DOI:
10.1039/C6PY01771A
(Paper)
Polym. Chem., 2016,
7, 7223-7229
Poly(amidoamine)–BSA conjugates synthesised by Michael addition reaction retained enzymatic activity†
Received
10th October 2016
, Accepted 7th November 2016
First published on 14th November 2016
Abstract
Polymer–protein conjugates are key to overcome some of the therapeutic protein limitations, including inefficient intracellular delivery. Poly(amidoamine)s are bioresponsive polyelectrolytes, which can form complexes with proteins and promote their delivery into the cytosol of cells. To investigate if conjugation would affect the activity of the protein, two poly(amidoamine)–BSA conjugates were synthesised using a “grafted to” method and Michael addition reaction. Following purification, the conjugates were characterised by electrophoresis, size exclusion chromatography (Mn(C1) = 140.7 kDa; Mn(C2) = 218.6 kDa) and light scattering (Dh(C1) = 37.5 nm; Dh(C2) = 75.1 nm). As a result of the conjugation with the cationic polymer, the conjugates had a positive zeta potential (ζ(C1) = +15.4 mV; ζ(C2) = +20.2 mV). TNBS assays demonstrated that 16% to 25% of the protein amine groups were modified and HPLC analysis indicated that the amount of protein in the conjugate was 0.76 mg of BSA mg−1 of PAA (C1) and 0.43 mg of BSA mg−1 of PAA (C2). Enzymatic assays indicated the conjugates displayed an esterase activity similar (C1) or reduced ∼35% (C2) compared to BSA. Altogether the results demonstrated that the conjugation of poly(amidoamine)s to a model protein can lead to the formation of bioconjugates that retain the enzymatic activity of the native protein. Such conjugates could have some application in protein delivery and enzyme engineering for biocatalysis and biosensors.
Introduction
Polymer–protein conjugates are hybrid materials that display properties generally derived from biological (i.e. the protein) and synthetic (i.e. the polymer) components. Since the 1970s1,2 interest in such bioconjugates has increased exponentially due to their ability to overcome some of the protein limitations. These semi-synthetic materials have been developed for a range of applications, including nanomedicine, biocatalysis and biosensors,3–5 to overcome issues mostly related to protein stability or delivery efficiencies. The physicochemical characteristics of the synthetic polymers can be tailored to obtain properties suitable to their applications. Polymers selected for the synthesis of biopharmaceuticals should be water-soluble, non-toxic, non-immunogenic, well defined, and be biodegradable and/or be easily eliminated from the body. In this context, one of the most commonly used polymers is poly(ethylene glycol) (PEG).6 PEG has been used to prepare numerous conjugates (e.g. Oncaspar®, Pegasys®, CIMZIA®) to treat different diseases with pegylated proteins exerting their pharmacological effect either in the blood circulation or on membrane receptors. However, in recent years, pegylation has faced a number of challenges7,8 and other polymers (e.g. polysialic acid, poly(2-oxazoline)s, PNIPAM, PEGMA) have been the subject of increasing research as potential alternative.
Polymer–protein conjugates are generally synthesised either via a “grafting to” or “grafting from” method. The former relies on coupling techniques between the protein and a preformed polymer, whereas the latter relies on in situ controlled radical polymerisation in which the polymer chains can grow directly from the protein.8,9 Both methods have advantages and disadvantages. “Grafting from” approaches usually use metal catalysts or unstable reagents which could favour protein denaturation and may limit future clinical applications of the conjugates due to cytotoxicity. “Grafting to” conjugations can lead to low grafting densities and usually require an excess of polymer which may imply extensive purification. However to date, the approved polymer–protein conjugates have all been obtained via a “grafting to” method.6,7
Poly(amidoamine)s (PAAs) are “smart”, water-soluble polymers known for their ability to enter the cells by endocytosis.10,11 They are pH-responsive and at low pH, protonation of amino groups arranged regularly along the polymer backbone induces conformational changes potentially facilitating interactions with biological membranes. In contrast to PEG, they are degradable and have been used as nanocarriers for the intracellular delivery of drugs12,13 and biomacromolecules, including proteins.14,15 Bioconjugation can lead to a decrease of the protein biological activity.6,7,16 If PAAs were to be used to develop polymer–protein conjugates, the resulting hybrids should retain some activity. In the present work, bioconjugates between a cationic poly(amidoamine) and bovine serum albumin (BSA) were synthesised using a “grafting to” method. The conjugation was carried out under mild conditions suitable for biologicals. The hydrogen-transfer reaction between amino groups and activated vinyl groups was carried out at different pH in an aqueous environment. The conjugates were purified by ionic exchange and size exclusion chromatography and the enzymatic activities were determined and compared with the native protein under non-denaturing and denaturing conditions.
Experimental
Materials
BSA, NaHCO3, TRIS, methanol, coomassie blue, H2O (HPLC grade), acetic acid, CH3COONa·3H2O, NaCl, NaN3, urea, SDS, Na2HPO4·7H2O, NaH2PO4·H2O were from Fisher. Piperazine hexahydrate, acetonitrile, glycerol and p-nitrophenyl acetate were from Acros Organics. Acryloyl chloride was from Alfa Aesar. NaOH, HCl, anhydrous MgSO4, hexane, dichloromethane, 2-methylpiperazine, N,N′-bis(2-hydroxyethyl)ethylenediamine), trinitrobenzene sulfonate (TNBS), glycine, thyroglobulin (669 kDa), apoferritin (443 kDa), alcohol dehydrogenase (150 kDa) and carbonic anhydrase (29 kDa) were from Sigma. Deuterated dimethyl sulfoxide (DMSO-d6) with tetramethylsilane (TMS), deuterated water (D2O) and 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) were from CIL. The NMR spectra were acquired on an ECA FT NMR spectrometer (Jeol) and the chemical shifts were determined relative to internal reference peak (i.e. TMS or DSS – 1H-NMR) or solvent peak (i.e.13C-NMR).
Synthesis of N,N′-bis(acryloyl)piperazine (BAP) (3)
10 g (0.051 mol) of piperazine hexahydrate (1) was dissolved in 51.55 mL of dichloromethane. A solution of NaOH (4.32 g, 0.11 mol) in H2O (10.3 mL) and a solution of acryloyl chloride (2) (8.77 mL, 0.11 mol) in dichloromethane (4.8 mL) were added dropwise and simultaneously. The mixture was stirred overnight at room temperature, filtered and the organic layer was successively washed with NaOH (0.1 M), HCl (0.1 M), H2O and dried under MgSO4. The solvent was removed under vacuum and the solid recrystallised in dichloromethane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane – 1:10 (v/v). BAP (3) was recovered by filtration and dried under vacuum at 40 °C. Yield: 5.19 g (52.4%). 1H-NMR (DMSO-d6) δ (ppm): 3.59 (s, 4H, –CH2–CH2–), 5.71 (dd, 1H, J = 2.3 Hz, J = 10.5 Hz, HC
:hexane – 1:10 (v/v). BAP (3) was recovered by filtration and dried under vacuum at 40 °C. Yield: 5.19 g (52.4%). 1H-NMR (DMSO-d6) δ (ppm): 3.59 (s, 4H, –CH2–CH2–), 5.71 (dd, 1H, J = 2.3 Hz, J = 10.5 Hz, HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHcisHtrans), 6.13 (dd, 1H, J = 2.3 Hz, J = 16.7 Hz, HCCHcisHtrans), 6.81 (dd, 1H, J = 10.5 Hz, J = 16.7 Hz, HCCHcisHtrans). 13C-NMR (DMSO-d6) δ (ppm): 41.36–45.59 (–CH2–CH2–), 127.98 (–CHCH2), 128.55 (–CHCH2), 164.82 (CO).
CHcisHtrans), 6.13 (dd, 1H, J = 2.3 Hz, J = 16.7 Hz, HCCHcisHtrans), 6.81 (dd, 1H, J = 10.5 Hz, J = 16.7 Hz, HCCHcisHtrans). 13C-NMR (DMSO-d6) δ (ppm): 41.36–45.59 (–CH2–CH2–), 127.98 (–CHCH2), 128.55 (–CHCH2), 164.82 (CO).
Synthesis of poly[(N,N′-bis(acryloyl)piperazine)-co-(2 methylpiperazine)-co-(N,N′-bis(2 hydroxyethyl)ethylenediamine)](PAA) (6)
Under nitrogen, BAP (3) (38.85 g, 0.2 mol), 2-methylpiperazine (15.02 g, 0.1 mol) (4) and N,N′-bis(2-hydroxyethyl)ethylenediamine) (15.25 g, 0.1 mol) (5) were added to 100 mL of H2O. The reaction was stirred for 3 days at 30 °C. A solution of BAP (3) (0.78 g, 0.4 mmol) in 5 mL of H2O was then added and the reaction stirred for one more day. 200 mL of H2O was added and the pH was adjusted to 2 with HCl (6 M). The PAA (6) was recovered by ultrafiltration (5 kDa MWCO) followed by freeze drying. The polymer was further characterised by size exclusion chromatography (SEC) using a PL-GPC 50 system (Varian) equipped with a RI detector. The sample (5 mg mL−1) was analysed at 37 °C using PL-aquagel-OH 30 and 40 columns (8 μm; 10 mm × 300 mm). The molecular weight was determined relative to PEG standards (116300–3930 g mol−1) [mobile phase: TRIS (0.1 M, pH = 8.0), 0.5 M NaCl; flow rate: 1 mL min−1]. Yield: 28.34 g (43.9%). 1H-NMR (D2O) δ (ppm): 1.51 (s, 3H, CH3), 3.05 (m, 7H, methylpiperazine), 3.47–3.65 (m, 8H, NCH2CH2N, NCH2CH2CO, NCH2CH2OH), 3.87 (m, 8H, piperazine), 3.96 (m, 2H, NCH2CH2OH), 5.82 (d, 1H, J = 10.86 Hz, CHCHcisHtrans) 6.16 (d, 1H, J = 16.96 Hz, CHCHcisHtrans), 6.70 (m, 1H, CHCHcisHtrans). SEC (buffer, RI): Mn = 7000 g mol−1; Mw = 13200 g mol−1; PDI = 1.9.
