
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multiple polymerization – formation of hybrid materials consisting of two or more polymers from one monomer†
T.
Ebert
 a,
A.
Wollbrink
b,
A.
Seifert
a,
R.
John
a and
S.
Spange
*a
a,
A.
Wollbrink
b,
A.
Seifert
a,
R.
John
a and
S.
Spange
*a
aPolymer Chemistry, Technische Universität Chemnitz, 09107 Chemnitz, Germany. E-mail: stefan.spange@chemie.tu-chemnitz.de
bInstitute of Physical Chemistry and Electrochemistry, Leibniz University Hannover, Callinstrasse 3A, D-30167 Hannover, Germany
First published on 13th October 2016
Abstract
Hybrid materials consisting of three different components were synthesized by the polymerization of one heterotrifunctional monomer in just one reaction step using, at the most, one catalyst. The polymerization of 2-furfuyloxy-2-methyl-4H-1,3,2-benzodioxasiline leading to a hybrid material consisting of phenolic resin, poly(furfuryl alcohol), and polymethylsilsesquioxane is, to the best of our knowledge, the first polymerization of this kind. The influence of different catalysts on the polymerization behavior and thus on the structure of the hybrid material was investigated. In accordance with the term “twin polymerization”, which is used for the polymerization of one monomer yielding two separate polymers, this type of polymerization could be called “triple polymerization”. The term “multiple polymerization” is introduced as a general term for the underlying concept of the synthesis of multiple polymers starting from one monomer in one process step.
Introduction
The importance of nanostructured materials has been reported for a variety of applications, such as the support of catalysts, energy storage, and conversion or separation processes.1–4 For this purpose the combination of more than two components and a precise structuring in the nanometer range are often required.5 Template-assisted polymerization and sol–gel chemistry are suitable procedures for this scope. However, most of the established synthetic methods require a work up procedure afterwards.In recent years, the synthetic concept of twin polymerization has been established as a strategy for hybrid material synthesis.6–8 During the twin polymerization process two different polymers are formed from just one monomer in a mechanistically coupled process. This concept differs from other established synthetic strategies for the synthesis of hybrid materials.9 Nanostructured hybrid materials can directly be synthesized by polymerizing a monomer melt, partly even without the need of a catalyst.10 For the synthesis of hybrid materials of multiple components and the tuning of the domain size the simultaneous twin polymerization has been developed.11–13 This concept describes the polymerization of two different twin monomers in one process step, and yields a hybrid material consisting of up to four homopolymers. If two twin monomers with one identical structural element are polymerized, either three homopolymers, or one homopolymer and one copolymer are obtained. A nanostructured hybrid material consisting of poly(furfuryl alcohol), phenolic resin and silicon dioxide has been synthesized by a simultaneous twin polymerization of 2,2′-spirobi[benzo-4H-1,3,2-dioxasiline] (Spiro) and tetrafurfuryloxysilane (TFOS).11
We herein demonstrate that a hybrid material consisting of three different polymers can also be synthesized by polymerizing only one monomer. Thus, the concept of twin polymerization is only an example of a more general procedure – the formation of several polymers within just one synthetic step, starting from only one monomer. When this concept is compared with other well-known polymerization procedures, it can be regarded as the opposite of a copolymerization where, starting from more than one monomer one copolymer is formed. In accordance with the term “twin polymerization”, the general term of multiple polymerization is suggested (Fig. 1).
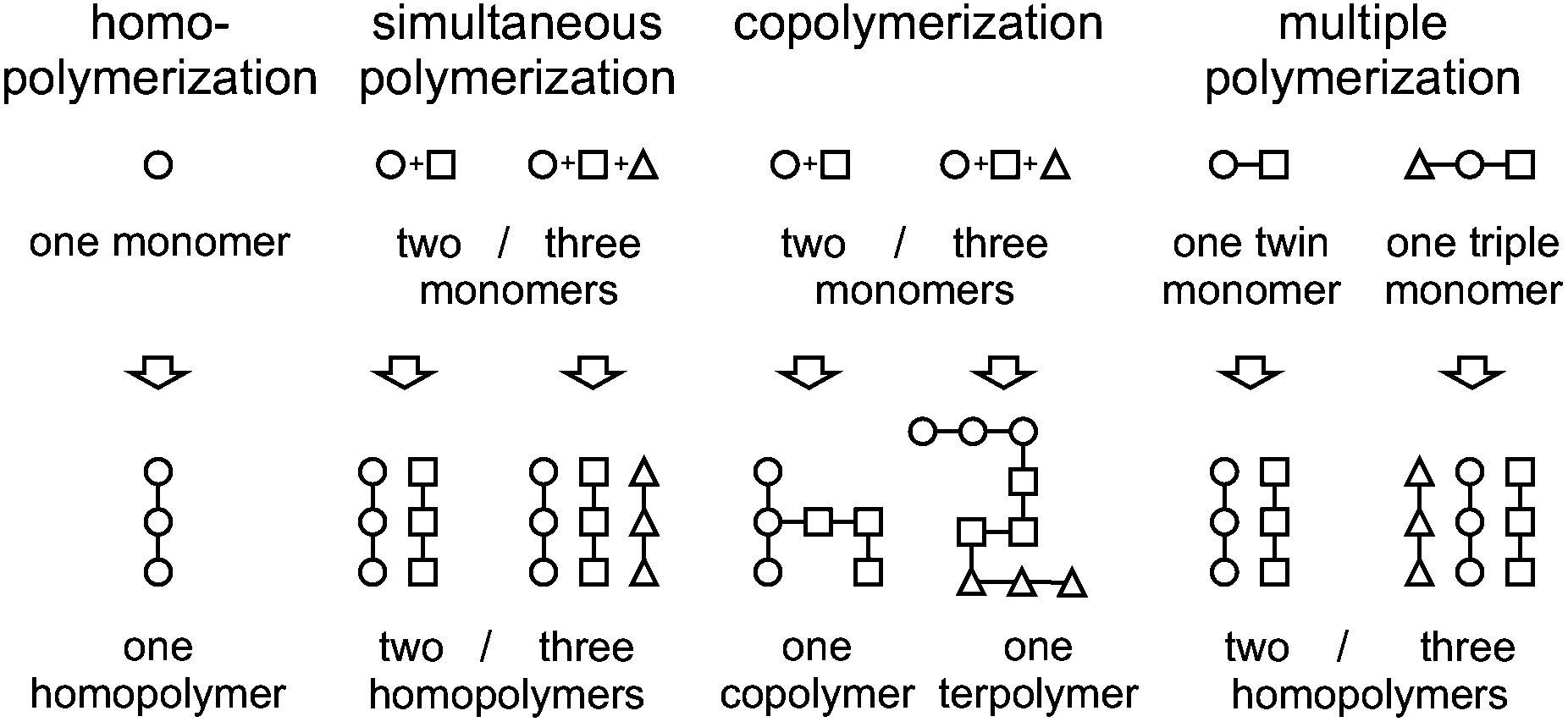 | ||
| Fig. 1 Comparison of multiple polymerization with the well-established polymerization concepts of homopolymerization, simultaneous polymerization and copolymerization. | ||
Homopolymerization and copolymerization are defined by IUPAC.14 The simultaneous polymerization is described in the literature11 and the related term “simultaneous interpenetrating polymer network” is also defined by IUPAC.15 So far, the twin polymerization is, to the best of our knowledge, the only example of a multiple polymerization. The concept is useful for the synthesis of nanostructured hybrid materials, and consequently also for nanoporous materials, which are accessible by the annealing and removing of one of the components of the hybrid material.9 For the formation of nanostructured phase domains it is crucial that two or more polymers are formed in the vicinity at the same time, so that the viscosity is increased fast and phase separation is reduced. Monomers for multiple polymerization can be constructed to yield more than two different polymers. Furthermore, as for twin polymerization, it is also possible to construct the monomers in such a way that functional polymers are obtained.9
To prove the concept, we synthesized and investigated the polymerization behavior of the heterotrifunctional monomer 2-furfuryloxy-2-methyl-4H-1,3,2-benzodioxasiline (1, see Scheme 1). To be consistent with the terminology of twin polymerization and twin monomers, this monomer could be regarded as a “triple monomer”, and the polymerization to three polymers could be called a “triple polymerization”. However, the term “heterotrifunctional monomer” should be used, as this expression is independent of its polymerization behavior during the differently catalyzed polymerizations.
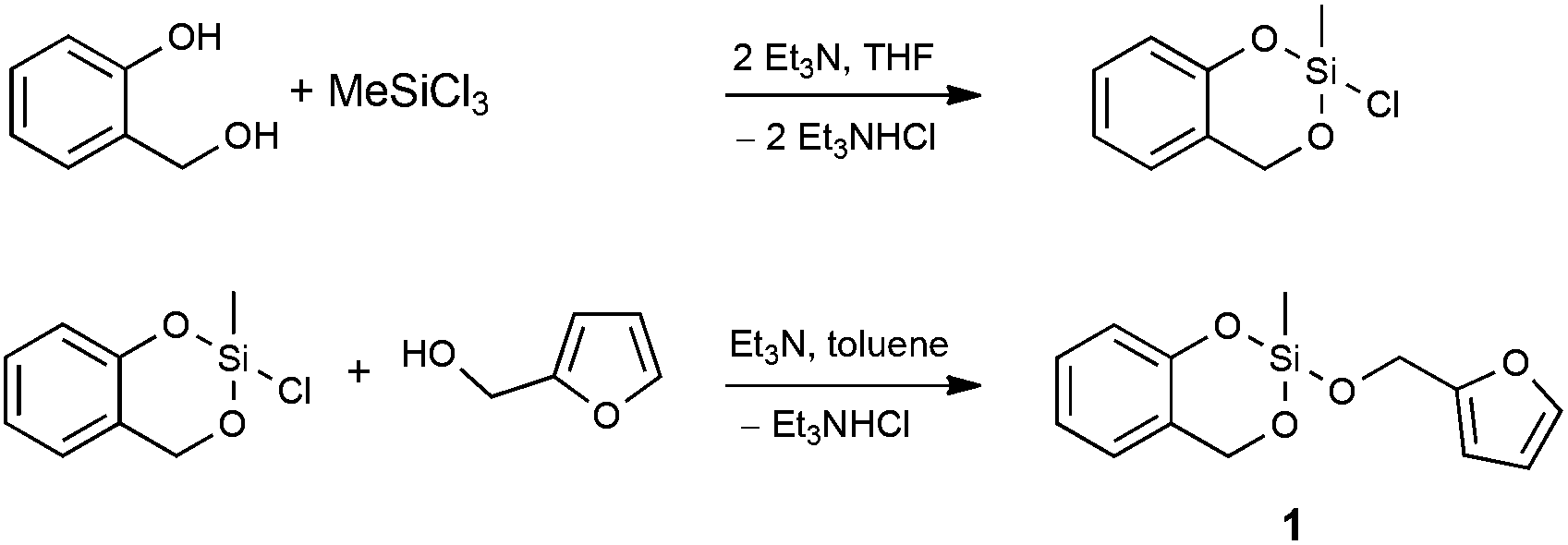 | ||
| Scheme 1 Synthesis of the heterotrifunctional monomer 2-furfuryloxy-2-methyl-4H-1,3,2-benzodioxasiline (1). | ||
Results and discussion
Preliminary synthetic work showed that monomer 1 cannot easily be obtained in a transesterification reaction using trialkoxysilanes, salicyl alcohol and furfuryl alcohol. However, a stepwise approach was developed, which depends on the synthesis of the key intermediate 2-chloro-2-methyl-4H-1,3,2-benzodioxasiline, which is obtained by a reaction of trichloromethylsilane with one equivalent of salicyl alcohol (see Scheme 1). It can be purified by distillation, and the chlorine atom can readily be substituted with different polymerizable groups. Therefore, 2-chloro-2-methyl-4H-1,3,2-benzodioxasiline can act as a precursor for many heteromultifunctional monomers. In this publication, we report on the reaction with furfuryl alcohol to yield the final heterotrifunctional monomer 2-furfuryloxy-2-methyl-4H-1,3,2-benzodioxasiline (1), which can easily be synthesized in a multigram scale. In all reaction steps water has to be excluded to prevent hydrolysis. 2-Chloro-2-methyl-4H-1,3,2-bezodioxasiline can react with moisture, leading to the formation of hydrochloric acid, which catalyzes its polymerization.The polymerization behavior of monomer 1 was studied with differential scanning calorimetry (DSC). The two related and already described twin monomers 2,2′-spirobi[benzo-4H-1,3,2-dioxasiline] (Spiro) and tetrafurfuryloxysilane (TFOS) differ in their polymerization behavior. Spiro can be polymerized by simple thermal annealing,10 or catalyzed by acids8 and bases.16TFOS can only be polymerized under acidic conditions.6 An annealing of this monomer does not lead to an exothermic peak in the thermogram of the differential scanning calorimetry (see Fig. S1, ESI†). Thus, no polymerization occurs (also not in the presence of a base) below a temperature of 350 °C.
When both the respective organic moieties, the furfuryl alcohol and the salicyl alcohol, are combined in one monomer (monomer 1), it is expected that, by the choice of the catalyst, the number of ongoing polymerization reactions can be controlled. An acid catalyst should be able to trigger the polymer formation of poly(furfuryl alcohol), phenolic resin, and polymethylsilsesquioxane, whereas a base catalyzed reaction is expected to lead to phenolic resin, and poly(methylfurfuryloxysiloxane), a so-called twin prepolymer. This is shown in Scheme 2 together with a schematic description, where X symbolizes the inorganic fragment and Y, and Z the two different organic moieties of the heterotrifunctional monomer. W mirrors the water released during the polymerization of the furfuryloxy fragment Z.
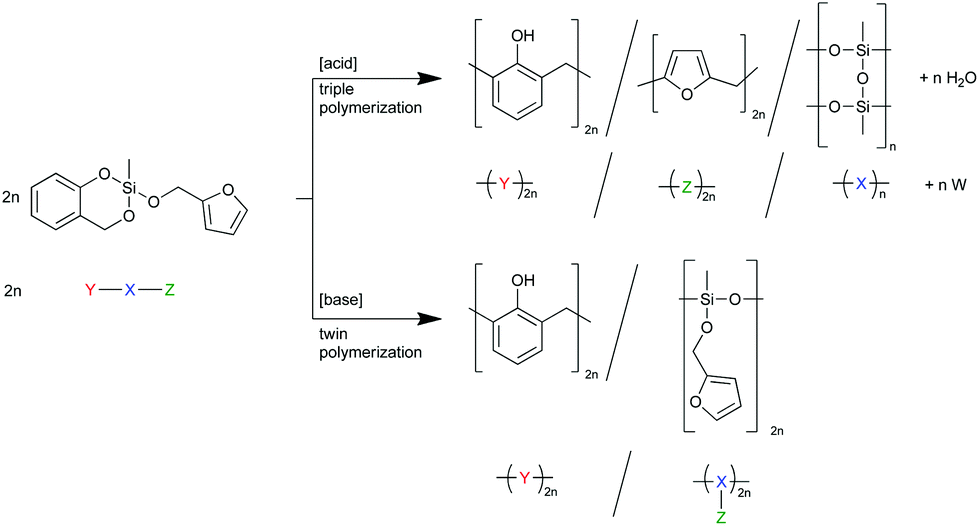 | ||
| Scheme 2 Schematic description and reaction equation of the expected polymerization scenarios of the heterotrifunctional monomer 2-furfuryloxy-2-methyl-4H-1,3,2-benzodioxasiline (1) either leading to three different polymers or one polymer and one twin prepolymer. | ||
In the course of the cationic multiple polymerization a copolymerization between the poly(furfuryl alcohol) and the phenolic resin cannot be excluded but is unlikely. Zhang and Solomon investigated the curing behavior of novolac/furfuryl alcohol resins and described their reaction with each other of minor importance. However, methylene linkages could be observed when hexamethylenetetramine was used for further crosslinking.17 Pizzi et al. and Abdalla et al. investigated the reaction of mimosa tannin extract and furfuryl alcohol and identified reaction products using MALDI-TOF. Besides the products of the reaction of fisetinidin flavonoid units with furfuryl alcohol, they always found oligomers derived from the self-condensation of furfuryl alcohol.18,19 As fisetinidin flavonoid units are much more reactive than phenol in Friedel–Crafts reactions, this shows that the self-condensation of furfuryl alcohol should be favored over the reaction with phenol. When comparing the π-nucleophilicity parameters according to Mayr,20 the value of furan (N = 1.33, sN = 1.29)21 is higher than the value of anisole (N = −1.18, sN = 1.20).20 This means that the reactivity of furan is 250 times higher than anisole. If we assume a similar effect of the additional methylol group and a comparability of the –OMe with the –OH substituent, the reactivity of furfuryl moieties should be higher and thus homopolymers should be formed.
The presence of different reaction scenarios can be supported by DSC measurements (see Fig. 2) of monomer 1 solely and together with potential catalysts. The catalysts 1,8-diazabicycloundec-7-ene (DBU) and trifluoroacetic (TFA) acid are used for the polymerization in bulk, as they are suitable catalysts for the twin polymerization of Spiro.8 During the annealing of monomer 1 (black line) up to 400 °C, an exothermic peak is visible at around 267 °C. When DBU is added as a catalyst, the temperature of this exothermic peak is reduced to 136 °C, indicating the catalytic activity of the base. The addition of TFA leads to a polymerization already starting at room temperature during the mixing of the monomer and the catalyst (see Fig. S2, ESI,† for 1 + TFA). To delay this reaction the room temperature solid trichloroacetic acid (TCA) was used as a catalyst in the DSC investigations. When the integrals of the exothermic peaks are compared, it is obvious that the acid catalyzed polymerization is more exothermic than the base catalyzed polymerization. Exact values cannot be obtained, as the endothermic melting of TCA and the beginning of the evaporation of DBU occur in the same temperature range as the exothermic reaction (see Fig. S1, ESI†). The curves are calculated per mol of monomer only, neglecting the effects from the catalyst. These first results were verified by reproducing these polymerizations on a multigram scale, and the subsequent analysis of the products. Monolithic hybrid materials were obtained by TFA (A), or DBU (B) catalyzed as well as thermal polymerization (T) of 1. BA describes the hybrid material from DBU catalyzed polymerization after post-treatment with methanesulfonic acid (MSA) (see Table 1).
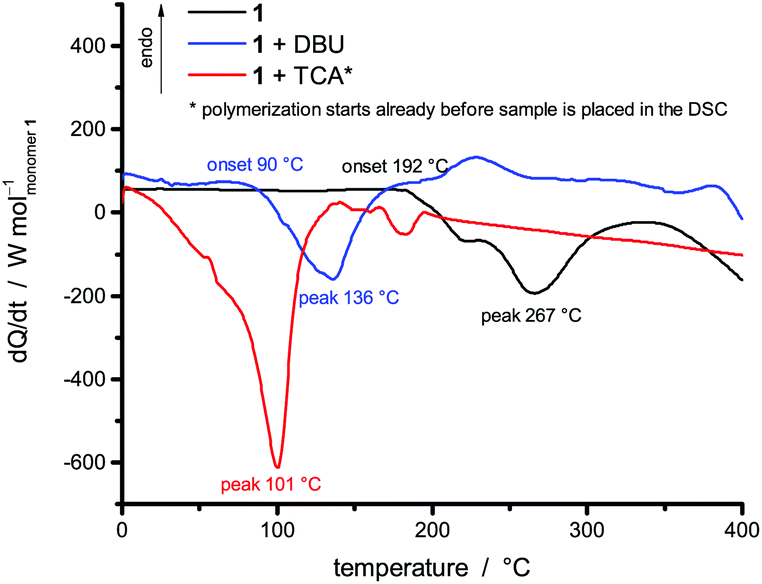 | ||
| Fig. 2 Differential scanning calorimetry measurements of the monomer 2-(furfuryloxy)-2-methyl-4H-1,3,2-benzodioxasiline (1) solely (black line) and together with 1,8-diazabicycloundec-7-ene (DBU, blue line) and trichloroacetic acid (TCA, red line) catalysts. | ||
| Sample | Catalyst (conc./mol%) | Temp./°C | Residue mass TGA/% | Carbon content/% | Picture | |
|---|---|---|---|---|---|---|
| Air | He | |||||
| A | TFA (10) | 50 | 21.3 | 50.1 | 57.0 |

|
| B | DBU (10) | 140 | 13.4 | 39.2 | 65.0 |

|
| T | — | 250 | 25.0 | 44.7 | 60.4 |

|
| BA | 1. DBU (10) | 140 | 23.2 | 59.5 | 55.2 |

|
| 2. MSA (10) | 25 | |||||
A first indication, if the furfuryloxy-fragment has been polymerized or not, is the color of the hybrid material. During the formation of poly(furfuryl alcohol) some conjugated structures are always formed, which explain its dark green almost black color.22 Only the hybrid material from the DBU catalyzed polymerization is orange and transparent, which indicates that the polymerization of the furfuryloxy-fragment did not take place.
Fig. 3 shows the 13C-{1H}-CP-MAS-NMR spectra of hybrid materials obtained from the polymerization of monomer 1 in the melt with different catalysts compared to the 13C-NMR of compound 1 in CDCl3 with the related chemical structures. All solid-state NMR spectra prove the formation of phenolic resin (signals a–e in Fig. 3). Only traces of unreacted CH2O–R-Groups can be found, which indicates a high conversion of the salicyl alcohol fragments. When the polymerization was carried out with DBU as the catalyst, the sharp signals at 57 ppm and 142 ppm indicate unbroken CH2–O–Si units, and the unsubstituted carbon in position 5 of the furan ring, respectively (see Fig. 3). This can be explained by the formation of a twin prepolymer and phenolic resin as suggested in Scheme 2. In the hybrid materials from acid catalyzed or thermally induced polymerization the isolated signals 4 and 13 of typical side products as well as the broad signals 5 and 6 show the formation of poly(furfuryl alcohol). The other signals overlap with the phenolic resin signals.23 The absence of a signal at 57 ppm and the very low intensity of the signal at 142 ppm indicate that the furfuryl alcohol fragment showed a high conversion during the polymerization, as well. Salabarria et al. describe a reaction between furfuryl alcohol and phenol occurring mainly in the para position of the phenol.24 Signals b and c represent unsubstituted ortho and para carbon atoms of the phenolic ring,23,25 which shows that a reaction of furfuryl alcohol with the phenolic resin is of minor importance. In the solid state 29Si-{1H}-CP-MAS-NMR spectra of the hybrid materials different T signals can be distinguished.26,27 They result from structural units shown in Fig. 4, which further supports the described polymerization reactions. When DBU is used as a base catalyst for a polymerization in bulk, two relatively sharp, separated peaks, indicating the presence of T2 and T3 structures, and one peak of minor intensity, indicating T1 units, are found. T3 signals in the hybrid material from the base catalyzed polymerization might result from already occurring condensation reactions. Structural units of the suggested twin prepolymer can explain T2 and T1 signals. For the thermal and acid catalyzed polymerization in bulk still T2 but mainly T3 units are found. The peaks are broader and less separated, which indicates a higher condensed network. When the polymerization was carried out with MSA as precipitation polymerization, almost only the T3 signal is found (see Fig. S4 ESI†). This shows that for the polymerization in bulk there might be a diffusion limitation leading to an incomplete polymerization of the inorganic fragment. It should be noted that, due to the cross polarization NMR experiments, the signal intensity of silicon atoms with hydrogen in the proximity is higher. Thus, the T1 and T2 structures give stronger signals than T3 structures.
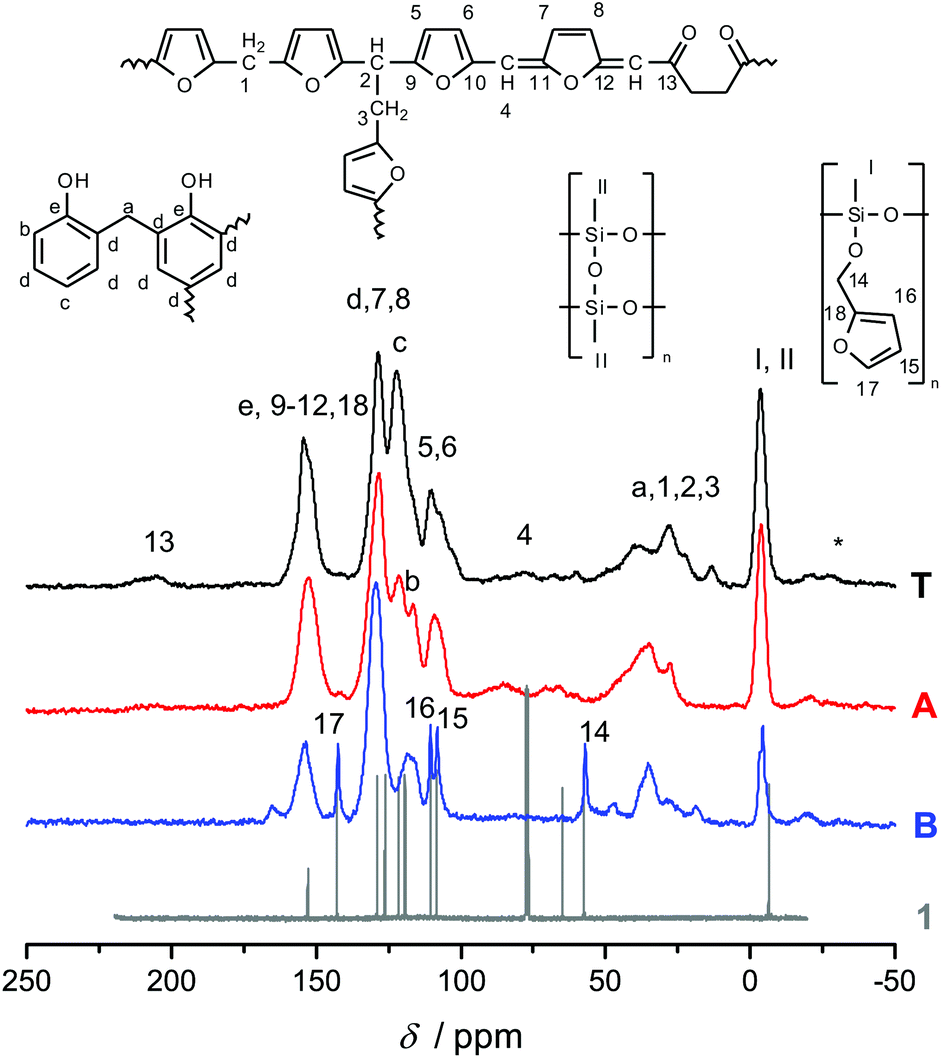 | ||
