., 1956, 48, 794–797 CrossRef CAS.
- S. Otto, US Pat, 2671082A, 1954 Search PubMed.
- L. Rand, A. B. Lateef and J. A. Reeder, J. Org. Chem., 1971, 36, 2295–2298 CrossRef CAS.
- J. W. Baker and J. Gaunt, J. Chem. Soc., 1949, 27–31 RSC.
- S. Mohanty and N. Krishnamurti, Eur. Polym. J., 1998, 34, 77–83 CrossRef CAS.
- G. Sankar and N. Yan, J. Macromol. Sci., Part A: Pure Appl. Chem., 2015, 52, 47–55 CrossRef CAS.
- H. Wu, Z. Li, L. Bai, L. Zhu and J. Gu, J. Macromol. Sci., Part A: Pure Appl. Chem., 2015, 52, 847–855 CrossRef CAS.
- H. J. Laas, R. Halpaap and J. Pedain, US Pat, 5237058A, 1993 Search PubMed.
- H. R. Kricheldorf, Handbook of Polymer Synthesis, Taylor & Francis, New York, 1991 Search PubMed.
- T. Engbert, J. Frohlich, W. Kubitza, P. Breidenbach and G. Mennicken, US Pat, 4929724A, 1990 Search PubMed.
- R. Lomölder, F. Plogmann and P. Speier, J. Coat. Technol., 1997, 69, 51–57 CrossRef.
- G. M. Carlson, C. M. Neag, C. Kuo and T. Provder, in Fourier Transform Infrared Characterization of Polymers, ed. H. Ishida, Springer US, Boston, MA, 1987, pp. 197–212 Search PubMed.
- D. A. Wicks and Z. W. Wicks, Prog. Org. Coat., 2001, 43, 131–140 CrossRef CAS.
- S. Petrak, V. Shadurka and W. H. Binder, Prog. Org. Coat., 2009, 66, 296–305 CrossRef CAS.
- W. J. Blank, Z. A. He and E. T. Hessell, Prog. Org. Coat., 1999, 35, 19–29 CrossRef CAS.
- W. J. Blank, Z. A. He and M. E. Picci, Polym. Mater. Sci. Eng., 1998, 79, 399 CAS.
- J. J. Florio and D. J. Miller, Handbook Of Coating Additives, Taylor & Francis, New York, 2004 Search PubMed.
- T. Shanmugam and A. S. Nasar, Macromol. Chem. Phys., 2008, 209, 651–665 CrossRef CAS.
- R. Spindler and J. M. J. Frechet, Macromolecules, 1993, 26, 4809–4813 CrossRef CAS.
- K. A. Günay, P. Theato and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1–28 CrossRef.
- S. Bode, M. Enke, H. Gorls, S. Hoeppener, R. Weberskirch, M. D. Hager and U. S. Schubert, Polym. Chem., 2014, 5, 2574–2582 RSC.
- D. Klinger, J. Y. Chang and P. Theato, Macromol. Rapid Commun., 2007, 28, 718–724 CrossRef CAS.
- H. Van Den Berghe, J. Coudane and M. Vert, J. Bioact. Compat. Polym., 2007, 22, 637–650 CrossRef CAS.
- G. Gody, C. Rossner, J. Moraes, P. Vana, T. Maschmeyer and S. Perrier, J. Am. Chem. Soc., 2012, 134, 12596–12603 CrossRef CAS PubMed.
- S. P. Le-Masurier, G. Gody, S. Perrier and A. M. Granville, Polym. Chem., 2014, 5, 2816–2823 RSC.
- G. Gody, D. A. Roberts, T. Maschmeyer and S. Perrier, J. Am. Chem. Soc., 2016, 138, 4061–4068 CrossRef CAS PubMed.
- M. Okaniwa, K. Takeuchi, M. Asai and M. Ueda, Macromolecules, 2002, 35, 6224–6231 CrossRef CAS.
- C. Sivakumar and A. S. Nasar, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3337–3351 CrossRef CAS.
- A. Sagi, R. Weinstain, N. Karton and D. Shabat, J. Am. Chem. Soc., 2008, 130, 5434–5435 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |