
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGrafting of functional methacrylate polymer brushes by photoinduced SET-LRP†
Mariia
Vorobii‡
a,
Ognen
Pop-Georgievski‡
b,
Andres
de los Santos Pereira
 b,
Nina Yu.
Kostina
a,
Ryan
Jezorek
c,
Zdeňka
Sedláková
b,
Virgil
Percec
*c and
Cesar
Rodriguez-Emmenegger
*ac
b,
Nina Yu.
Kostina
a,
Ryan
Jezorek
c,
Zdeňka
Sedláková
b,
Virgil
Percec
*c and
Cesar
Rodriguez-Emmenegger
*ac
aDWI - Leibniz-Institute for Interactive Materials and Institute of Technical and Macromolecular Chemistry, RWTH Aachen University, Forckenbeckstraße 50, 52074 Aachen, Germany. E-mail: rodriguez@dwi.rwth-aachen.de
bDepartment of Chemistry and Physics of Surfaces and Biointerfaces, Institute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic, v.v.i., Heyrovsky sq. 2, 162 06 Prague, Czech Republic
cRoy & Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6323, USA. E-mail: percec@sas.upenn.edu
First published on 19th October 2016
Abstract
Photoinduced surface-initiated single electron transfer living radical polymerization (SET-LRP) is a versatile technique for the preparation of polymer brushes. The vast diversity of compatible functional groups, together with a high end-group fidelity that enables precise control of the architecture, makes this approach an effective tool for tuning the properties of surfaces. We report the application of photoinduced SET-LRP for the surface-initiated grafting of polymer brushes from a wide range of methacrylate monomers for the first time. The living character of the process was demonstrated by the linear evolution of the polymer brush thickness in time, the ability to reinitiate the polymerization for the preparation of well-defined block copolymers, and also by X-ray photoelectron spectroscopy depth profiling. The surface patterning with these brushes could be achieved simply by restricting the irradiated area. The ability of poly(methacrylate) brushes prepared in this way to prevent non-specific protein adsorption is also demonstrated, indicating the suitability of this procedure for advanced applications.
Introduction
Polymer brushes are ultrathin polymer coatings, formed by highly packed polymer chains that are tethered by one end to a surface or interface.1,2 The diversity of polymers and their functional groups makes them a powerful tool to achieve control of the properties of surfaces.3–5 As an example, covering the surface with polymer brushes could suppress non-specific protein adsorption (protein fouling) from biological media6–8 as well as decrease the forces of bacterial adhesion on the surface.9 These properties are interesting for use in medical devices, such as artificial heart valves, catheters, or other indwelling devices to prevent bacterial infections and to avoid the formation of blood clots. Particularly in affinity biosensors, antifouling polymer brushes are used to eliminate the non-specific response in complex biological matrices,10,11 including blood plasma,12 to allow for label-free detection.13,14 At the same time, polymer brushes have also been employed as a platform for selective protein binding and applied for protein purification.15There are two main strategies for the preparation of polymer brushes: “grafting to” and “grafting from”, which are compatible with a wide diversity of substrates and nanomaterials.16 Since the grafting-to approach is based on the attachment of large end-functionalized polymer chains that must diffuse through the formed polymer film to reach the reactive groups on the surface, this strategy cannot yield very dense and thick polymer brushes.2,15,17 On the other hand, the grafting-from approach, based on direct polymerization typically from initiator-functionalized surfaces, allows achieving very dense polymer brushes and good control over the thickness, composition and architecture.18,19 Among different living polymerization techniques employed for this purpose, including cationic and anionic polymerizations18 and ring-opening polymerization,20 the most frequently used methods are controlled radical polymerization techniques:17 surface-initiated atom transfer radical polymerization (SI-ATRP),21,22 surface reversible addition–fragmentation chain transfer polymerization (S-RAFT),20,23 surface-initiated nitroxide-mediated polymerization (SI-NMP),24–26 and surface-initiated photoiniferter-mediated polymerization (SI-PIMP).27,28 A promising new technique for the preparation of polymer brushes is single-electron transfer living radical polymerization (SET-LRP).29–34 Typical for this approach are very fast polymerization kinetics at room temperature29,35,36 and below37,38 with very low catalyst concentrations.39–41 SET-LRP is compatible with monomers containing various functional groups,42 as well as with different solvents such as dimethyl sulfoxide (DMSO),43,44 alcohols, fluorinated alcohols,45,46 ionic liquids, water, and mixtures.39,47,48 Moreover, it was recently extended to mixtures of non-disproportionating solvents with water.49 SET-LRP is characterized by perfect or near-perfect chain-end fidelity33,50 which is critical for the control of the polymer architecture via end group modification, re-initiation and preparation of multiblocks.40 Interestingly, even RAFT agents could be used as initiators for SET-LRP instead of the typical alkyl halides.51
In the past decade, great interest has been raised over surface modifications, enabling the patterning of polymers, and also development of multifunctional surfaces. Among the applications that require such capabilities are biosensors, presenting areas of varying functionality or multiple sensing regions.52 The most commonly used strategies for the preparation of patterns are: photo and interference lithography, electron-beam lithography, scanning probe lithography, soft lithography, and other lithographic approaches.53 Photolithography is particularly attractive for several of its advantages,17 since relatively large areas can be easily and rapidly patterned. Furthermore, the decreased contact time lowers the risk of contamination in comparison with contact writing techniques. One way to use this approach is in combination with surface-initiated polymerization to prepare binary polymer brush patterns.53,54 Binary polymer brushes are interesting because of their ability to adjust their morphology in response to changes in their surrounding environment.55–58
Recently, photoinduced surface-initiated SET-LRP was introduced, merging the advancements in living radical polymerization of activated monomers with the enhanced control over the process that is enabled by the use of UV light as a polymerization trigger.39,41,59–63 This has several benefits, including direct spatial control over polymer growth and regulation of the molecular weight by control of the intensity of illumination.64 The translation of this promising approach to the surface-initiated grafting of polymer brushes has led to micropatterns, while preserving the living character of the polymerization and enabling the formation of block copolymers.41,65 This technique was applied to a wide range of acrylate monomers of various functionalities, constituting the fastest surface-initiated polymerization reported thus far and reaching unprecedented thicknesses of up to 1 μm within 1 h of polymerization.41 Moreover, using this protocol, polymer brushes of N-(2-hydroxypropyl)methacrylamide (HPMA) could be obtained, while its surface-initiated polymerization proceeded in a living manner for the first time.65 Consequently, this methodology holds vast potential for the controlled grafting of various types of functional polymer brushes.
Particularly important families of monomers that have been used to tailor surface functionality are those including a methacrylate polymerizable group.10,11,66–68 Several such monomers have already been used for the modification of surfaces. For example, polymer brushes obtained from oligo(ethylene glycol) methyl ether methacrylate (MeOEGMA), 2-hydroxypropyl methacrylate (HPM),6 and 2-hydroxyethyl methacrylate (HEMA)11,69 are well known for their antifouling properties. They prevent nonspecific protein adsorption from single-protein solutions and reduce fouling of human blood plasma up to 90% in comparison with bare gold. Poly[2-(dimethylamino)ethyl methacrylate] (DMAEMA) was grafted from cellulose paper and the surfaces obtained were used to detect DNA hybridization.70 Moreover, poly(DMAEMA) brushes can be used to prepare stimuli-responsive surfaces, as they are able to change their conformation in response to temperature changes in a pH-dependent manner.71,72 Thus, there is considerable interest in the development of improved surface-initiated polymerization protocols for such monomers. Additionally, introducing light as a control mechanism to spatially confine the polymerization would enable the facile fabrication of (micro)patterned poly(methacrylate) brushes.
In this report, we present the use of photoinduced SET-LRP for the surface-initiated grafting of polymer brushes from a wide range of methacrylate monomers presenting various functionalities. We investigated the influence of varying the solvent composition and the concentration of the Cu-based catalyst on the polymerization process. Additionally, we demonstrate the livingness of the process when applied to methacrylate monomers, as well as the ability to create surface patterns of polymer brushes by a simple photolithographic approach.
Results and discussion
Grafting of methacrylate polymer brushes
Photoinduced SET-LRP was used for the preparation of polymer brushes from a variety of methacrylate monomers on silicon wafer chips as a model substrate. The process can be divided in two steps: firstly, a self-assembled monolayer (SAM) of the initiator is formed, and secondly, the photoinduced polymerization of each monomer is carried out.For the preparation of the SAM, 11-(trichlorosilyl)undecyl 2-bromo-2-methylpropanoate was chosen as the initiator, as it has been shown to lead to the formation of well-defined monolayers. This is an essential requirement for the stable grafting of dense brushes. The silicon substrates were freshly activated with air plasma and immersed in a solution of the initiator in dry toluene to form the SAM (Scheme 1).
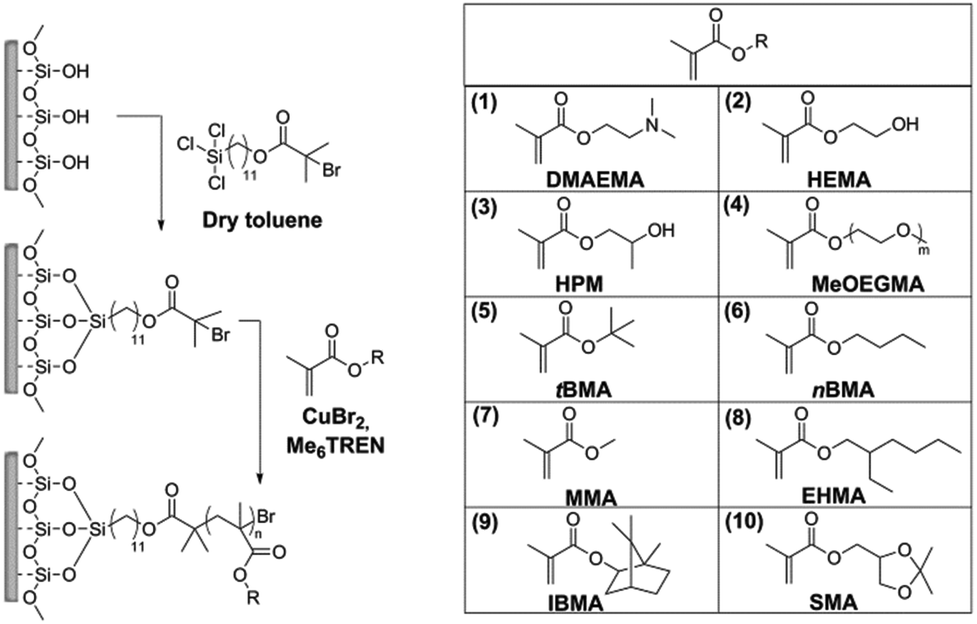 | ||
| Scheme 1 Surface-initiated photoinduced SET-LRP of methacrylates. (1) 2-(Dimethylamino)ethyl methacrylate (DMAEMA), (2) 2-hydroxyethyl methacrylate (HEMA), (3) 2-hydroxypropyl methacrylate (HPM), (4) oligo(ethylene glycol) methyl ether methacrylate (MeOEGMA), (5) t-butyl methacrylate (tBMA), (6) n-butyl methacrylate (nBMA), (7) methyl methacrylate (MMA), (8) 2-ethylhexyl methacrylate (EHMA), (9) isobornyl methacrylate (IBMA), and (10) solketal methacrylate (SMA). | ||
Dynamic water contact angle, atomic force microscopy (AFM), X-ray photoelectron spectroscopy (XPS), and spectroscopic ellipsometry were used to confirm the successful grafting of the initiator layer. The formation of the SAM is confirmed by a change in the surface wettability as measured by sessile-drop dynamic water contact angle goniometry, from fully wettable freshly plasma-cleaned silicon substrates to markedly more hydrophobic for the SAM (θadv = 83° and θrec = 77°). This result is expected due to the chemical structure of the initiator, containing a long alkyl backbone. The thickness of the initiator layer was determined to be (0.97 ± 0.04) nm by ellipsometry, which is consistent with the presence of a monolayer. AFM topography images show homogeneous coverage and a low roughness of Rq = 1.14 nm (see the ESI, Fig. SF1†). The chemical composition of the SAM was confirmed by XPS. The XPS spectrum of the C 1s region (Fig. 1a) shows the predominance of the ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) –C, –H component at 285.0 eV, attributed to the alkane backbone of the initiator SAM. The resolved contributions of the envelope at 284.2, 286.7 and 289.2 eV correspond to the –Si bond, the carbon atoms of the –O–(C
–C, –H component at 285.0 eV, attributed to the alkane backbone of the initiator SAM. The resolved contributions of the envelope at 284.2, 286.7 and 289.2 eV correspond to the –Si bond, the carbon atoms of the –O–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and Br––(CO)–O, and the ester group ((O)–O), respectively. The presence of Br groups is further confirmed by the high resolution spectrum of the Br 3d region (see the ESI, Fig. SF2†).
O) and Br––(CO)–O, and the ester group ((O)–O), respectively. The presence of Br groups is further confirmed by the high resolution spectrum of the Br 3d region (see the ESI, Fig. SF2†).
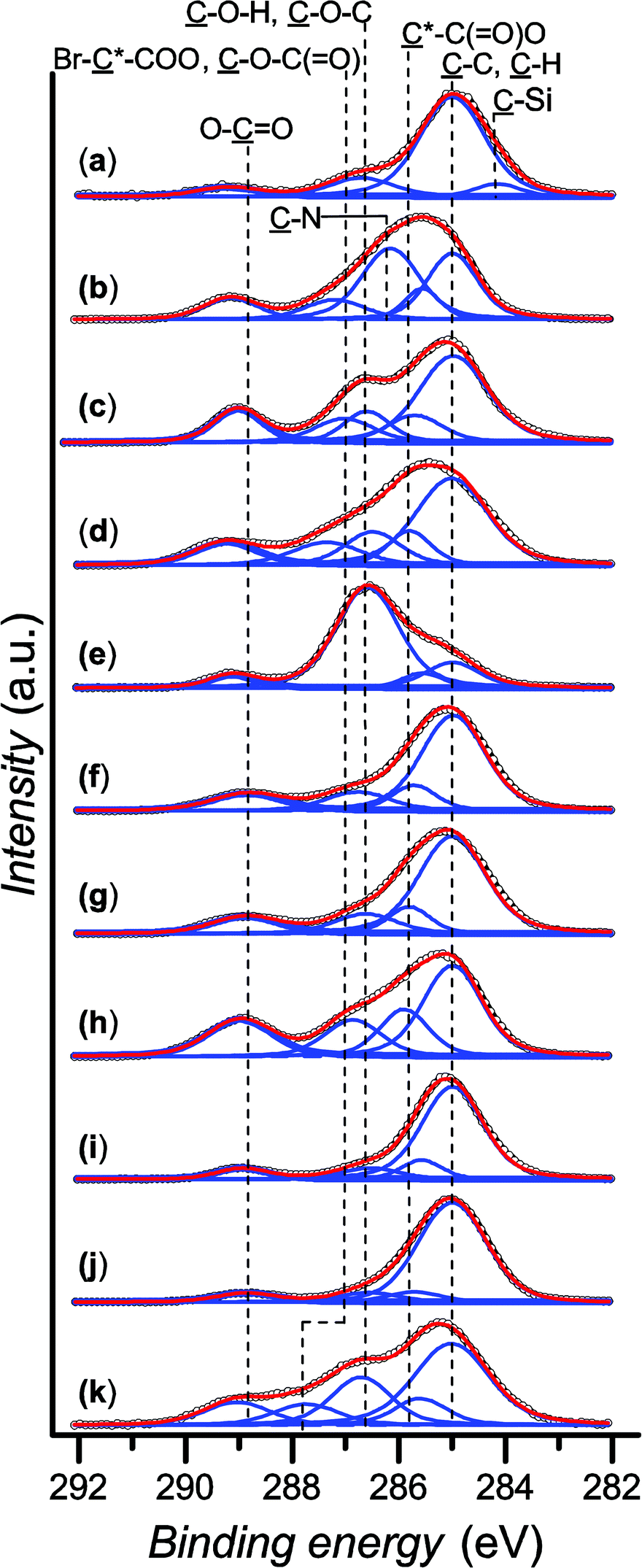 | ||
| Fig. 1 High-resolution XPS spectra of the C 1s region of (a) the SAM of initiator and the obtained polymer brushes: (b) poly(DMAEMA), (c) poly(HEMA), (d) poly(HPM), (e) poly(MeOEGMA), (f) poly(tBMA), (g) poly(nBMA), (h) poly(MMA), (i) poly(EHMA), (j) poly(IBMA), (k) poly(SMA). | ||
Polymer brushes from various methacrylate monomers were grafted from the obtained initiator layer: 2-(dimethylamino)ethyl methacrylate (DMAEMA), 2-hydroxyethyl methacrylate (HEMA), 2-hydroxypropyl methacrylate (HPM), oligo(ethylene glycol) methyl ether methacrylate (MeOEGMA), t-butyl methacrylate (tBMA), n-butyl methacrylate (nBMA), methyl methacrylate (MMA), 2-ethylhexyl methacrylate (EHMA), isobornyl methacrylate (IBMA), and solketal methacrylate (SMA). The polymer brushes were prepared by immersing the initiator-SAM-coated substrates in a degassed solution of the monomer in DMSO containing CuBr2 and tris[2-(dimethylamino)ethyl]amine (Me6TREN) in sealed reactors and subsequently exposing the system to UV light irradiation at ambient temperature (Scheme 1) for a set amount of time. The substrates were subsequently removed from the reactors and rinsed with solvents and then dried. The chemical structures of the obtained polymer brushes were confirmed by XPS. Fig. 1(b)–(k) show the high-resolution C 1s regions of the spectra. The spectra of all methacrylate polymer brushes show a –C, –H contribution at 285.0 eV. This peak dominates the C 1s spectra of poly(HEMA), poly(HPM), poly(tBMA), poly(nBMA), poly(MMA), poly(IBMA) and poly(SMA) brushes. The –C, –H contributions are accompanied by a component at (285.7 ± 0.1) eV arising from the secondary chemical shift, i.e. the effect of the ester group on the tertiary carbon atom in the *–(CO)–O–R structure. Furthermore, all polymer brushes show a contribution at (286.9 ± 0.4) eV from –O–(CO) and a well-resolved ester peak (O–O) at (289.0 ± 0.1) eV. In addition to these components, the spectrum of poly(DMAEMA) shows a dominant –N peak at (286.2 ± 0.1) eV. Interestingly, the brushes of monomers having hydroxyl groups in their covalent structure, i.e. HEMA and HPM, exhibit a C–O–H peak at (286.6 ± 0.1) eV which is well resolved from the –O–(CO) contributions. The presence of oligo(ethylene glycol) side chains gives rise to the strongest contribution in the C 1s spectrum of poly(MeOEGMA) brushes at (286.6 ± 0.1) eV which for this type of brush seems to overlap with the –O–(CO) contributions. However, the C–O–C and the –O–(CO) contributions are well resolved for the poly(SMA) brushes with a separation between the peaks of about 1 eV. Notably, the measured ratios between the individual contributions of the C 1s envelopes for the initiator molecules and polymer brushes match the expected theoretical values within the experimental errors of XPS (see the ESI, Table ST1†). With the exception of poly(DMAEMA), the high resolution N 1s spectra of all polymer brushes lack contributions from nitrogen (see ESI, Fig. SF4†) as expected from their chemical structures. The N 1s spectrum of poly(DMAEMA) could be resolved with two contributions at 399.1 eV and 399.9 eV arising from the uncharged and charged dimethylamino groups. For all investigated substrates, no signals were observed around 933 eV (see the ESI, Fig. SF5†), demonstrating a quantity of copper on the surfaces below the detection limit of XPS, which is important for any potential biological applications that would be limited by toxicity arising from such contamination.
The dynamic water contact angles were measured for all of the grafted polymer brushes. The changes in wettability correspond to hydrophobicity/hydrophilicity expected from the structure of the polymers. A marked decrease in the advancing and especially in the receding water contact angles is observed for HEMA, HPM, MeOEGMA, and DMAEMA, while on the other hand, polymer brushes of tBMA, nBMA, MMA, EHMA, IBMA, and SMA show poor wettability (see the ESI, Table ST2†). The observed contact angle hysteresis is caused by the swelling of the polymer layers upon contact with the water droplet.
The homogeneity of the obtained polymer brush layers was assessed by AFM. The thicknesses of the prepared polymer brushes were found to be homogeneous with low mean square roughness (see the ESI, Table ST3†).
Optimization of the polymerization conditions
Since the polymerization is confined to the surface, the monomer conversion is negligible even for relatively thick layers of over 100 nm. For this reason, the polymerization kinetics were studied by following the evolution of the brush thickness, as measured by ellipsometry, with polymerization time. DMAEMA was chosen as a model monomer to study the polymerization using various catalyst concentrations and solvent compositions (Fig. 2). The influence of the catalyst concentration (Fig. 2a) was studied in DMSO. The results obtained show that the linear character of the thickness growth is maintained even with a very low concentration of copper (77 ppb). Decreasing the catalyst concentration leads to a reduction in the rate of polymerization. | ||
Fig. 2 Polymer thickness of dry poly(DMAEMA) brushes as a function of polymerization time. (a) Effect of Cu concentration. (b) Effect of the solvent. Polymerization conditions: (a) [DMAEMA] = 1.68 M, solvent DMSO. Catalyst concentration: [CuBr2] = 166 μM (7.7 ppm) (black squares), [CuBr2] = 16.6 μM (770 ppb) (red circles), [CuBr2] = 16.6 μM (770 ppb) (blue triangles), [Me6TREN] = 6 × [CuBr2]; (b) [DMAEMA] = 1.68 M, [CuBr2] = 166 μM (7.7 ppm), [Me6TREN] = 995 μM. Solvents: DMSO (black squares), DMSO/acetone 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (red circles), DMSO/2,2,2-trifluoroethanol (TFE) 3:1 (blue triangles), DMSO/water 2:1 (brown diamonds). :1 (red circles), DMSO/2,2,2-trifluoroethanol (TFE) 3:1 (blue triangles), DMSO/water 2:1 (brown diamonds). | ||
While photoinduced living radical polymerization of methacrylates has been reported previously, the requirement for catalyst systems based on rare metals such as Ir may be problematic due to availability concerns, potentially impairing its wide adoption.64,73 In this regard, copper catalysts are already commonly used in established Cu-mediated living polymerizations. The possibility to perform the polymerization at ppb-level concentrations of catalyst brings this protocol even closer to applications.41
The effect of the solvent was studied by comparing the kinetics in DMSO, DMSO/acetone 2:1, DMSO/2,2,2-trifluoroethanol (TFE) 3:1, and DMSO/water 2:1 while using 7.7 ppm of copper (Fig. 2b). A linear evolution of the thickness was observed for all conditions in the first 15 min of irradiation. In the following 15 min for DMSO/acetone 2:1 and DMSO/TFE 3:1 a reduction in the rate of polymerization was observed, suggesting the presence of termination processes during the polymerization. Thus, these conditions were not employed for other monomers. The highest rate of polymerization was observed in DMSO/water 2:1, leading to a brush thickness of more than 100 nm in 30 min. However, the obtained brush thicknesses were less homogeneous over the surfaces in comparison with brushes grown using neat DMSO as a solvent, detected by a higher deviation of the thickness measured in different spots on individual samples (up to 13 nm for DMSO/water vs. less than 2.8 nm in pure DMSO). Linear polymerization kinetics were observed in DMSO. A thickness of 100 nm was reached in only 30 min. AFM topography images reveal that the surface was smooth and homogeneous (see ESI, Table ST3†).
In order to demonstrate the importance of each component of the polymerization system, a series of blank experiments was carried out. The individual removal of either the UV irradiation, the initiator SAM on the substrates, or the catalyst prevented the polymerization from taking place (see ESI,† blank experiments), as no polymer growth was detected. Concomitantly, this proves that the polymerization only occurs from initiators grafted on the surfaces.
The polymerization kinetics were studied for the full range of monomers employed to graft polymer brushes (see Scheme 1) using DMSO as the solvent for all monomers except EHMA and IBMA. In these two cases the polymerization took place in DMSO/toluene 1:2 to achieve monomer miscibility. All polymerization solutions were exposed to irradiation from 2.5 min to 30 min. The evolution of the obtained thickness is presented in Fig. 3. During that time the rate of growth was close to constant for each monomer. The observed linear growth implies that the propagation rate is constant, evidencing the (near) absence of termination processes during the polymerization and the preservation of the growing polymerization centres. The SET-LRP polymerization of SMA was performed for the first time. It was considerably slower than the other methacrylates. This was in agreement with previous reports showing less than full conversion and low rates even at high temperatures (65 and 85 °C).74–76 It is interesting to note that photoinduced SET-LRP has also been successfully applied to acrylate41 and methacrylamide65 monomers, achieving in both cases linear polymerization kinetics. The application of this photoinduced SET-LRP procedure to methacrylates is also effective to achieve a high thickness in short polymerization times with a low catalyst concentration.
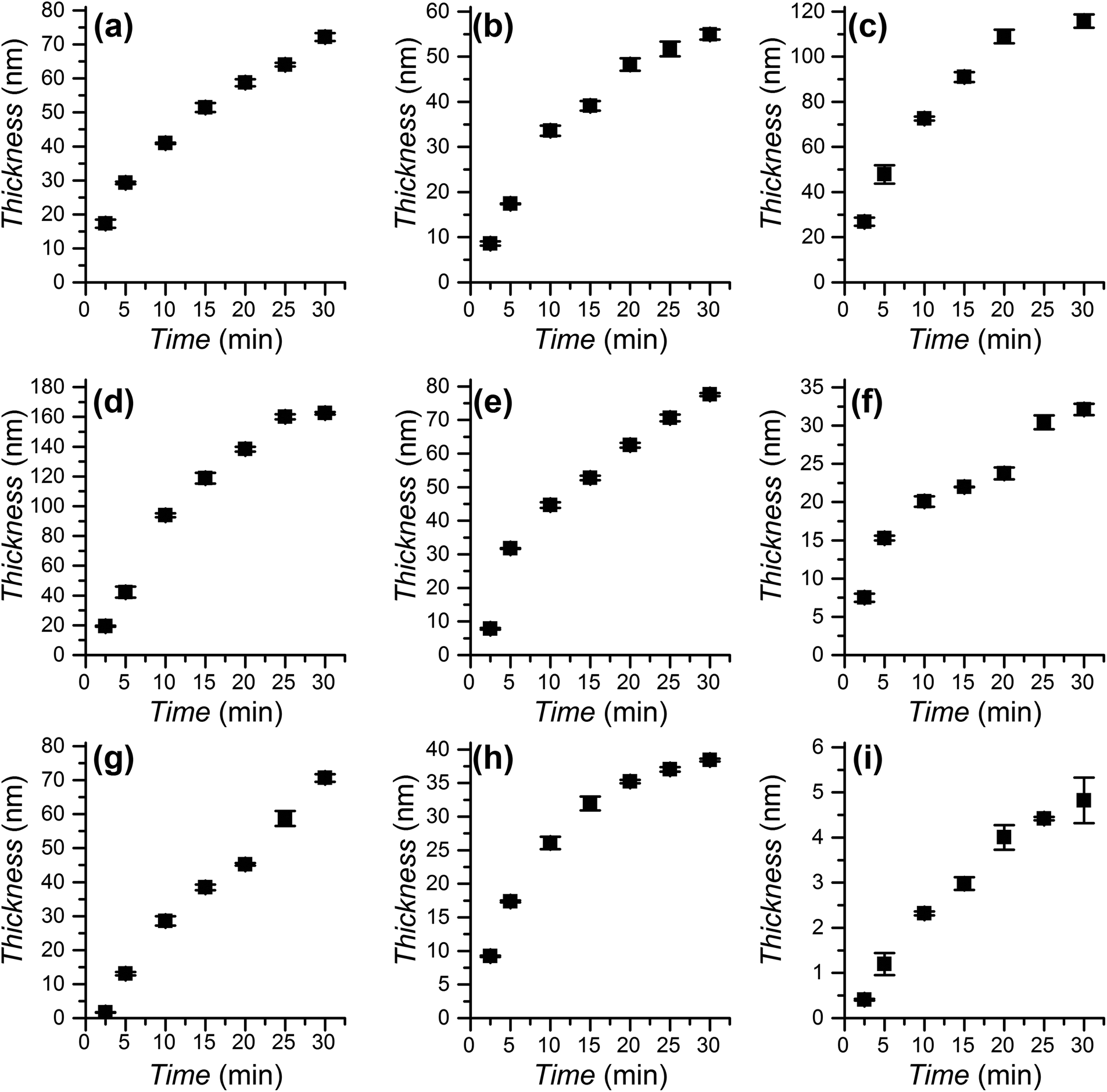 | ||
| Fig. 3 Dry ellipsometric thickness of polymethacrylate brushes: (a) HEMA, (b) HPM, (c) MeOEGMA, (d) tBMA, (e) nBMA, (f) MMA, (g) EHMA, (h) IBMA, and (i) SMA. Conditions: [Monomer] = 1.68 M, [CuBr2] = 166 μM, [Me6TREN] = 995 μM. Solvents: (a)–(f), (i) DMSO, (g), (h) DMSO/toluene 1:2. | ||
Livingness
The presence of the dormant species on the polymer chain ends, able to be re-initiated, was demonstrated by performing re-initiation experiments and confirming the ability to synthesize diblock copolymer brushes. EHMA was selected as the bottom block to be used as a macroinitiator, prepared by immersing the initiator-coated substrates in the polymerization solution and irradiating under the same conditions used to study the polymerization kinetics.41,65 Using these poly(EHMA) brushes as substrates, the polymerization was re-initiated for simple chain extension with EHMA or alternatively, a second block of poly(MeOEGMA). For this, MeOEGMA was chosen due to the significant difference with poly(EHMA) in the expected composition ratios that can be observed by XPS, specifically in the C 1s region between C–O and C*–O–(CO) and C–C, C–H and (C*O)O.
For the re-initiation experiment, the first poly(EHMA) brush was polymerized for 10 min in DMSO/toluene, ([EHMA] = 1.68 M, [CuBr2] = 166 μM, [Me6TREN] = 995 μM) reaching a thickness which corresponds well to the polymerization kinetics. Subsequently, the same sample was immersed in a freshly prepared polymerization solution containing EHMA as the monomer and irradiated for a further 20 min. The total polymer layer thickness shows that the growth after re-initiation proceeded at the same rate as on a continuously irradiated sample (Fig. 4a). The ability of the polymerization to be re-initiated indicates the preservation of the dormant polymerization centers after the first polymerization step.
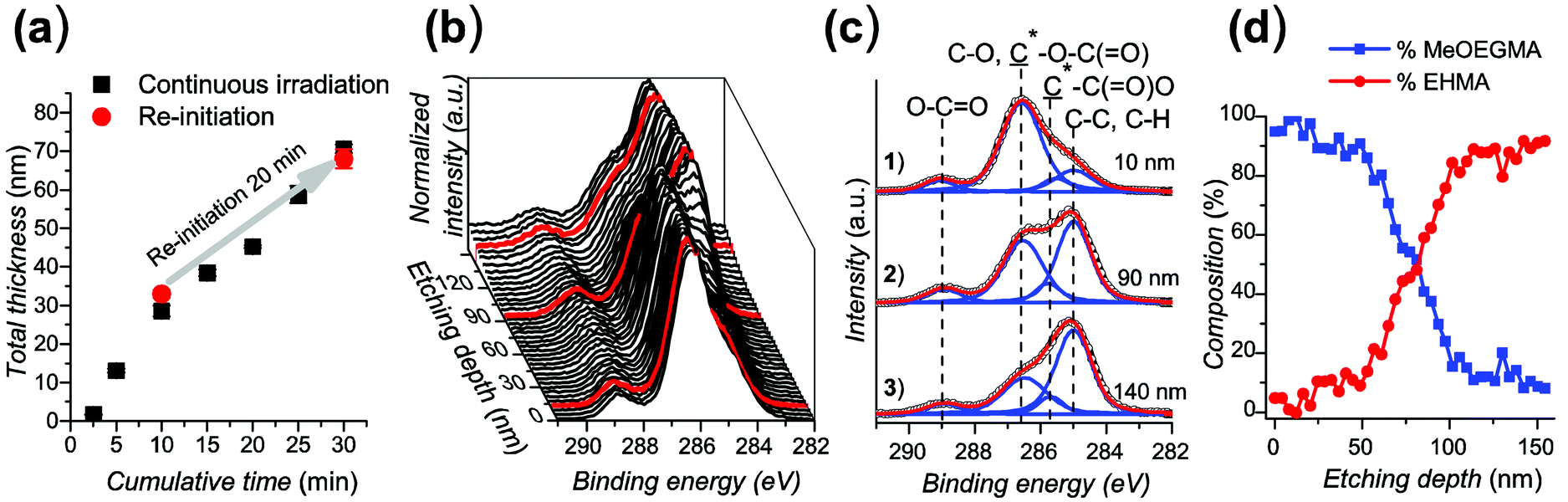 | ||
| Fig. 4 (a) Dry ellipsometric thickness of poly(EHMA) before and after re-initiation of the polymerization, carried out in DMSO/toluene (1:2) (red dots). [EHMA] = 1.68 M, [CuBr2] = 166 μM, [Me6TREN] = 995 μM. Initial irradiation for 10 min yielded poly(EHMA) brushes of (32.9 ± 1.0) nm thickness, which were used as macroinitiators for re-initiation of the polymerization (for an additional 20 min) to yield poly(EHMA) brushes of (68.1 ± 2.5) nm thickness. The polymerization kinetics with continuous irradiation are shown for comparison (black squares). (b)–(d) XPS depth profile of poly(EHMA-b-MeOEGMA). (b) Evolution of the C 1s XPS spectrum with etch depth. (c) Deconvolution of representative C 1s XPS spectra at three etch depths. (d) Approximate ratio of MeOEGMA (blue squares) and EHMA (red dots) monomer units in poly(EHMA-b-MeOEGMA) brushes over etch depth. The total thickness of the poly(EHMA-b-MeOEGMA) was (150.4 ± 4.0) nm. | ||
Additionally, a diblock copolymer was prepared by re-initiating the substrates coated with poly(EHMA) of (52.2 ± 0.4) nm thickness with MeOEGMA ([MeOEGMA] = 1.68 M, [CuBr2] = 166 μM, [Me6TREN] = 995 μM) to reach a total thickness of (150.4 ± 4.0) nm. This corresponds to a thickness of 98.2 nm for the second block. The changes in the chemical composition of the diblock copolymer layer were confirmed by depth profiling performed by XPS, i.e. acquiring spectra after increasing the etching time with Ar+ clusters. Fig. 4b shows the evolution of the C 1s region of the XPS spectrum of poly(EHMA-b-MeOEGMA) with increasing etch depth. The uppermost 50-nm-thick brush layer shows a clear predominance of the peak at (286.6 ± 0.1) eV originating from the –O–C contributions of MeOEGMA. The intensity of this peak rapidly decreases throughout the following 40 nm in the direction of the substrate. The 60-nm-thick bottom layer is dominated by the (–C, –H) contributions at 285.0 eV of the EHMA block. A detailed analysis by deconvolution of the contributions constituting the C 1s envelope (Fig. 4c) further proved the intuitive observation presented in the waterfall graph. Based on the measured ratios between the components within the high resolution C 1s XPS spectra of poly(MeOEGMA) and poly(EHMA) homopolymers (see the ESI, Table ST1†) and the measured depth evolution of the (–O–C, –O–(CO)):(–C, –H), (–C, –H):(O–O) and *–(CO)–O:O–O ratios (see the ESI, Fig. SF6†) we have approximated the average atomic composition of the poly(EHMA-b-MeOEGMA) brushes over etch depth (Fig. 4d). As can be seen from the compositional profile, the top layer is almost exclusively composed of MeOEGMA (concentration of more than 90 atomic%), whereas the bottom layer, close to the substrate, has a clear predominance of EHMA units (concentration of more than 85 atomic%). These results clearly confirm not only the successful chain extension of poly(EHMA) by MeOEGMA, but also prove the block copolymer structure of the end-tethered poly(EHMA-b-MeOEGMA) brushes.
Spatial control
Since the polymerization is photo-triggered (see the ESI,† blank experiment), a simple restriction of the irradiation area on the substrate using a shadow mask (Fig. 5b) can be employed to confine the growth of the brushes. After 10 min of polymerization of DMAEMA, the pattern obtained on the surface was visible to the naked eye. Stylus profilometry (Fig. 5c) and ellipsometric mapping (Fig. 5d) confirmed the successful patterning on the surface. Both techniques yield the same thickness in examined areas, which corresponds to the expected value from the studied polymerization kinetics of this monomer.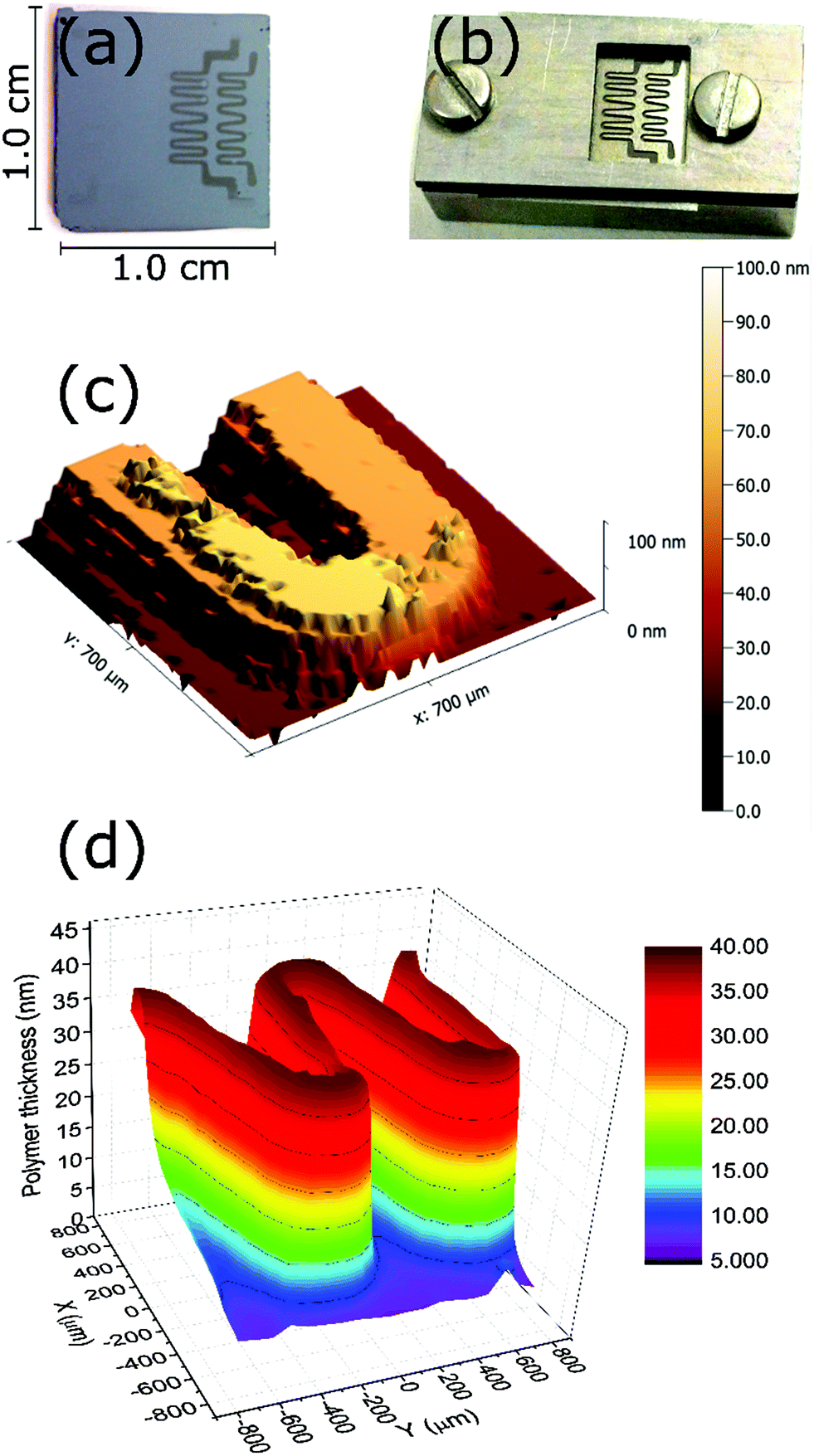 | ||
| Fig. 5 Patterning of poly(DMAEMA) brushes using photoinduced SET-LRP. (a) Optical image of the pattern, (b) picture of the sample holder featuring the shadow mask used, (c) profilometry of a small area of the pattern and (d) ellipsometric mapping of the pattern. Conditions: [DMAEMA] = 1.68 M, [CuBr2] = 166 μM (7.7 ppm), [Me6TREN] = 995 μM, DMSO. | ||
Antifouling properties
One of the main problems in label-free affinity biosensing is the inability to discriminate between signals arising from the specific response to the binding of the analyte and the nonspecific response to the deposition of other compounds from the tested media.7,77 Therefore, it is critical to control the interactions occurring at the interface in applications involving surfaces making contact with biological media. Poly(MeOEGMA) has been widely studied for its ability to suppress fouling.6,67,69,78As photoinduced SET-LRP provides a practical way to create patterned surfaces, we studied the ability of the brushes grown by this technique to reduce protein fouling from model solutions in buffer as well as real biological media. The antifouling properties for the obtained poly(MeOEGMA) brushes were studied by surface plasmon resonance (SPR). The optimum dry thickness previously reported for poly(MeOEGMA) brushes to reach the maximum resistance to protein fouling is around 30 nm.6 Polymer brushes of 12 nm, 28 nm and 51 nm thickness were grown by photoinduced SET-LRP in less than 5 min, from gold-coated glass substrates (SPR sensor chips) modified with a SAM of ω-mercaptoundecyl bromoisobutyrate. The adsorption from model solutions of the main plasma proteins, human serum albumin (HSA) and fibrinogen (Fbg), as well as from undiluted human blood plasma (BP) and fetal bovine serum (FBS) was studied. The brushes were brought into contact with the solutions for 15 min. No fouling could be detected for HSA and Fbg on any of the poly(MeOEGMA) brushes evaluated. For more challenging complex media FBS and BP the fouling was 1.95 ng cm−2 and 16.2 ng cm−2 respectively for the 28-nm-thick brushes (Fig. 6 and Table ST4 in the ESI†). The thinner 12 nm-thick poly(MeOEGMA) brushes showed diminished antifouling performance in accordance with previous results, at 4.35 ng cm−2 and 28.6 ng cm−2 for FBS and BP, respectively (Table ST4 in the ESI†).79 The brushes of 51 nm thickness performed similarly as those of 28 nm thickness (fouling of 2.5 ng cm−2 and 16.3 ng cm−2 for FBS and BP respectively). The obtained surfaces of optimum thickness (28 nm) reduced 95% of the fouling in comparison with the fouling on a bare gold surface. The antifouling properties displayed by the poly(MeOEGMA) brushes achieved by the present technique are on the same level as those of brushes prepared from the same monomer by other previously reported polymerization protocols.6
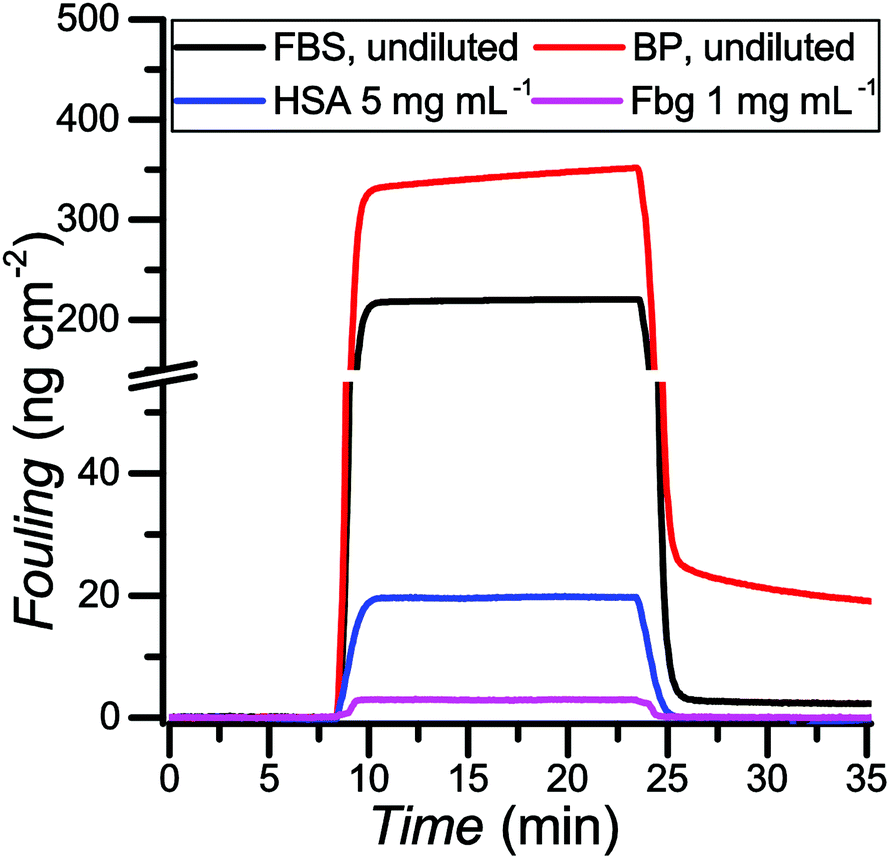 | ||
| Fig. 6 Protein fouling measured by SPR on a gold SPR chip coated with poly(MeOEGMA) brushes (28 nm). Black: fetal bovine serum (FBS, undiluted); red: undiluted blood plasma (BP, undiluted); blue: human serum albumin (HSA, 5 mg mL−1 in PBS); pink: fibrinogen (Fbg, 1 mg mL−1 in PBS). | ||
Conclusions
The successful surface-confined photoinduced SET-LRP of a wide range of methacrylate monomers was achieved with only small amounts of a copper catalyst. In particular, the surface grafting of solketal methacrylate is reported for the first time. The living nature of polymerization was demonstrated via formation of a diblock copolymer brush. Successful spatial control over polymerization of methacrylate monomers was also demonstrated. The antifouling properties of poly[oligo(ethylene glycol) methyl ether methacrylate] brushes, critical for biomedical devices and biosensing, were demonstrated by surface plasmon resonance.These features make this procedure highly promising for advanced applications requiring precise control over the properties of surfaces as well as the ability to create patterns.
Experimental
Materials
Oligo(ethylene glycol) methyl ether methacrylate (Mn = 300 g mol−1, MeOEGMA), 2-hydroxyethyl methacrylate (≥99%, HEMA), t-butyl methacrylate (98%, tBMA), n-butyl methacrylate (99%, nBMA), methyl methacrylate (99%, MMA), 2-ethylhexyl methacrylate (98%, EHMA), 2-(dimethylamino)ethyl methacrylate (98%, DMAEMA), solketal methacrylate (50 wt% in dichloromethane, SMA), isobornyl methacrylate (98.5%, IBMA), CuBr2 (99.999% trace metal basis), and tris[2-(dimethylamino)ethyl]amine (97%, Me6TREN) were obtained from Sigma-Aldrich, Czech Republic. 2-Hydroxypropyl methacrylate, toluene (extra-dry over molecular sieves, 99.85%) and dry dimethyl sulfoxide (extra-dry over molecular sieves 99.7%, DMSO) were purchased from Acros, Czech Republic. Ethanol (99.8%, EtOH), acetone (99.5%), tetrahydrofuran (99.5%, THF), toluene (99%), and DMSO (99%) were purchased from Lach-Ner, Czech Republic. Deionized water was obtained from a Milli-Q system (Merck-Millipore, Czech Republic). Silicon wafers were purchased from Siegert Wafer GmbH, Germany. 11-(Trichlorosilyl)undecyl 2-bromo-2-methylpropanoate9 and ω-mercaptoundecyl bromoisobutyrate80 were synthesized according to procedures published earlier. Human blood plasma (mix from 5 donors) was provided by the Institute of Hematology and Blood Transfusion, Czech Republic. Fetal bovine serum, fibrinogen and human serum albumin were purchased from Sigma-Aldrich, Czech Republic.Self-assembled monolayer of initiator
Silicon wafer substrates (1 × 1 cm2) were rinsed twice with EtOH and deionized water. All substrates were exposed to air plasma for 20 min to generate silanol groups on the surface. The freshly activated surfaces were immediately immersed in a 1 mg mL−1 solution of 11-(trichlorosilyl)undecyl 2-bromo-2-methylpropanoate in dry toluene. The silanization of the substrates to form the self-assembled monolayer was allowed to proceed for 3 h at room temperature in a dry environment. The substrates were subsequently rinsed with toluene, acetone, EtOH, and deionized water and blow-dried with N2.The SPR chips (gold-coated glass) and gold-coated Si-wafer chips were cleaned by rinsing twice with EtOH and deionized water, blow dried with N2, and cleaned in a UV/Ozone cleaner (Jelight) for 20 min. Directly after cleaning, the chips were immersed in a 1 mM solution of ω-mercaptoundecyl bromoisobutyrate in EtOH and kept overnight in the dark at room temperature. Subsequently, they were rinsed twice with EtOH and deionized water and blow-dried with N2.
Photoinduced SET-LRP of methacrylates
A stock solution (S) of CuBr2 (3.9 mM) and Me6TREN (23.4 mM) was freshly prepared in dry DMSO. An aliquot of the stock solution S (273 μL) was transferred to a round-bottom flask containing dry DMSO (3.15 mL) to give the solution A. In another round-bottom flask, a solution of methacrylate monomer (10.8 mmol) in DMSO (3.00 mL) was prepared (solution B). Solutions A and B were degassed by bubbling Ar for 60 min while stirring and kept in the dark by wrapping them in Al-foil. Subsequently, solution A was transferred using a gas-tight syringe under Ar protection to solution B. The combined polymerization solution C was stirred and transferred to individual crimped vials containing the initiator-coated Si wafer substrates, which had been previously degassed by purging with Ar for 15 min. The polymerization was conducted by irradiating the vials inside a UV-reactor, consisting of a nail-curing device (four 9 W lamps, λmax = 365 nm) placed on top of a shaker plate (90 min−1) and kept at room temperature. After irradiation for a preset amount of time the vials were removed from the reactor, quickly opened, and filled with DMSO to quench the polymerization. The samples were removed from the solution and rinsed with DMSO, acetone, EtOH, and deionized water in the case of hydrophilic monomers, or DMSO, acetone, THF, and EtOH for hydrophobic monomers, and dried by blowing with N2.For the polymerization of EHMA and IBMA, the same procedure was employed but using a 1:2 mixture of dry DMSO/toluene as the solvent instead of pure DMSO. For the polymerization of methyl methacrylate, a volume of stock solution S (273 μL) was transferred to a round bottom flask containing DMSO (6.15 mL) to form solution A, while the neat monomer was degassed separately by bubbling Ar for 1 h. After degassing, the appropriate amount of monomer (10.8 mmol, 1.15 mL) was transferred to solution A by using a gas-tight syringe under Ar protection to obtain solution C. The polymerization procedure was continued in the same way as described above.