Synthesis of the poly(amidoamine)–BSA conjugates C1 and C2
PAA (6) (8.92 g) was dissolved in H2O (10 mL) and the pH was adjusted to 8 (conjugate 1 (C1) (7)) or 9 (conjugate 2 (C2) (8)) with NaOH (6 M). BSA (150 mg) in 5 mL of H2O was added and the reactions were stirred for 8 days at 30 °C. The pH was maintained (at 8 or 9) throughout. 50 mL of H2O was then added and the solutions were ultrafiltrated (10 kDa MWCO). The conjugates were further purified by ionic exchange chromatography (IEX) (Q-Sepharose) and SEC (Superdex 200) using an ÄKTA Explorer FPLC system (GE Healthcare) equipped with a fraction collector. Detection was at 280 nm. [IEX: 0–60 min: PBS (0.1 M, pH = 7); 60–90 min: 0% to 100% PBS (0.1 M, pH = 7), 1 M NaCl; 90–140 min: PBS (0.1 M, pH = 7), 1 M NaCl; flow rate: 2 mL min−1 – SEC: PBS (10 mM, pH = 7) at 1 mL min−1]. The fractions containing the conjugates were pooled, dialysed against H2O (5 kDa MWCO) and freeze dried. BSA and PAA were used as controls. Yields: C1: 47.8 mg, 36.7%; C2: 42.0 mg, 32.2%.
Size exclusion chromatography
The conjugates (100 μL, 1 mg mL−1) were analysed at 37 °C using the PL-GPC 50 system described previously. Molecular weights were determined using a PL-aquagel-OH 40 and a PL-aquagel-OH MIXED-H (8 μm; 10 mm × 300 mm) columns and protein standards (thyroglobulin (669 kDa), apoferritin (443 kDa), alcohol dehydrogenase (150 kDa) and carbonic anhydrase (29 kDa)) [mobile phase: TRIS (0.1 M, pH = 8), 0.5 M NaCl; flow rate: 1 mL min−1].
Size and zeta potential
Measurements were carried out at 25 °C using a nanoZS zetasizer (Malvern). Zeta potentials were measured using the Smoluchowski analysis. All samples were prepared in H2O at the following concentrations: 1 mg mL−1 for size measurements and 3 mg mL−1 (BSA) or 1 mg mL−1 (PAA and conjugates) for zeta potential measurements.
Native PAGE electrophoresis
Samples in loading buffer (10 μL) were applied onto 4%–12% native PAGE gels at the following concentrations: BSA (0.5 mg mL−1), PAA (10 mg mL−1), conjugates (1 and 10 mg mL−1). Electrophoresis were run for 2 h at 120 V. The gels were stained for 30 min using solution I, de-stained for 1 h with solution II and overnight with solution III. Pictures were recorded using a GBox transilluminator (Syngene). NativeMark™ unstained proteins (5 μL, 0.2–1 mg mL−1) were used as standards [loading buffer: TRIS (0.05 M, pH = 6.8), 2% (v/v) glycerol; running buffer: TRIS (0.125 M, pH = 8.3), 0.96 M glycine; solution I: 8% (w/v) coomassie in methanol:acetic acid 5:1 (v/v); solution II: H2O:methanol:acetic acid 65:25:10 (v/v); solution III: H2O:methanol:acetic acid 88:5:7 (v/v)].
Protein quantification
The amount of BSA in each conjugate was determined by quantification of the tryptophan residue using HPLC (Waters 600 pump and Waters 717plus auto sampler connected to a RF-535 Shimadzu fluorescence detector). The conjugates (0.5 mg) were first hydrolysed for 2 h in NaOH (3 mL, 6 M) and at 100 °C. The samples (50 μL) were then analysed using a C18 Eclipse Plus column (5 μm; 4.6 × 100 mm) with detection at 427 nm (λexc = 280 nm) [mobile phase: acetate buffer (0.2 M; pH = 8), 0.02% (w/v) NaN3; flow rate: 2 mL min−1]. The amount of protein in the conjugates was determined relative to a calibration curve obtained from BSA samples (0.05–0.5 mg) subjected to the same treatment.
TNBS assay
50 μL of TNBS (0.34 mM) were added to 100 μL of NaHCO3 (0.1 M, pH = 8.5) containing BSA or a conjugate (0.5 mg mL−1 BSA eq.). The samples were incubated at 37 °C for 4 h and SDS (50 μL, 0.35 M) and HCl (25 μL, 1 M) were added to stop the reaction. The absorbances were measured at 335 nm using an Infinite M200 Pro microplate reader (TECAN).
Enzymatic assay
To 100 μL of native protein or conjugates (2.5 mg mL−1 BSA eq.) in PBS (0.1 M, pH = 8) was added 10 μL of p-nitrophenyl acetate (20–60 mM) in acetonitrile and 890 μL of PBS (0.1 M, pH = 8). The formation of p-nitrophenol at 37 °C was followed by recording the absorbance at 400 nm every 10 s for 10 min using appropriate blank reference (UV-1700 Pharma spectrophotometer (Shimadzu), PCB 150 peltier system (Varian)). The assay was also carried out after subjecting the protein or the conjugates to denaturing conditions such as incubation in the presence of 4 M urea (30 min at room temperature) or incubation at 50 °C for 10 min.
Results and discussion
Poly(amidoamine) and poly(amidoamine)–BSA conjugates synthesis
Poly(amidoamine)s are synthesised by step-growth polymerisation via Michael-type addition of primary or bis(secondary)amines to bisacrylamides (Scheme 1).11 They have been used to promote the delivery of proteins via polymer complexation.15,17 However, few attempts have been made to develop bioactive protein–PAA conjugates. Albumin–PAAs have been prepared via both convergent and divergent methods but minimal characterisation of the conjugates was provided and the residual biological activity was not investigated.18,19 Melittin–PAAs were synthesised using a “grafted from” approach in order to improve the translocation capacity of the polymers.20 One conjugate demonstrated improved efficiency but the results were mitigated by the associated increased cytotoxicity. To synthesise the protein–PAA conjugates, we selected a poly(amidoamine) based on the structure of ISA1. Earlier studies reported low cytotoxicity compare to most polycations (e.g. PEI) and its capacity to deliver non permeant proteins to the cytosol.14 The structure of the polymer, synthesised with vinyl terminal groups (5–7 ppm), was confirmed by 1H-NMR.† The molecular weight, relative to PEG standards, was 13000 Da and in solution the polymer formed nanoparticles with an average hydrodynamic diameter (Dh) of 75 nm (Table 1). The PAA was subsequently conjugated to BSA by Michael addition using a “grafted to” methodology which involved hydrogen-transfer between the available amino groups of the protein and the PAA's acrylamido bond (Scheme 1). BSA has ∼30 lysines accessible for conjugation.21 To react with the activated acrylamido bond of the polymer, the ε-amino group of the lysine residues must be unprotonated.22 These ε-amino groups have a pKa of ∼10.8.23 However, in the protein, the specific micro environment surrounding the lysine residues is responsible for decreasing this value to a pKa range of 9.3–9.5.21,24 The pH of the reaction was initially maintained at 8, but to favour the conjugation, the reaction was also carried out at pH 9. Zhang et al. demonstrated that increasing the amount of polymer favoured the formation of the protein–polymer conjugate.25 We therefore carried out the conjugation reactions using a 10:1 molar ratio between the PAA and the ε-amino groups.
 |
| | Scheme 1 Synthesis of poly[(N,N′-bis(acryloyl) piperazine)-co-(2-methylpiperazine)-co-(N,N′-bis(2-hydroxyethyl) ethylenediamine) and the BSA conjugates. | |
Table 1 Physicochemical characterisation of the conjugates
| |
M
n (Da) |
M
w (Da) |
Đ
|
D
ha (nm) |
Đ
|
ζ (mV) |
BSAb (mg mg−1) |
NH2c (%) |
|
M
n: number average molecular weight. Mw: weight average molecular weight. Đ: polydispersity. Mn, Mw and Đ were determined by SEC relative to protein standards for the conjugates and BSA and relative to PEG standards for PAA. Dh: hydrodynamic diameter. ζ: zeta potential. Determined by DLS. mg of BSA per mg of conjugate. % of primary amine modified following conjugation to PAA. Mean ± SD, n = 3. |
| C1 |
140700 |
208900 |
1.5 |
37.5 ± 1.7 |
0.47 ± 0.02 |
+15.4 ± 0.4 |
0.76 ± 0.03 |
15.9 ± 0.8 |
| C2 |
218600 |
530400 |
2.4 |
75.1 ± 1.8 |
0.54 ± 0.02 |
+20.2 ± 1.0 |
0.43 ± 0.09 |
24.8 ± 1.0 |
| BSA |
102500 |
120200 |
1.2 |
5.9 ± 0.3 |
0.31 ± 0.01 |
−11.7 ± 1.9 |
— |
— |
| PAA |
7000 |
13200 |
1.9 |
73.5 ± 10.7 |
0.47 ± 0.07 |
+44.8 ± 2.3 |
— |
— |
Purification of the poly(amidoamine)–BSA conjugates
The “grafted to” conjugation leads to a combination of conjugates, unreacted polymer and free protein. The recovered mixtures were ultrafiltrated against deionised water and freeze dried. The conjugates were purified by ion exchange chromatography (IEX) (Fig. 1a) to remove any free BSA (∼100 min). The collected fractions (∼20–50 min) were dialysed against water, freeze dried and further purified by size exclusion chromatography (SEC) to remove the remaining unreacted PAA (Fig. 1b). The conjugates eluted at ∼10–15 min while the polymer eluted at ∼20–30 min.
 |
| | Fig. 1 Purification of the conjugates. Panel (a): Free BSA was removed by IEX (0–60 min (PBS); 60–90 min (0 to 100% PBS/NaCl); 90–140 min (PBS/NaCl); 2 mL min−1). Panel (b): Collected fractions were further purified by SEC (PBS; 1 mL min−1) to remove free PAA. Collected fractions were dialysed against H2O and freeze dried before further analysis. BSA and PAA were used as control for IEX and SEC, respectively. | |
Physicochemical characterisation of the poly(amidoamine)–BSA conjugates
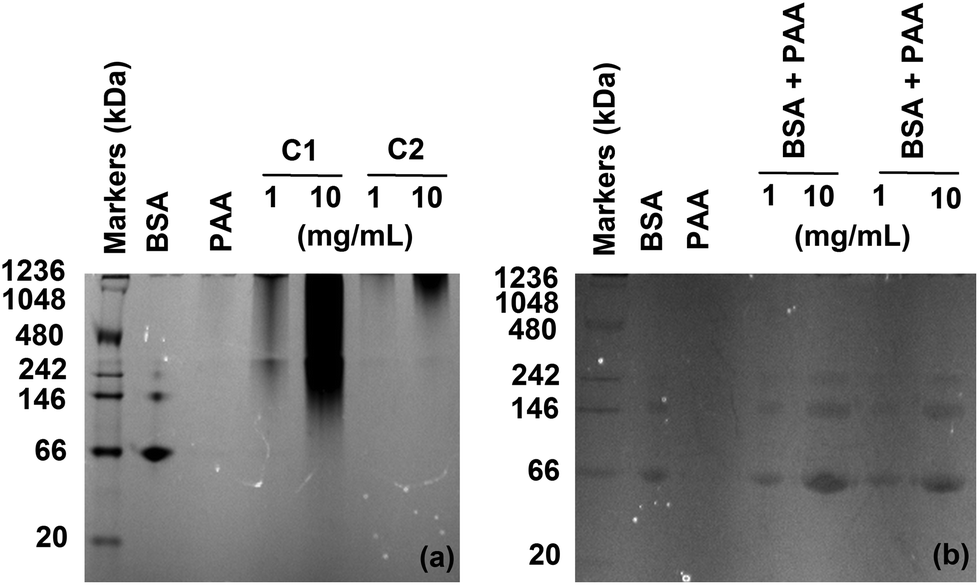
The analysis of the samples by native PAGE electrophoresis clearly demonstrated the presence of some conjugates with higher molecular weight in comparison to BSA, while no free protein could be detected (Fig. 2a). The broadness of the bands reflected the polydispersity of the samples, due to the presence of different conjugates and has been previously observed.25–27 BSA has been found to adsorb onto nanoparticles coated with amphiphilic polymers28 and we have demonstrated the formation of complexes with poly(amidoamine)s which could induce the retardation of the protein.15,29 Control experiments were therefore carried out by mixing free PAA and free BSA. No bands at higher molecular weight were observed (Fig. 2b) confirming the formation and detection of the bioconjugates. The conjugates were further characterised by SEC and dynamic light scattering (DLS). An increase in the hydrodynamic diameter and molecular weight was noticed for both conjugates in comparison to BSA (Table 1). C2 displayed larger values compare to C1, potentially indicating the presence of a higher number of PAA chains attached to the protein. The polydispersity of the samples observed in the acrylamide gels (Fig. 2) was also apparent from these results and is due to the conjugation method which did not allow control of the conjugation. BSA has two tryptophan residues. The amount of protein per milligrams of conjugate was determined by quantifying the tryptophan content of the samples.
 |
| | Fig. 2 Native PAGE analysis of the purified conjugates at low and high concentrations (a) and a mixture of BSA and PAA corresponding to the composition of C1 and C2 at low and high concentrations (b). BSA (0.5 mg mL−1) and PAA (10 mg mL−1) were used as control. Free PAA did not elute. | |
Both conjugates were subjected to alkaline hydrolysis and analysed by HPLC. C1 possessed the highest amount of BSA per mg of conjugate (Table 1). These results were correlated to the percentage of modified amine following conjugation with the polymer. The remaining percentage of free primary amines, relative to unconjugated BSA, was determined using a TNBS assay (Table 1).30 C1 had ∼84% of unmodified amines while C2 had only ∼75%. These results are in agreement with the conjugates BSA content and the conjugates relative size obtained by SEC and DLS (Table 1). As expected, the zeta potential measurements demonstrated that conjugation of the polymer chains to BSA, modified the protein surface charge.31 The poly(amidoamine) used in the conjugation being cationic (+44.8 mV), both conjugates displayed a positive potential (+15.4 mV and +20.2 mV) in comparison to BSA (−11.7 mV) (Table 1).
Enzymatic activity
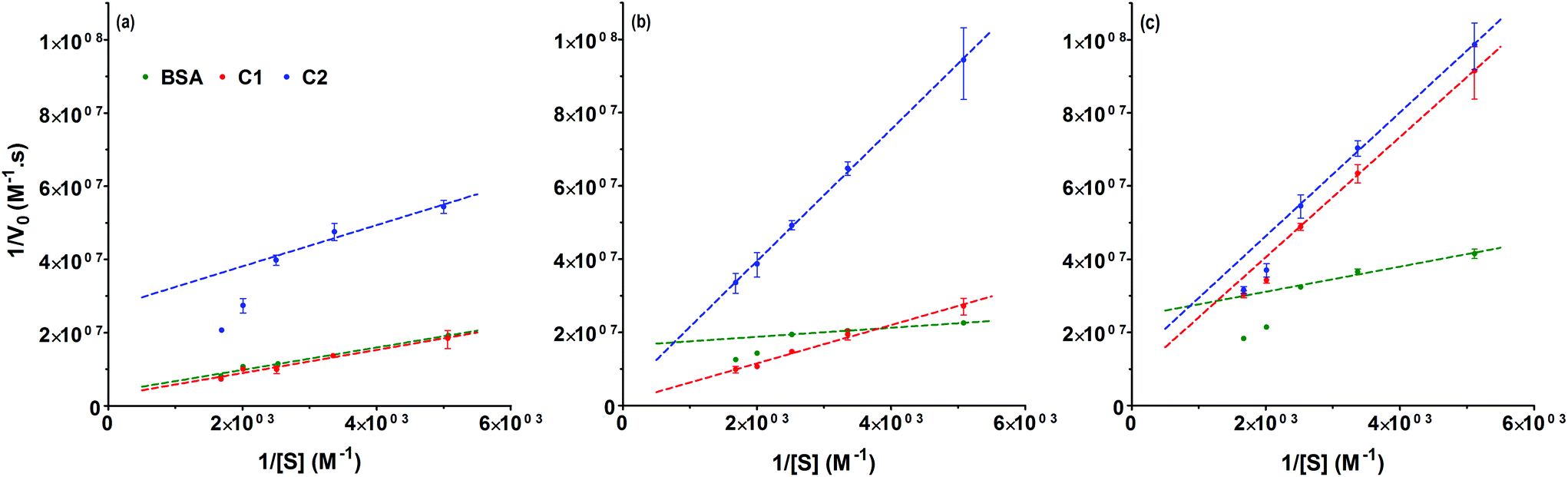
BSA has been frequently used as a model protein to investigate different bioconjugation strategies.11,18,32–37 The conjugation of polymers to proteins can affect the protein stability and/or bioactivity. Factors such as the nature of the modified amino acids, the structure and physicochemical properties of the polymer or the method used for the conjugation or the ligation strategy can influence the protein activity either positively (i.e. increased activity) or negatively (i.e. decreased activity).16 BSA catalyses the hydrolysis of p-nitrophenyl acetate to nitrophenol, the reaction can be monitored by following product formation at 400 nm.38 Previous studies have reported the synthesis of polymer–BSA conjugates which, at room temperature, had a residual activity between 63 to 94% of the native protein.39–42 The esterase-like enzymatic activities of the conjugates were determined at 37 °C (Fig. 3). Under non-denaturing condition, C1 displayed 104% (±3%) activity of the non-modified BSA, while C2 retained only 35% (±0.3%). The decreased activity of C2 could be linked to the higher degree of amine modification obtained at pH 9 (Table 1). This correlation has been previously observed for other conjugates.42,43 Seo et al. developed PMPC–BSA conjugates which retained full activity after incubation at 64 °C for 5 min.44 Under similar stress conditions (10 min at 50 °C) C1 and C2 retained 71% (±1%) and 20% (±0.5%) activity, respectively. In the presence of 4 M urea both conjugates displayed an activity around 20% (±0.4%). Upon conjugation, modification of the affinity between the substrate and the enzyme has been reported for different conjugates.45–50 The enzyme kinetics of BSA, C1 and C2 was investigated and the kinetic parameters (Vmax, KM and kcat) were determined from Lineweaver–Burk plots (Fig. 4). Under normal conditions, BSA and C1 showed similar linear curves (Fig. 4a). A small increase of KM was observed for C1 indicating a potential decrease of the affinity for the substrate (Table 2). However, this did not significantly affect kcat/KM as C1 displayed a catalytic efficiency (kcat/KM = 84.5 (M s)−1) similar to BSA (kcat/KM = 87.5 (M s)−1). Following incubation at 50 °C for 10 min, the Lineweaver–Burk plot for BSA deviated from linearity, whereas C1 still displayed a linear plot (Fig. 4b). Similar deviation has been observed for C2 at 37 °C. Such deviation has been observed for other enzymes and has been attributed to substrate activation, cooperative interactions, presence of isoenzymes or multiple enzymes catalysing the same reaction.51,52 When subjected to an increase in temperature, BSA is denatured and upon quenching of the temperature, only a fraction of the protein will refold correctly.53 This could lead to the presence of different protein structures in solution, each with different affinity towards the substrate, and could potentially explain the deviation from linearity. In this case, the kinetic parameters extracted from the low substrate concentrations of the Lineweaver–Burk plots should be considered cautiously as they will only characterise the enzyme with the highest affinity for the substrate.53 This is reflected by the 10-fold decrease of the KM for BSA. In the case of C1, we noted a ∼4-fold decrease of the conjugate affinity corresponding to a lower catalytic efficiency (kcat/KM = 50.8 (M s)−1) but we did not detect a deviation from linearity, which may indicate some protecting effect. In the presence of 4 M urea (Fig. 4c), all the Lineweaver–Burk plots deviated from linearity potentially indicating the presence of different protein and conjugate conformation and no protection against urea denaturation.
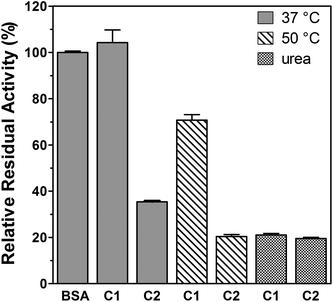 |
| | Fig. 3 Relative residual enzymatic activities under non-denaturing (37 °C) and denaturing conditions (50 °C and urea). All residual activities are relative to the enzymatic activity of the non-modified BSA, which was 12.5 μmol min−1 g−1 protein. Mean ± SD, n = 3. | |
 |
| | Fig. 4 Lineweaver–Burk plots for BSA (green), C1 (red) and C2 (blue). (a) 37 °C (b) 50 °C and (c) urea. The Michaelis–Menten kinetic parameters were obtained by nonlinear regression using the Lineweaver–Burk equation. Mean ± SD, n = 3. | |
Table 2 Kinetic parameters for the hydrolysis of p-nitrophenyl acetate
| |
|
V
max (10−9 M s−1) |
K
M (10−3 M) |
k
cat (10−3 s−1) |
|
V
max: maximum velocity; KM: affinity constant; kcat: turnover number. High affinity binding sites at low substrate concentrations. |
| 37 °C |
BSA |
269 |
0.82 |
71.7 |
| C1 |
371 |
1.17 |
98.8 |
| C2a |
37 |
0.21 |
9.9 |
| 50 °C |
BSAa |
61 |
0.08 |
16.3 |
| C1 |
937 |
4.91 |
249.3 |
| C2 |
286 |
5.14 |
75.8 |
| Urea |
BSAa |
41 |
0.14 |
10.9 |
| C1a |
129 |
2.12 |
34.3 |
| C2a |
80 |
1.34 |
21.2 |
Conclusions
Poly(amidoamine)–BSA conjugates were successfully synthesised using a “grafted to” method. The conjugation to lysine residues was carried out using mild conditions at room temperature and in an aqueous environment. Following purification by chromatography, the conjugates were fully characterised. Under non denaturing conditions, the residual enzymatic activity of the bioconjugates correlated with the percentage of conjugated lysine residues. Exposure to increased temperature led to a decrease of the catalytic efficiency but, as opposed to the native protein, without evidence of formation of intermediates. To date poly(amidoamine)s have been essentially used for biomedical applications to promote cellular delivery of biomacromolecules. The same methodology could be used for therapeutic proteins with an intracellular pharmacological target but could also have potential applications in the field of biocatalysis and biosensors.
Acknowledgements
We would like to thank the Medway School of Pharmacy and the University of Kent for financial support and Dr B. Nielsen (University of Greenwich) for providing access to the FPLC.
References
- T. Yoshimura, A. Imanishi and T. Isemura, J. Biochem., 1968, 63, 730 CAS.
- A. Abuchowski, T. Vanes, N. C. Palczuk and F. F. Davis, J. Biol. Chem., 1977, 252, 3578 CAS.
- J.-F. Lutz and H. G. Boerner, Prog. Polym. Sci., 2008, 33, 1 CrossRef CAS.
- S. Basak, V. D. Punetha, G. Bisht, S. S. Bisht, N. G. Sahoo and J. W. Cho, Polym. Rev., 2015, 55, 163 CrossRef CAS.
- W. Zhao, F. Liu, Y. Chen, J. Bai and W. Gao, Polymer, 2015, 66, A1 CrossRef CAS.
- G. Pasut, Polymer, 2014, 6, 160 Search PubMed.
-
A. Besheer, R. Liebner, M. Meyer and G. Winter, in Tailored Polymer Architectures for Pharmaceutical and Biomedical Applications, ed. C. Scholz and J. Kressler, 2013, p. 215 Search PubMed.
- E. M. Pelegri-O'Day, E. W. Lin and H. D. Maynard, J. Am. Chem. Soc., 2014, 136, 14323 CrossRef PubMed.
- J. D. Wallat, K. A. Rose and J. K. Pokorski, Polym. Chem., 2014, 5, 1545 RSC.
- S. C. W. Richardson, N. G. Pattrick, N. Lavignac, P. Ferruti and R. Duncan, J. Controlled Release, 2010, 142, 78 CrossRef CAS PubMed.
- P. Ferruti, J. Polym. Sci., Polym. Chem., 2013, 51, 2319 CrossRef CAS.
- N. Lavignac, J. L. Nicholls, P. Ferruti and R. Duncan, Macromol. Biosci., 2009, 9, 480 CrossRef CAS PubMed.
- P. Urban, J. Jose Valle-Delgado, N. Mauro, J. Marques, A. Manfredi, M. Rottmann, E. Ranucci, P. Ferruti and X. Fernandez-Busquets, J. Controlled Release, 2014, 177, 84 CrossRef CAS PubMed.
- N. Lavignac, M. Lazenby, P. Foka, B. Malgesini, I. Verpilio, P. Ferruti and R. Duncan, Macromol. Biosci., 2004, 4, 922 CrossRef CAS PubMed.
- J. L. N. Dubois and N. Lavignac, J. Mater. Chem. B, 2015, 3, 6501 RSC.
- M. A. Gauthier and H. A. Klok, Polym. Chem., 2010, 1, 1352 RSC.
- G. Coué, C. Freese, R. E. Unger, C. James Kirkpatrick and J. F. J. Engbersen, Acta Biomater., 2013, 9, 6062 CrossRef PubMed.
- E. Ranucci, F. Bignotti, P. L. Paderno and P. Ferruti, Polymer, 1995, 36, 2989 CrossRef CAS.
- P. Ferruti, N. Mauro, A. Manfredi and E. Ranucci, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4947 CrossRef CAS.
- N. Lavignac, M. Lazenby, J. Franchini, P. Ferruti and R. Duncan, Int. J. Pharm., 2005, 300, 102 CrossRef CAS PubMed.
-
G. T. Hermanson, Bioconjugate Techniques, Academic Press, SanDiego, CA, 1996 Search PubMed.
- B. Malgesini, I. Verpilio, R. Duncan and P. Ferruti, Macromol. Biosci., 2003, 3, 59 CrossRef CAS.
-
D. Voet and J. G. Voet, Biochemistry, Wiley, Hoboken, NJ, 3rd edn, 2004 Search PubMed.
- D. G. Isom, C. A. Castaneda, B. R. Cannon and B. E. Garcia-Moreno, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5260 CrossRef CAS PubMed.
- Q. L. Zhang, N. Vanparijs, B. Louage, B. G. De Geest and R. Hoogenboom, Polym. Chem., 2014, 5, 1140 RSC.
- K. L. Heredia, L. Tao, G. N. Grover and H. D. Maynard, Polym. Chem., 2010, 1, 168 RSC.
- J. Nicolas, V. San Miguel, G. Mantovani and D. M. Haddleton, Chem. Commun., 2006, 4697, 10.1039/b609935a.
- Y. S. Liu, R. B. Zhong, P. Zhang, Y. X. Ma, X. L. Yun, P. Gong, J. M. Wei, X. M. Zhao and F. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 2478 CAS.
- J. L. N. Dubois and N. Lavignac, Polym. Chem., 2014, 5, 1586 RSC.
- P. Caliceti, O. Schiavon, T. Hirano, S. Ohashi and F. M. Veronese, J. Controlled Release, 1996, 39, 27 CrossRef CAS.
- V. Thilakarathne, V. A. Briand, Y. X. Zhou, R. M. Kasi and C. V. Kumar, Langmuir, 2011, 27, 7663 CrossRef CAS PubMed.
- P. C. Nauka, J. Lee and H. D. Maynard, Polym. Chem., 2016, 7, 2352 RSC.
- Q. Zhang, M. Li, C. Zhu, G. Nurumbetov, Z. Li, P. Wilson, K. Kempe and D. M. Haddleton, J. Am. Chem. Soc., 2015, 137, 9344 CrossRef CAS PubMed.
- N. Vanparijs, S. Maji, B. Louage, L. Voorhaar, D. Laplace, Q. Zhang, Y. Shi, W. E. Hennink, R. Hoogenboom and B. G. De Geest, Polym. Chem., 2015, 6, 5602 RSC.
- Z. Y. Liu, C. H. Dong, X. M. Wang, H. J. Wang, W. Li, J. Tan and J. Chang, ACS Appl. Mater. Interfaces, 2014, 6, 2393 CAS.
- A. Simakova, S. E. Averick, D. Konkolewicz and K. Matyjaszewski, Macromolecules, 2012, 45, 6371 CrossRef CAS.
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955 CrossRef CAS PubMed.
- Y. Sakurai, S. F. Ma, H. Watanabe, N. Yamaotsu, S. Hirono, Y. Kurono, U. Kragh-Hansen and M. Otagiri, Pharm. Res., 2004, 21, 285 CrossRef CAS.
- G. N. Grover, S. N. S. Alconcel, N. M. Matsumoto and H. D. Maynard, Macromolecules, 2009, 42, 7657 CrossRef CAS PubMed.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2008, 130, 11288 CrossRef CAS PubMed.
- C. Boyer, V. Bulmus, J. Q. Liu, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Am. Chem. Soc., 2007, 129, 7145 CrossRef CAS PubMed.
- H. Sasaki, Y. Ohtake, A. Matsushima, M. Hiroto, Y. Kodera and Y. Inada, Biochem. Biophys. Res. Commun., 1993, 197, 287 CrossRef CAS PubMed.
- D. Miyamoto, J. Watanabe and K. Ishihara, Biomaterials, 2004, 25, 71 CrossRef CAS PubMed.
- J. H. Seo, R. Matsuno, Y. Lee, M. Takai and K. Ishihara, Biomaterials, 2009, 30, 4859 CrossRef CAS PubMed.
- N. Suthiwangcharoen and R. Nagarajan, Biomacromolecules, 2014, 15, 1142 CrossRef CAS PubMed.
- Z. Zarafshani, T. Obata and J.-F. Lutz, Biomacromolecules, 2010, 11, 2130 CrossRef CAS PubMed.
- B. Treetharnmathurot, C. Ovartlarnporn, J. Wungsintaweekul, R. Duncan and R. Wiwattanapatapee, Int. J. Pharm., 2008, 357, 252 CrossRef CAS PubMed.
- T. Shimoboji, E. Larenas, T. Fowler, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 2003, 14, 517 CrossRef CAS PubMed.
- Z. R. Lu, P. Kopeckova, Z. C. Wu and J. Kopecek, Bioconjugate Chem., 1998, 9, 793 CrossRef CAS PubMed.
- G. Chen and A. S. Hoffman, Bioconjugate Chem., 1993, 4, 509 CrossRef CAS PubMed.
-
I. H. Segel, Enzyme kinetics: Behavior and analysis of rapid equilibrium and steady-state enzyme systems, John Wiley & Sons, New York, 1993 Search PubMed.
-
J. E. Bell and E. T. Bell, in Proteins and enzymes, Prentice-Hall, University of Michigan, 1988, p. 498 Search PubMed.
- Y. Moriyama, E. Watanabe, K. Kobayashi, H. Harano, E. Inui and K. Takeda, J. Phys. Chem. B, 2008, 112, 16585 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra. See DOI: 10.1039/c6py01771a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*