| Fig. 3 13C-NMR spectrum of compound 1 in CDCl3 compared with the solid state 13C-{1H}-CP-MAS-NMR spectra of hybrid materials, obtained from the polymerization of monomer 1 thermally induced (black line) or catalyzed with DBU (blue line) and TCA (red line), together with the attributed chemical structures. | ||
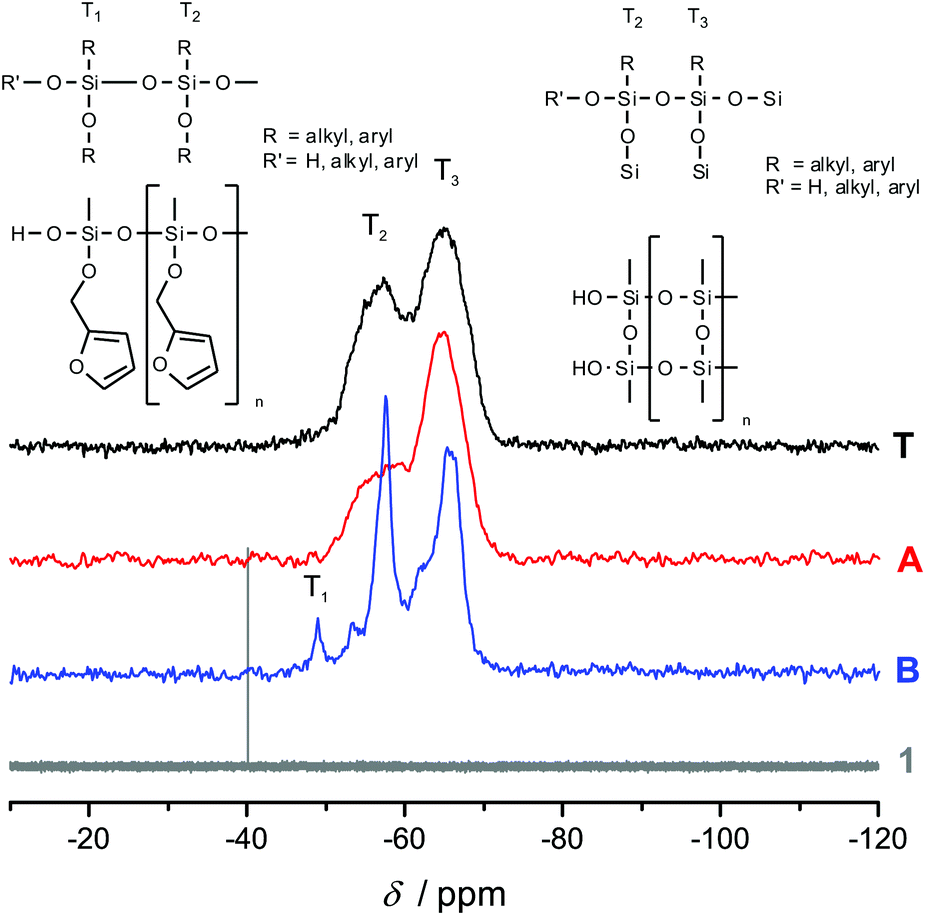 | ||
| Fig. 4 29Si-NMR of compound 1 in CDCl3 compared with the solid state 29Si-{1H}-CP-MAS-NMR spectra of hybrid materials, obtained from the polymerization of monomer 1 with different catalysts, together with the attributed chemical structures. | ||
To investigate the accessibility of the furfuryloxy moieties for further reactions, the hybrid material from the DBU catalyzed twin polymerization of monomer 1 was ground and examined in the DSC. When solely the hybrid material is measured, no heat flow is observed. The same measurement, after addition of TCA, clearly shows an exothermal signal that indicates a polymerization (see Fig. S5, ESI†). Thus, the ground hybrid material B was suspended in CH2Cl2, and MSA was added as an acid catalyst, which should trigger a further polymerization as shown in Scheme 3. MSA is chosen as it is a good catalyst for twin polymerization of Spiro in solution.8 Immediately after the addition of the acid the color becomes darker, which indicates the polymerization of the furanic moieties. The final hybrid material was isolated by filtration and investigated with solid state NMR spectroscopy to prove the molecular structure. It should be emphasized that soluble components are washed off during this process and do not remain in the hybrid material. It was found that the signals for the monomeric furfuryloxy moieties (signals 17 and 14 in Fig. 5) are drastically reduced, and poly(furfuryl) alcohol is found in the final hybrid material as evidenced by the newly occurring peaks 4, 5, 6 and 13. Furthermore, after the treatment with an acid, the T2 signal in the 29Si-{1H}-CP-MAS-NMR spectra (see Fig. 6) is reduced compared to the T3 signal, which indicates the formation of a fully condensed polymethylsilsesquioxane network. This shows that the hybrid material is still flexible enough to allow the polymerization reactions of the furfuryloxy moieties and the inorganic network.
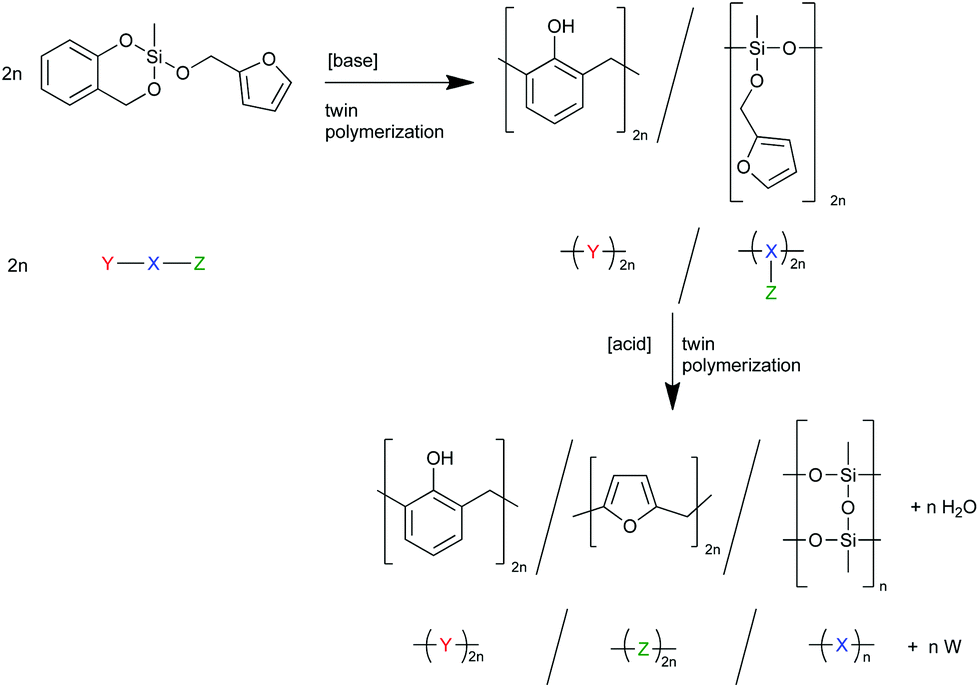 | ||
| Scheme 3 Schematic description and reaction equation of the twin polymerization of monomer 1 yielding a twin prepolymer which can then undergo further twin polymerization to the final hybrid material. | ||
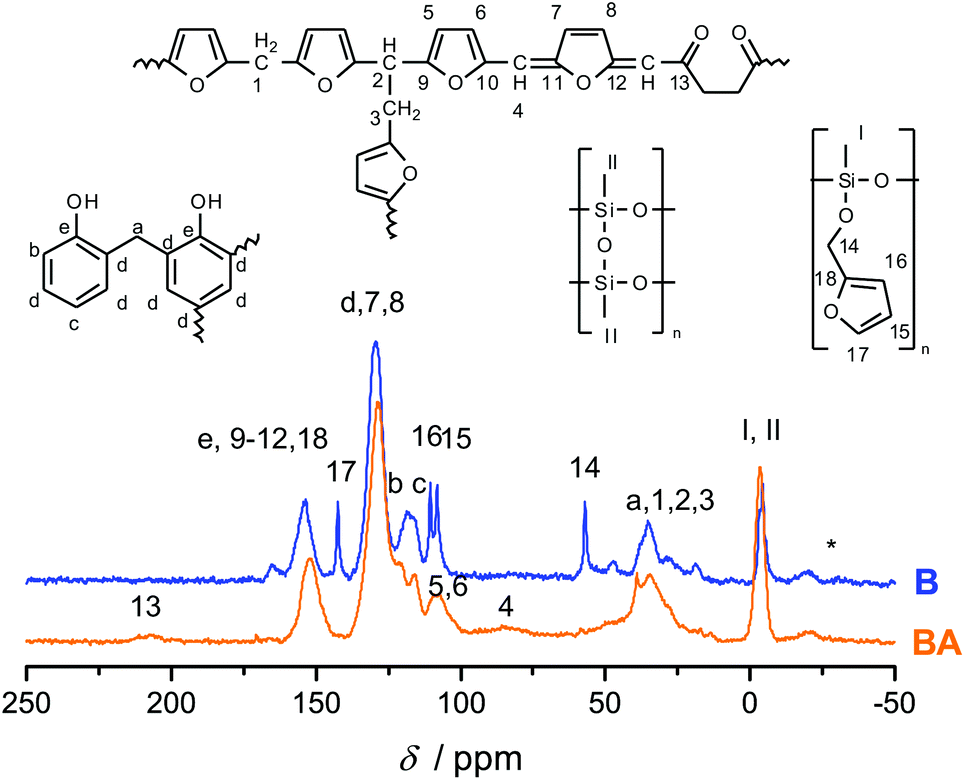 | ||
| Fig. 5 13C-{1H}-CP-MAS-NMR spectra of hybrid materials, obtained from the polymerization of monomer 1 with DBU as a catalyst compared with the acid post treated material, together with the attributed chemical structures. | ||
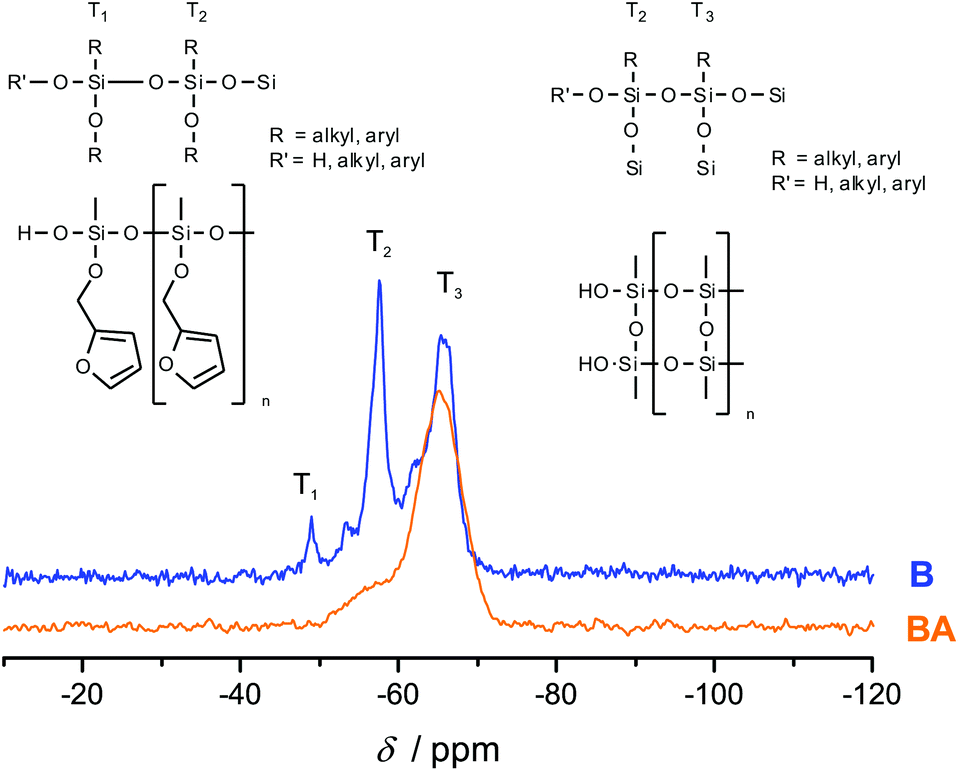 | ||
| Fig. 6 29Si-{1H}-CP-MAS-NMR spectra of hybrid materials, obtained from the polymerization of monomer 1 with DBU as a catalyst compared with the acid post treated material, together with the attributed chemical structures. | ||
For information on the size of the phase domains high angle annular dark field scanning transmission electron microscopy (HAADF-STEM) investigations were carried out. This technique shows an elemental contrast besides the contrast attributed to sample thickness and density. With an increasing atomic number, higher signal intensity is observed.28 Thus, silicon-rich domains of the hybrid material are shown brighter than the organic polymers. When the HAADF-STEM images of the different hybrid materials are compared (Fig. 7), it can be seen that the materials from the TFA catalyzed and thermally induced multiple polymerization show bright SiO2 clusters with a rather similar size of 1–3 nm (see Fig. 7a and b). These SiO2 clusters are very similar to the inorganic domains described for the phenolic resin/SiO2 hybrid material obtained by twin polymerization of Spiro.8 In contrast, the hybrid material derived from the DBU catalyzed twin polymerization of monomer 1 shows only a little contrast (Fig. 7c). This is attributed to the fact that the furfuryloxy unit is still connected to the silicon of the inorganic polymer and thus the elemental contrast is reduced. Remarkably, after the post treatment with MSA, the silicon rich domains can clearly be seen. They show a similar size to the hybrid materials from acid catalyzed and thermally induced multiple polymerization of monomer 1 (Fig. 7d). This leads to the conclusion that the hybrid material B also consists of co-continuous nanodomains. By treating this hybrid material with MSA, the Si–O–CH2 bond is cleaved and poly(furfuryl) alcohol is formed, leading to poylmethylsilsesquioxane as an inorganic phase with a lower content of carbon, hydrogen, and oxygen.
 | ||
| Fig. 7 HAADF-STEM images from the hybrid material synthesized by acid catalyzed (a), thermally induced (b) and base catalyzed (c) multiple polymerization of monomer 1 compared with the hybrid material of the base catalyzed polymerization with additional treatment with methanesulfonic acid (d). | ||
The synthesized hybrid materials can be converted into porous carbon by annealing in an inert atmosphere and the removal of the inorganic network. To obtain porous SiO2 the organic parts can be removed by annealing in an air atmosphere. During the oxidation the polymethylsilsesquioxane is oxidized to SiO2.29 As the organic and inorganic networks are interpenetrating, the occurring pores give an indication of the domain size of their respective counterparts in the hybrid materials.
The annealing in air leads to porous SiO2, which shows specific surface areas between 423 and 621 m2 g−1. After oxidation, the theoretic amount of the residue is 22.9 wt% if the furfuryloxy moieties are polymerized, and 23.7 wt% if they remain unpolymerized. In contrast to the other hybrid materials B shows a residue of only 13.4 wt% (see Table 1, Fig. S7, ESI†). This is explained by a partial removal of the inorganic polymer and is known for polydimethylsiloxanes,30–32 especially in the presence of base catalysts.33 This result further proves the lower extend of condensation of the inorganic polymer.
The obtained SiO2 materials A_ox, B_ox and T_ox show different adsorption properties (see Fig. 8a and b; Table 2). The isotherms of the SiO2, obtained from the hybrid material of acid or base catalyzed polymerization, can be classified as type IV isotherms according to IUPAC.34 In contrast, the isotherm of the SiO2 from thermal polymerization is similar to type I. This behavior is reflected in the calculated pore size distribution. The type I isotherm is caused by smaller pores including some amount of micropores. In contrast, the hybrid materials A_ox and B_ox show only mesopores mainly between 3.5 and 10 nm and 3 and 12 nm for TFA and DBU catalyzed polymerization, respectively.
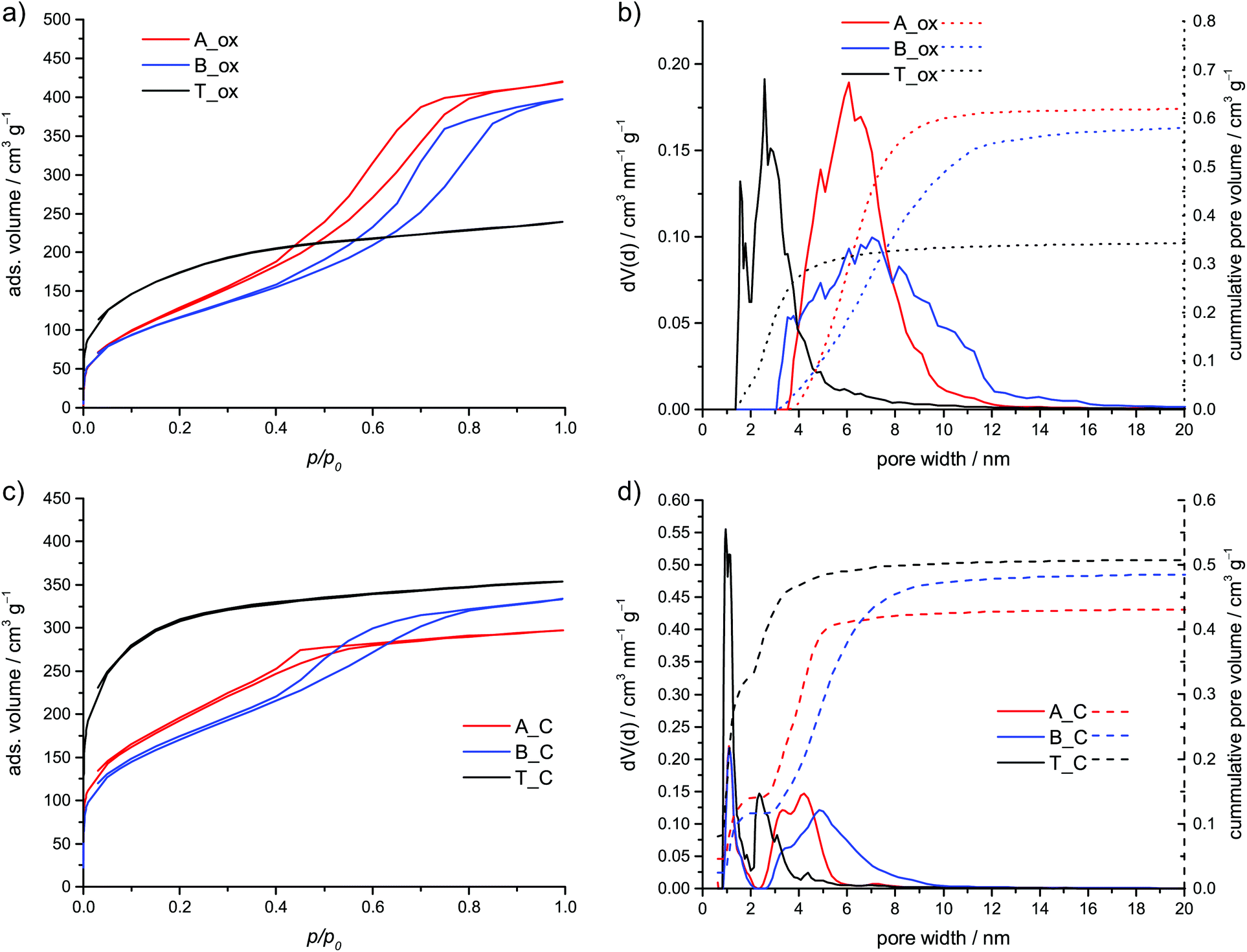 | ||
| Fig. 8 Nitrogen sorption isotherms measured at 77 K and pore size distribution (NLDFT, adsorption branch, cylindrical pores) of the oxidized hybrid materials (a, b) and nitrogen sorption isotherms and pore size distribution (QSDFT, adsorption branch, slit pores + cylindrical pores) of the carbon materials obtained by inert annealing and subsequent HF etching of the hybrid materials (c, d). | ||
| Sample | BET surface area/m2 g−1 | DFT surface area/m2 g−1 (method) | Pore volume/cm3 g−1 | Micropore volume/cm3 g−1 |
|---|---|---|---|---|
| A_ox | 473 | 406 (NLDFT) | 0.627 | 0 |
| B_ox | 423 | 346 (NLDFT) | 0.595 | 0 |
| T_ox | 621 | 481 (NLDFT) | 0.349 | 0.053 |
| A_C | 638 | 688 (QSDFT) | 0.433 | 0.140 |
| B_C | 603 | 547(QSDFT) | 0.489 | 0.116 |
| T_C | 1114 | 1038(QSDFT) | 0.510 | 0.329 |
A similar trend is observed for the carbon materials obtained by carbonization of the hybrid material and the removal of the inorganic component. The inert annealing of polymethylsilsesquioxane leads to a mixture of SiO2, carbon and silicon oxycarbide35–37 with SiO2 as the main component.36 Thus, it can be removed with hydrofluoric acid solution. During inert annealing the hybrid material B shows the lowest amount of residue (see Fig. S8, ESI†). Furthermore, material B, after carbonization, shows the highest mass loss in an air atmosphere. Thus, it has the lowest inorganic content (see Fig. S9, ESI†) compared to the other carbonized materials due to a partial decomposition of the inorganic network. The final carbon materials were investigated in TGA and show mass losses of 98.8 wt% and more in an air atmosphere, proving the almost complete removal of the inorganic network (see Fig. S6, ESI†). The isotherm of the porous carbon T_C can be classified as type I isotherm, whereas the carbon materials A_C and B_C show a hysteresis (see Fig. 8c). While the latter shows a type IV isotherm, it is difficult to assign an isotherm type for the carbon A_C. The calculated pore size distributions can be seen in Fig. 8d. Though, in all cases, some micropores are present, the content of micropores is the highest for the carbon obtained from the hybrid material of the thermal polymerization. Both, the content and the size of the remaining mesopores are the highest for carbon from the DBU catalyzed hybrid material. Together with the HAADF-STEM investigation, these results prove that in all cases nanostructured hybrid materials are obtained.
Conclusions
The heterotrifunctional monomer 2-(furfuryloxy)-2-methyl-4H-1,3,2-benzodioxasiline was synthesized and investigated for its polymerization behavior. It could be shown that, by the choice of the polymerization conditions, either two or three polymers are formed in one reaction step using, at the most, one catalyst. It is the first single-source monomer, which can be polymerized to three different polymers. The formation of the different networks occurs on the same time scale. Therefore, phase separation is very small and very similar to the already known hybrid materials from twin polymerization. This shows that regardless of whether two or three different polymers are formed, the underlying concept of the simultaneous formation of two or more polymers from only one monomer can lead to hybrid materials with very small nanodomains of its components. Future work will focus on the synthesis of other heteromultifunctional monomers for the multiple polymerization. Replacing furfuryl alcohol with 2-thiophenemethanol leads to a monomer, which might be polymerized in similar ways. Due to the additional sulfur, a more detailed investigation of the distribution of the individual polymers can be conducted.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft for (DFG SP 392/34-2, SP 392/35-2, Cluster of Excellence EXC 1075 “MERGE”) is greatly appreciated. We thank P. Dill for her experimental work and Dr. P. Formánek for the preparation of the microtome cut. We also thank Prof. Dr. A. Feldhoff and F. Steinbach for TEM maintenance. The authors thank Dr. K. Schreiter and Dr. S. Höhne for the fruitful discussions.Notes and references
- M. Antonietti, Nat. Mater., 2003, 2, 9–10 CrossRef CAS PubMed.
- A. S. Aricò, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed.
- A. Thomas, F. Goettmann and M. Antonietti, Chem. Mater., 2008, 20, 738–755 CrossRef CAS.
- H. S. Thiam, W. R. W. Daud, S. K. Kamarudin, A. B. Mohammad, A. A. H. Kadhum, K. S. Loh and E. H. Majlan, Int. J. Hydrogen Energy, 2011, 36, 3187–3205 CrossRef CAS.
- G. Kickelbick, Hybrid materials: synthesis, characterization, and applications, Wiley-VCH, Weinheim, 2007 Search PubMed.
- S. Grund, P. Kempe, G. Baumann, A. Seifert and S. Spange, Angew. Chem., Int. Ed., 2007, 46, 628–632 CrossRef CAS PubMed.
- S. Spange and S. Grund, Adv. Mater., 2009, 21, 2111–2116 CrossRef CAS.
- S. Spange, P. Kempe, A. Seifert, A. A. Auer, P. Ecorchard, H. Lang, M. Falke, M. Hietschold, A. Pohlers, W. Hoyer, G. Cox, E. Kockrick and S. Kaskel, Angew. Chem., Int. Ed., 2009, 48, 8254–8258 CrossRef CAS PubMed.
- T. Ebert, A. Seifert and S. Spange, Macromol. Rapid Commun., 2015, 36, 1623–1639 CrossRef CAS PubMed.
- P. Kempe, T. Löschner, A. A. Auer, A. Seifert, G. Cox and S. Spange, Chem. – Eur. J., 2014, 20, 8040–8053 CrossRef CAS PubMed.
- T. Löschner, A. Mehner, S. Grund, A. Seifert, A. Pohlers, A. Lange, G. Cox, H.-J. Hähnle and S. Spange, Angew. Chem., Int. Ed., 2012, 51, 3258–3261 CrossRef PubMed.
- T. Löschner, A. Seifert, R. Lungwitz, G. Cox, A. Lange, H.-J. Hähnle and S. Spange, Macromol. Rapid Commun., 2011, 32, F60–F61 Search PubMed.
- M. Göring, A. Seifert, K. Schreiter, P. Müller and S. Spange, Chem. Commun., 2014, 50, 9753–9756 RSC.
- A. D. Jenkins, P. Kratochvíl, R. F. T. Stepto and U. W. Suter, Pure Appl. Chem., 1996, 68, 2287–2311 CAS.
- J. V. Alemán, A. V. Chadwick, J. He, M. Hess, K. Horie, R. G. Jones, P. Kratochvíl, I. Meisel, I. Mita, G. Moad, S. Penczek and R. F. T. Stepto, Pure Appl. Chem., 2007, 79, 1801–1829 CrossRef.
- T. Ebert, G. Cox, E. Sheremet, O. Gordan, D. R. T. Zahn, F. Simon and S. Spange, Chem. Commun., 2012, 48, 9867 RSC.
- X. Zhang and D. H. Solomon, Chem. Mater., 1998, 10, 1833–1840 CrossRef CAS.
- A. Pizzi, G. Tondi, H. Pasch and A. Celzard, J. Appl. Polym. Sci., 2008, 110, 1451–1456 CrossRef CAS.
- S. Abdalla, A. Pizzi, F. Bahabri and A. Ganash, Maderas: Cienc. Tecnol., 2015, 99–104 Search PubMed.
- H. Mayr, B. Kempf and A. R. Ofial, Acc. Chem. Res., 2003, 36, 66–77 CrossRef CAS PubMed.
- J. Ammer, C. Nolte and H. Mayr, J. Am. Chem. Soc., 2012, 134, 13902–13911 CrossRef CAS PubMed.
- M. Choura, N. M. Belgacem and A. Gandini, Macromolecules, 1996, 29, 3839–3850 CrossRef CAS.
- G. A. Webb, Annual reports on NMR spectroscopy, Academic Press, San Diego, London, 1994, vol. 29 Search PubMed.
- I. R. Salabarría, P. A. Oyanguren, D. P. Fasce and R. J. J. Williams, Polym. Int., 1995, 37, 21–26 CrossRef.
- R. Rego, P. J. Adriaensens, R. A. Carleer and J. M. Gelan, Polymer, 2004, 45, 33–38 CrossRef CAS.
- R. Joseph, S. Zhang and W. T. Ford, Macromolecules, 1996, 29, 1305–1312 CrossRef CAS.
- Z. L. Da, eXPRESS Polym. Lett., 2007, 1, 698–703 CrossRef CAS.
- S. Pennycook and D. Jesson, Phys. Rev. Lett., 1990, 64, 938–941 CrossRef CAS PubMed.
- V. V. Kireev, B. I. D'yachenko and V. P. Rybalko, Polym. Sci., Ser. A, 2008, 50, 394–402 CrossRef.
- C. W. Lewis, J. Polym. Sci., 1958, 33, 153–159 CrossRef CAS.
- T. H. Thomas and T. C. Kendrick, J. Polym. Sci., Part B: Polym. Phys., 1969, 7, 537–549 CrossRef CAS.
- T. H. Thomas and T. C. Kendrick, J. Polym. Sci., Part B: Polym. Phys., 1970, 8, 1823–1830 CrossRef CAS.
- M. Kučera, J. Láníková and M. Jelínek, J. Polym. Sci., 1961, 53, 301–310 CrossRef.
- K. S. W. Sing, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- V. Belot, R. J. P. Corriu, D. Leclercq, P. H. Mutin and A. Vioux, J. Mater. Sci. Lett., 1990, 9, 1052–1054 CrossRef CAS.
- J. Ma, L. Shi, Y. Shi, S. Luo and J. Xu, J. Appl. Polym. Sci., 2002, 85, 1077–1086 CrossRef CAS.
- G. Trimmel, R. Badheka, F. Babonneau, J. Latournerie, P. Dempsey, D. Bahloul-Houlier, J. Parmentier and G. D. Soraru, J. Sol–Gel Sci. Technol., 2003, 26, 279–283 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, monomer characterization and supporting figures. See DOI: 10.1039/c6py01619g |
| This journal is © The Royal Society of Chemistry 2016 |