Patterning of DMAEMA
The pattern of DMAEMA was prepared by the photoinduced SET-LRP procedure in DMSO ([DMAEMA] = 1.68 M, [CuBr2] = 166 μM, and [Me6TREN] = 995 μM) as was described above. The Si wafer was mounted in a shadow mask holder and covered with a shadow mask (Fig. 5b). The polymerization was conducted by irradiating the vial inside the UV-reactor for 10 min. The sample was removed from the shadow mask holder, rinsed with DMSO, acetone, THF, and EtOH, and dried by blowing with N2.Re-initiation of EHMA
The polymer brushes of EHMA were prepared by the photoinduced SET-LRP procedure in DMSO/toluene 1:2 ([EHMA] = 1.68 M, [CuBr2] = 166 μM, and [Me6TREN] = 995 μM). After UV irradiation for 10 min, the polymerization was stopped, the samples were rinsed with DMSO, acetone, THF, and EtOH, and blow-dried with N2. The obtained surfaces were characterized by ellipsometry. Subsequently, these surfaces were immersed in a freshly prepared polymerization solution of EHMA and irradiated for an additional 20 min. The samples were rinsed with solvents and dried with N2. The thickness of the samples was measured by ellipsometry.
Grafting of poly(EHMA-b-MeOEGMA)
The first block of poly(EHMA) brushes was prepared by photoinduced SET-LRP in DMSO/toluene 1:2 as described above. The polymerization proceeded under UV-irradiation, and subsequently the samples were removed from the solution and rinsed with DMSO, acetone, THF, and EtOH, and dried by blowing with N2.
The second block of poly(MeOEGMA) was prepared by photoinduced SET-LRP in DMSO ([MeOEGMA] = 1.68 M, [CuBr2] = 166 μM, and [Me6TREN] = 995 μM) as described above, using the substrates with a grafted first block of poly(EHMA) as macroinitiators. The polymerization was conducted by irradiating the vials inside the UV-reactor for 25 min. The samples were rinsed with DMSO, acetone, EtOH, and deionized water, and dried by blowing with N2.
Surface characterization
The obtained high resolution spectra were fitted/deconvoluted with Voigt profiles (binding energy uncertainty: ±0.2 eV). The analyzer transmission function, Scofield sensitivity factors, and effective attenuation lengths (EALs) for photoelectrons were applied for quantification. EALs were calculated using the standard TPP-2M formalism.81,82 All spectra were referenced to the C 1s peak attributed to C–C, C–H at 285.0 eV binding energy, which was controlled by means of the well-known photoelectron peaks of metallic Cu, Ag, and Au.
The reconstruction of the average atomic concentration profile of MeOEGMA (![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) ) and 2-EHMA (ȳ = 1 − ) monomers in the poly(EHMA-b-MeOEGMA) brush was performed on basis of the measured individual component ratios in the high resolution C 1s XPS spectra:
) and 2-EHMA (ȳ = 1 − ) monomers in the poly(EHMA-b-MeOEGMA) brush was performed on basis of the measured individual component ratios in the high resolution C 1s XPS spectra:

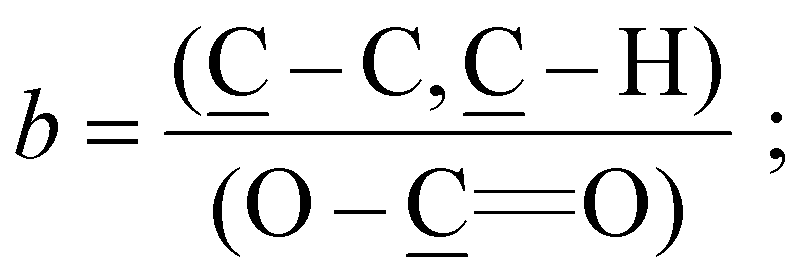
between the components of poly(MeOEGMA) and poly(EHMA) homopolymers and the measured depth evolution of the respective ratios in the block copolymer utilizing the equation:


The experimental uncertainties in the quantitative analysis of XPS are estimated to be below 7%. The value covers the overall uncertainties of the method that are typically introduced by the background subtraction.
Acknowledgements
This work was partially performed at the Center for Chemical Polymer Technology (CPT) under the support of the EU and the federal state of North Rhine-Westphalia (Grant EFRE 30 00 883 02). The work was also supported by the Grant Agency of the Czech Republic (GACR) under contract no. 15-09368Y and by OPPK CZ.2.16/3.1.00/21545. Financial support by the National Science Foundation (DMR-1066116 and DMR-1120901), the Humboldt Foundation and the P. Roy Vagelos Chair at Penn (all to VP) is gratefully acknowledged.Notes and references
- B. Zhao and W. J. Brittain, Prog. Polym. Sci., 2000, 25, 677–710 CrossRef CAS.
- S. T. Milner, Science, 1991, 251, 905–914 CrossRef CAS PubMed.
- F. Denes, V. Percec, M. Totolin and J. P. Kennedy, Polym. Bull., 1980, 2, 499–504 CrossRef CAS.
- S. Minko, J. Macromol. Sci., Polym. Rev., 2006, 46, 397–420 CrossRef CAS.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- C. Rodriguez-Emmenegger, E. Brynda, T. Riedel, M. Houska, V. Subr, A. B. Alles, E. Hasan, J. E. Gautrot and W. T. Huck, Macromol. Rapid Commun., 2011, 32, 952–957 CrossRef CAS PubMed.
- C. Rodriguez-Emmenegger, E. Brynda, T. Riedel, Z. Sedlakova, M. Houska and A. B. Alles, Langmuir, 2009, 25, 6328–6333 CrossRef CAS PubMed.
- M. Krishnamoorthy, S. Hakobyan, M. Ramstedt and J. E. Gautrot, Chem. Rev., 2014, 114, 10976–11026 CrossRef CAS PubMed.
- C. Rodriguez-Emmenegger, S. Janel, A. de los Santos Pereira, M. Bruns and F. Lafont, Polym. Chem., 2015, 6, 5740–5751 RSC.
- A. de los Santos Pereira, T. Riedel, E. Brynda and C. Rodriguez-Emmenegger, Sens. Actuators, B, 2014, 202, 1313–1321 CrossRef CAS.
- C. Rodriguez-Emmenegger, M. Houska, A. B. Alles and E. Brynda, Macromol. Biosci., 2012, 12, 1413–1422 CrossRef CAS PubMed.
- A. de los Santos Pereira, C. Rodriguez-Emmenegger, F. Surman, T. Riedel, A. B. Alles and E. Brynda, RSC Adv., 2014, 4, 2318 RSC.
- T. Riedel, C. Rodriguez-Emmenegger, A. de los Santos Pereira, A. Bědajánková, P. Jinoch, P. M. Boltovets and E. Brynda, Biosens. Bioelectron., 2014, 55, 278–284 CrossRef CAS PubMed.
- T. Riedel, F. Surman, S. Hageneder, O. Pop-Georgievski, C. Noehammer, M. Hofner, E. Brynda, C. Rodriguez-Emmenegger and J. Dostalek, Biosens. Bioelectron., 2016, 85, 272–279 CrossRef CAS PubMed.
- P. Jain, G. L. Baker and M. L. Bruening, Annu. Rev. Anal. Chem., 2009, 2, 387–408 CrossRef CAS PubMed.
- O. Azzaroni, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3225–3258 CrossRef CAS.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS PubMed.
- R. Advincula, in Surface-Initiated Polymerization I, 2006, vol. 197, pp. 107–136 Search PubMed.
- W. J. Brittain and S. Minko, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3505–3512 CrossRef CAS.
- S. Edmondson, V. L. Osborne and W. T. S. Huck, Chem. Soc. Rev., 2004, 33, 14–22 RSC.
- C. J. Fristrup, K. Jankova and S. Hvilsted, Soft Matter, 2009, 5, 4623–4634 RSC.
- U. Yildiz, B. Hazer and K. Tauer, Polym. Chem., 2012, 3, 1107 RSC.
- M. Zamfir, C. Rodriguez-Emmenegger, S. Bauer, L. Barner, A. Rosenhahn and C. Barner-Kowollik, J. Mater. Chem. B, 2013, 1, 6027–6034 RSC.
- K. J. Bian and M. F. Cunningham, Polymer, 2006, 47, 5744–5753 CrossRef CAS.
- L. Ghannam, J. Parvole, G. Laruelle, J. Francois and L. Billon, Polym. Int., 2006, 55, 1199–1207 CrossRef CAS.
- Z. Chen, Q. Yang, K. Peng and Y. Guo, J. Appl. Polym. Sci., 2011, 119, 3582–3590 CrossRef CAS.
- S. B. Rahane, A. T. Metters and S. M. Kilbey, Macromolecules, 2006, 39, 8987–8991 CrossRef CAS.
- J. Ma, S. Luan, L. Song, J. Jin, S. Yuan, S. Yan, H. Yang, H. Shi and J. Yin, ACS Appl. Mater. Interfaces, 2014, 6, 1971–1978 CAS.
- V. Percec, A. V. Popov, E. Ramirez-Castillo, M. Monteiro, B. Barboiu, O. Weichold, A. D. Asandei and C. M. Mitchell, J. Am. Chem. Soc., 2002, 124, 4940–4941 CrossRef CAS PubMed.
- V. Percec, A. V. Popov, E. Ramirez-Castillo and O. Weichold, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3283–3299 CrossRef CAS.
- N. Zhang, S. R. Samanta, B. M. Rosen and V. Percec, Chem. Rev., 2014, 114, 5848–5958 CrossRef CAS PubMed.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS PubMed.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4684–4695 CrossRef CAS.
- N. H. Nguyen, C. Rodriguez-Emmenegger, E. Brynda, Z. Sedlakova and V. Percec, Polym. Chem., 2013, 4, 2424–2427 RSC.
- A. H. Soeriyadi, C. Boyer, F. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 11128–11131 CrossRef CAS PubMed.
- G. Lligadas and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2745–2754 CrossRef CAS.
- S. R. Samanta, V. Nikolaou, S. Keller, M. J. Monteiro, D. A. Wilson, D. M. Haddleton and V. Percec, Polym. Chem., 2015, 6, 2084–2097 RSC.
- F. Alsubaie, A. Anastasaki, P. Wilson and D. M. Haddleton, Polym. Chem., 2015, 6, 406–417 RSC.
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, P. Wilson, K. Kempe, J. F. Quinn, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Rev., 2016, 116, 835–877 CrossRef CAS PubMed.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS PubMed.
- J. Laun, M. Vorobii, A. de Los Santos Pereira, O. Pop-Georgievski, V. Trouillet, A. Welle, C. Barner-Kowollik, C. Rodriguez-Emmenegger and T. Junkers, Macromol. Rapid Commun., 2015, 36, 1681–1686 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2016, 7, 1002–1026 RSC.
- T. Hatano, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 164–172 CrossRef CAS.
- N. H. Nguyen, X. Jiang, S. Fleischmann, B. M. Rosen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5629–5638 CrossRef CAS.
- S. R. Samanta, M. E. Levere and V. Percec, Polym. Chem., 2013, 4, 3212–3224 RSC.
- S. R. Samanta, A. Anastasaki, C. Waldron, D. M. Haddleton and V. Percec, Polym. Chem., 2013, 4, 5563–5569 RSC.
- M. Enayati, R. L. Jezorek, M. J. Monteiro and V. Percec, Polym. Chem., 2016, 7, 3608–3621 RSC.
- M. Gavrilov, T. J. Zerk, P. V. Bernhardt, V. Percec and M. J. Monteiro, Polym. Chem., 2016, 7, 933–939 RSC.
- M. Enayati, R. L. Jezorek, R. B. Smail, M. J. Monteiro and V. Percec, Polym. Chem., 2016, 7, 5930–5942 RSC.
- N. H. Nguyen, M. E. Levere, J. Kulis, M. J. Monteiro and V. Percec, Macromolecules, 2012, 45, 4606–4622 CrossRef CAS.
- Z. Zhang, W. Wang, H. Xia, J. Zhu, W. Zhang and X. Zhu, Macromolecules, 2009, 42, 7360–7366 CrossRef CAS.
- M. E. Welch, Y. Xu, H. Chen, N. Smith, M. E. Tague, H. D. Abruna, B. Baird and C. K. Ober, J. Photopolym. Sci. Technol., 2013, 25, 53–56 CrossRef PubMed.
- T. Chen, I. Amin and R. Jordan, Chem. Soc. Rev., 2012, 41, 3280–3296 RSC.
- X. Jia, X. Jiang, R. Liu and J. Yin, ACS Appl. Mater. Interfaces, 2010, 2, 1200–1205 CAS.
- S. Minko, M. Müller, M. Motornov, M. Nitschke, K. Grundke and M. Stamm, J. Am. Chem. Soc., 2003, 125, 3896–3900 CrossRef CAS PubMed.
- D. Usov, V. Gruzdev, M. Nitschke, M. Stamm, O. Hoy, I. Luzinov, I. Tokarev and S. Minko, Macromolecules, 2007, 40, 8774–8783 CrossRef CAS.
- F. J. Xu, L. Y. Liu, W. T. Yang, E. T. Kang and K. G. Neoh, Biomacromolecules, 2009, 10, 1665–1674 CrossRef CAS PubMed.
- S. Burkert, E. Bittrich, M. Kuntzsch, M. Muller, K. J. Eichhorn, C. Bellmann, P. Uhlmann and M. Stamm, Langmuir, 2010, 26, 1786–1795 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, N. P. Truong, G. S. Pappas, N. G. Engelis, J. F. Quinn, M. R. Whittaker, T. P. Davis and D. M. Haddleton, Macromolecules, 2015, 48, 5140–5147 CrossRef CAS.
- A. Anastasaki, V. Nikolaou, G. S. Pappas, Q. Zhang, C. Wan, P. Wilson, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Sci., 2014, 5, 3536–3542 RSC.
- A. Anastasaki, V. Nikolaou, F. Brandford-Adams, G. Nurumbetov, Q. Zhang, G. J. Clarkson, D. J. Fox, P. Wilson, K. Kempe and D. M. Haddleton, Chem. Commun., 2015, 51, 5626–5629 RSC.
- A. Anastasaki, V. Nikolaou, N. W. McCaul, A. Simula, J. Godfrey, C. Waldron, P. Wilson, K. Kempe and D. M. Haddleton, Macromolecules, 2015, 48, 1404–1411 CrossRef CAS.
- G. R. Jones, R. Whitfield, A. Anastasaki and D. M. Haddleton, J. Am. Chem. Soc., 2016, 138, 7346–7352 CrossRef CAS PubMed.
- J. E. Poelma, B. P. Fors, G. F. Meyers, J. W. Kramer and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 52, 6844–6848 CrossRef CAS PubMed.
- M. Vorobii, A. de los Santos Pereira, O. Pop-Georgievski, N. Y. Kostina, C. Rodriguez-Emmenegger and V. Percec, Polym. Chem., 2015, 6, 4210–4220 RSC.
- S. Desseaux and H.-A. Klok, Biomaterials, 2015, 44, 24–35 CrossRef CAS PubMed.
- J. E. Gautrot, B. Trappmann, F. Oceguera-Yanez, J. Connelly, X. He, F. M. Watt and W. T. S. Huck, Biomaterials, 2010, 31, 5030–5041 CrossRef CAS PubMed.
- B. Hazer, Makromol. Chem., 1992, 193, 1081–1086 CrossRef CAS.
- C. Rodriguez-Emmenegger, O. A. Avramenko, E. Brynda, J. Skvor and A. B. Alles, Biosens. Bioelectron., 2011, 26, 4545–4551 CrossRef CAS PubMed.
- A. Aied, Y. Zheng, A. Pandit and W. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 826–831 CAS.
- M. Thomas, M. Gajda, C. Amiri Naini, S. Franzka, M. Ulbricht and N. Hartmann, Langmuir, 2015, 31, 13426–13432 CrossRef CAS PubMed.
- T. Şanal, O. Oruç, T. Öztürk and B. Hazer, J. Polym. Res., 2015, 22, 3 CrossRef.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed.
- S. O. Kyeremateng, E. Amado and J. Kressler, Eur. Polym. J., 2007, 43, 3380–3391 CrossRef CAS.
- S. O. Kyeremateng, E. Amado, A. Blume and J. Kressler, Macromol. Rapid Commun., 2008, 29, 1140–1146 CrossRef CAS.
- Z. G. Zhang, G. J. Lin and S. Bell, Macromolecules, 2000, 33, 7877–7883 CrossRef CAS.
- M. Thompson, S. Sheikh, C. Blaszykowski and A. Romaschin, in Detection Challenges in Clinical Diagnostics, ed. P. Vadgama and S. Peteu, Royal Society of Chemistry, Cambridge, 2013, ch. 1, pp. 1–34 Search PubMed.
- H. Ma, J. Hyun, P. Stiller and A. Chilkoti, Adv. Mater., 2004, 16, 338–341 CrossRef CAS.
- C. Rodriguez-Emmenegger, E. Hasan, O. Pop-Georgievski, M. Houska, E. Brynda and A. B. Alles, Macromol. Biosci., 2012, 12, 525–532 CrossRef CAS PubMed.
- A. A. B. Darren, M. Jones and W. T. S. Huck, Langmuir, 2002, 18, 1265–1269 CrossRef.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1993, 21, 165–176 CrossRef.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py01730d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |