
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel PEG–haloperidol conjugate with a non-degradable linker shows the feasibility of using polymer–drug conjugates in a non-prodrug fashion
Felicity
Heath
a,
Amy
Newman
a,
Chiara
Clementi
b,
Gianfranco
Pasut
b,
Hong
Lin
a,
Gary J.
Stephens
a,
Benjamin J.
Whalley
a,
Helen M. I.
Osborn
a and
Francesca
Greco
*a
aReading School of Pharmacy, PO Box 224, Reading, RG6 6AD, UK. E-mail: f.greco@reading.ac.uk
bDept. of Pharmaceutical Sciences, Via F. Marzolo 5, University of Padua, 35100, Padova, Italy
First published on 15th November 2016
Abstract
A PEG–haloperidol conjugate containing a non-biodegradable linker was synthesised. Incubation with rat plasma demonstrated excellent linker stability, and competition radioligand binding assays demonstrated retained binding to the D2-receptor. In silico studies predicted that the conjugate will not cross the blood–brain barrier (BBB), thus potentially restricting haloperidol action to one side of the BBB.
1. Introduction
Many drugs display an unfavourable body distribution, which results in unwanted exposure of healthy tissues to the drug. One such case applies to drugs that have their desired therapeutic target in the central nervous system (CNS) but display unwanted effects due to their peripheral action. An example is L-DOPA, used in the treatment of Parkinson's disease, which can cause nausea and vomiting as a result of stimulation of the peripheral dopamine receptors by its metabolite dopamine.1,2 The opposite is also true, in that some drugs acting in the peripheral tissues display unwanted effects centrally. First generation anti-histamines that are able to cross the blood brain barrier (BBB) are widely known to cause side effects of drowsiness.3,4 In both scenarios, preventing drug permeation across the BBB would effectively localise drug action to a single compartment: periphery following peripheral administration, or central, following central administration. Many factors affect drug permeability across the BBB (e.g. lipophilicity and hydrogen bonding capacity), but size is known to play a particularly crucial role.5–7 Conjugation of a small, BBB-penetrating drug molecule to a polymer could restrict its transit across this barrier, thus removing any undesired effects in either compartment. Indeed, Satchi-Fainaro et al. have demonstrated that conjugation of an anti-tumour agent, TNP-470, to an HPMA-copolymer resulted in a significant reduction in neurotoxicity compared to the free drug. These findings suggested that conjugation to the polymer prevented its BBB passage.8 However, the system was still not ideal as the drug (TNP-470) was conjugated via a degradable linker (a peptidyl linker). As such, this conjugate still had the potential for drug-related toxicity once the drug is released. More recently, a PEGylated opioid antagonist (Movantik™) has been marketed as a peripherally-acting opioid antagonist for the treatment of opioid-induced constipation.9 Further, Kopecek and colleagues have proposed “drug free” polymer–drug conjugates, in which the polymer is an active agent per se’.10,11The concept of restricting the action of a drug selectively to the periphery or to the CNS, by its covalent conjugation to a polymer, is scientifically appealing, but for this strategy to work two conditions must be met. First, the polymer must be able to prevent the drug from crossing the BBB. Second, the polymer conjugated drug must retain (at least partially) its biological activity. Meeting the first condition is very straightforward as it is well known that size is a key modulator of BBB permeation.12 The second condition is much more challenging and requires careful selection of an appropriate ligand.
This study explores, for the first time, application of this strategy to the dopaminergic system. In particular, we propose a polymer–drug conjugate that will: (a) be stable in biological fluids, thus preventing unwanted drug release from the conjugate, (b) retain some ability to modulate the dopaminergic system, (c) have a size compatible with what has been reported to be BBB non-permeable. To explore this hypothesis, the D2 receptor antagonist, haloperidol (Fig. 1) was selected for conjugation to a linear bifunctional PEG polymer.
 | ||
| Fig. 1 Chemical structure of the D2 receptor antagonist haloperidol. | ||
This ligand presents a series of characteristics rendering it suitable for the proposed application, namely: (i) it is an antagonist of the chosen system (dopaminergic); (ii) as a free drug, it is able to cross the BBB; (iii) the hydroxyl group is a suitable point for conjugation; (iv) importantly, previous studies showing conjugation to affinity columns had shown an indication of ability to maintain its binding capacity to the D2 receptor after conjugation to sepharose solid supports as affinity column ligand.13
The system proposed in this study differs dramatically from most of the polymer-small-drug conjugates being developed, as the latter typically act as prodrugs and generally require a biodegradable bond or linker between polymer and drug to exert their action.14–18 Conversely, here we suggest a non degradable polymer–drug conjugate, in which the size of the polymer matches that needed to prevent BBB crossing, and in which the conjugation has been carried out at a point that allows (at least partial) retention of activity. The pharmacological target of the conjugate presented in this work (the dopamine D2 receptor) has the advantage of being expressed both centrally19–21 and peripherally,21–24 and also of being easily accessible to polymer–drug conjugates as a result of its location, for instance, on the plasma membrane of neuronal cells.
In this paper we report the synthesis, characterisation and in vitro biological assessment of a polymer–haloperidol conjugate designed to inhibit passage across the BBB. First, we report PEGylation of the chosen drug. PEG was chosen as the polymeric component due to its favourable properties such as: prolonged residence in the body, decreased metabolic degradation of the attached drug and improved solubility.25,26 Then, stability of the bond between the polymer and the drug was assessed in vitro using rat plasma. The ability of the conjugate to bind to D2 receptors was determined using radioligand binding assays. Finally, a theoretical model was employed to investigate the likelihood of PEG conjugation restricting the passage of haloperidol across the BBB. As such, this study can be considered as the first stage in determining the feasibility of the polymer–haloperidol conjugate for selective delivery to the central or peripheral system, after which it will be pertinent to conclude whether in vivo studies would be warranted.
2. Results and discussion
2.1. Synthesis and characterisation
To synthesise the PEG–haloperidol conjugate, direct conjugation of haloperidol to PEG via its tertiary hydroxyl group was initially attempted using the methane sulfonate ester of bis-hydroxy PEG. However, no reaction was observed with haloperidol, presumably due to the sterically hindered nature of the hydroxyl group. To aid conjugation of haloperidol to PEG, an ethylenediamine linker was attached to the hydroxyl group through a carbamate bond, introducing an accessible amine handle for subsequent conjugation to a polymer (Fig. 2).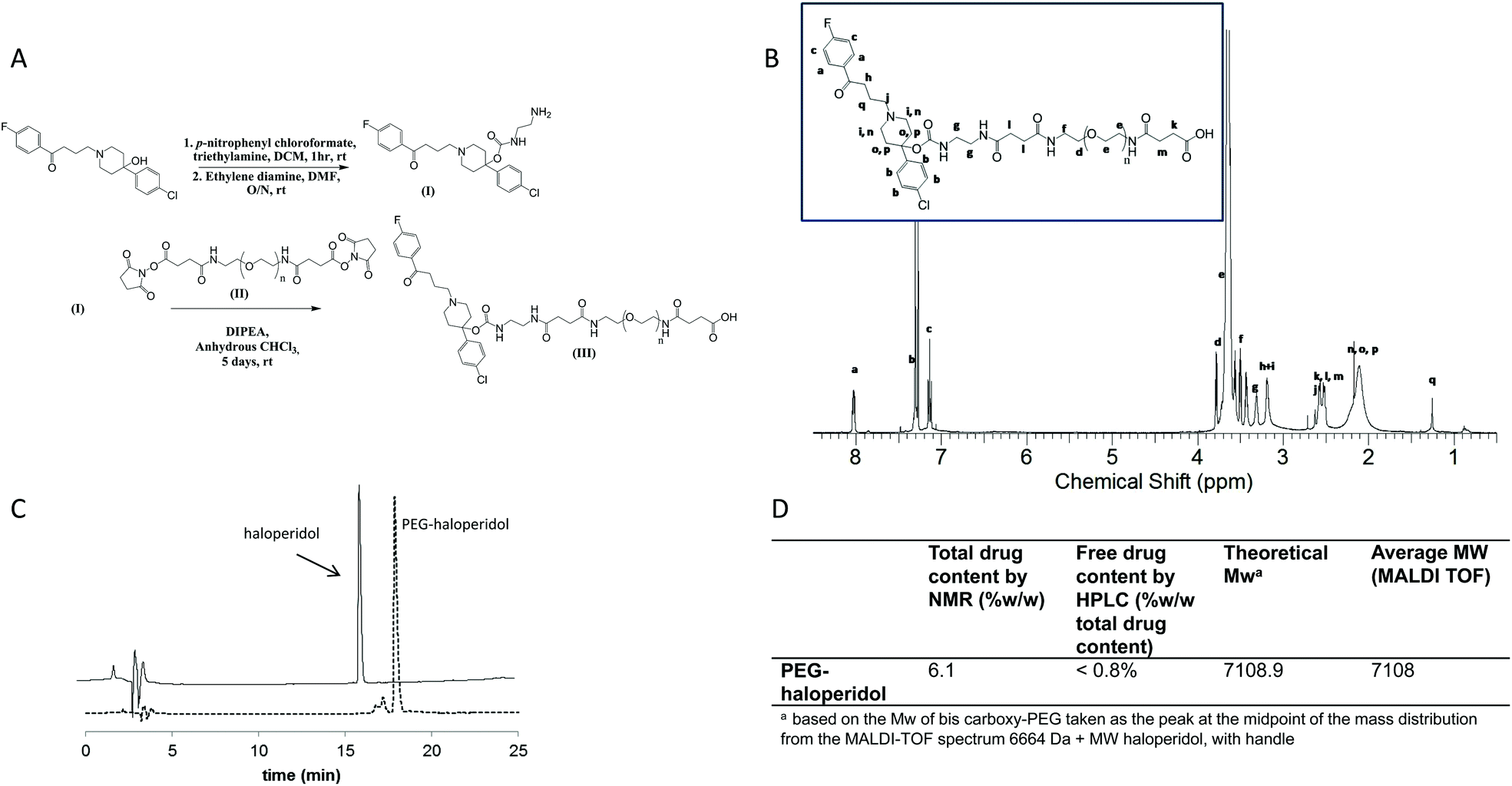 | ||
| Fig. 2 Panel A: Synthetic scheme of haloperidol-amino ethyl carbamate (haloperidol-AEC) (I), and PEG–haloperidol (III), Panel B: 1H-NMR spectrum of PEG–haloperidol, Panel C: C18 HPLC chromatogram of PEG–haloperidol (tR = 17.2 min) and free haloperidol (tR = 16.1), Panel D: Characterisation of PEG–haloperidol. | ||
The conjugation of haloperidol to a drug delivery carrier has not been extensively explored within the literature. Most examples of derivatisation of haloperidol at the hydroxyl group involve the formation of an ester bond.27–30 This is likely to be susceptible to degradation by esterases in vivo31 and hence would not be suitable for this study. Therefore we examined a carbamate linkage which was anticipated to be more stable.31,32
A method was developed for synthesis of the haloperidol–polymer conjugate that involved activation of haloperidol with p-nitrophenyl chloroformate, prior to reaction with ethylenediamine to give haloperidol amino-ethyl carbamate (haloperidol-AEC) (Fig. 2A). An excess of ethylene diamine was used to limit coupling of haloperidol to both ends of the linker. This reaction produced the target compound in a good yield of 49%.
The subsequent conjugation strategy involved reaction of the derivatised ligand with bis-carboxy PEG through the use of widely exploited carbodiimide coupling chemistry (Fig. 2). PEG–haloperidol was successfully synthesised, as verified by 1H and 13C NMR spectroscopic analysis, which showed evidence of the characteristic peaks of haloperidol and PEG (Fig. 2B). Also, MALDI TOF analysis showed a shift in the mass distribution of the conjugates to higher masses compared to unmodified PEG, mirroring the degree of ligand loading (Fig. 2D).
Total haloperidol content was determined using 1H NMR spectroscopic analysis. For the NMR analysis, ratios were determined for the peak areas representing the aromatic protons of haloperidol between 8.2–6 ppm and the peak areas of PEG protons from 3.8–3.4 ppm (Fig. 2B). PEG–haloperidol showed a loading of 6.1% w/w. The polymer conjugate was purified by precipitation in diethyl ether, followed by dialysis against water which gave a low residual free drug content of less than 0.8% w/w (of total drug), determined by RP-HPLC.
2.2. Stability of conjugate in plasma
For our proposed strategy to be effective, formation of a stable bond between the drug and the polymer is essential, as a biodegradable bond would result in drug release and subsequent crossing of the BBB, thus defeating the purpose of conjugation. Conjugate stability was tested in vitro upon incubation with rat plasma in order to probe stability under physiological conditions. Stability was assessed using HPLC to measure the amount of haloperidol released after incubation for varying lengths of time. Pleasingly, the conjugate proved stable when challenged with biological fluids. In particular PEG–haloperidol showed release of only 1% free haloperidol at 72 h, increasing up to only 2% after 1 week (Fig. 3). Interestingly, it was observed that the point of cleavage was at the carbamate bond in the haloperidol-AEC structure, shown by RP-HPLC with the release corresponding to free haloperidol, rather than free haloperidol-AEC (data not shown).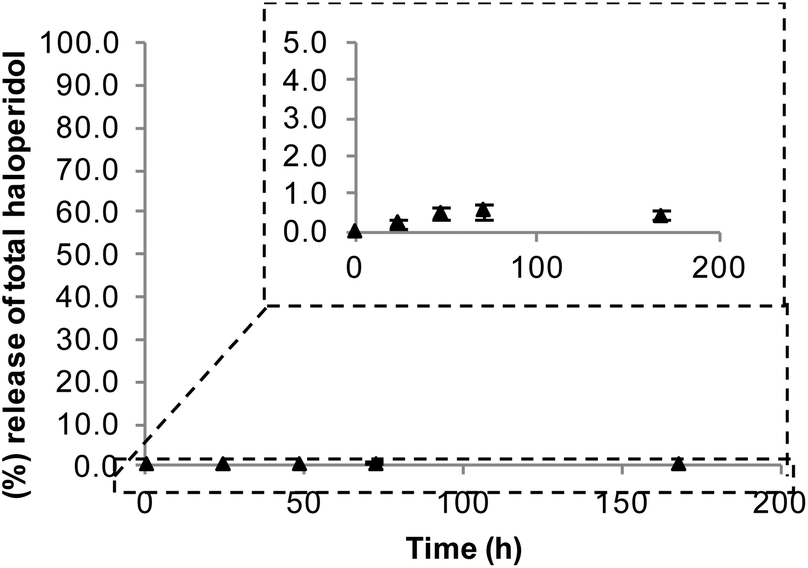 | ||
| Fig. 3 Stability of PEG–haloperidol conjugate on incubation with rat plasma. Data expressed as mean ± SEM (n = 3). Error bars hidden by the symbols when not visible. | ||
2.3. Assessment of the binding to the dopamine D2 receptor in vitro
The ability of the ligands to maintain some binding to D2 receptors after conjugation to PEG was crucial for our hypothesis to be viable, and hence worthy of future in vivo studies. The vast majority of polymer–drug conjugates rely on drug release to trigger biological effects. Therefore, it was essential to determine whether affinity of the conjugate for the D2 receptor was retained, and this was assessed by quantifying PEG–haloperidol's ability to displace the [3H] spiperone agonist binding in membrane preparations from Chinese hamster ovary (CHO) cells stably expressing D2 receptors. Dopamine was used as a positive control (pKi = 6.22 ± 0.09, n = 3) and PEG as a negative control (Fig. 4a). | ||
| Fig. 4 Binding curves for compounds competing with [3H] spiperone in D2 receptors from CHO cells: (a) PEG, negative control and dopamine, positive control, (b) PEG–haloperidol and free haloperidol. Data expressed as the mean ± SEM, (n = 3). pKi values were derived from data fitted to a single-binding site model. | ||
PEG–haloperidol was able to retain affinity for the D2 receptor, albeit with a lower pKi (6.92 ± 0.06, n = 3) compared to free haloperidol (8.05 ± 0.19, n = 3) (Fig. 4b). This reduction is in line with a previous report measuring BSA-conjugated haloperidol binding to the D2 receptor13 and as the binding is still sub-micromolar, it indicates potential for future applications.
2.4. Theoretical modelling to assess the ability of PEG conjugation to restrict passage of D2 receptor ligands across the BBB
It has long been known that large molecules are generally unable to cross the BBB. In particular, a MW of 400–600 is normally considered the upper limit for crossing the BBB.33–35 As the conjugate described in this study has a MW >than 6000 g mol−1 and is therefore well above this threshold, a gross restriction in its passive diffusion across the BBB is anticipated. However, to support this further, theoretical calculations were carried out using a model, proposed by Fu et al.36 (see section 4.10). The model predicts a log value of the ratio of concentration of compound in the brain![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :blood and has been employed in order to examine the hypothesis proposed in this paper; that the conjugation of haloperidol to PEG 6000 will inhibit its ability to cross the BBB.
:blood and has been employed in order to examine the hypothesis proposed in this paper; that the conjugation of haloperidol to PEG 6000 will inhibit its ability to cross the BBB.
The model predicts that haloperidol would be able to cross the BBB, which is well known and supported by experimental evidence.37 However, conjugation of haloperidol to PEG leads to a large reduction in the blood to brain concentration ratio predicted by the model (logBB shifting from 0.11 for haloperidol to −484.32 for the conjugate, Table 1), which means an overwhelming bias towards localisation within the blood.
BB values for haloperidol and PEG–haloperidol conjugate
It should be noted that the model used by Fu et al. was derived from a training set of 78 small molecule compounds using stepwise multiple regression analysis. The leave one out procedure was used to cross validate the model. While the model was developed from a range of small molecules, Fu et al. did not apply any restrictions with respect to the validity of their system for larger molecules, and indeed, molecular weight is one of the key parameters in their equation. Conversely, Fu et al. highlight that their equation only accounts for passive diffusion and that any compounds that are affected by P-glycoprotein efflux transporter are likely to be less well predicted. However, these limitations are unlikely to affect the predictions obtained for our system because haloperidol itself is known to be an inhibitor and not a substrate for P-glycoprotein efflux38,39 and PEG is thought to also have some inhibitory properties on P-glycoprotein.40
These theoretical predictions highlight the significant change in properties (with respect to size, hydrogen bond forming potential and lipophilicity) conferred to haloperidol upon conjugation to PEG. The theoretical modelling indicates that the PEG bound haloperidol would substantially restrict the passage of the haloperidol across the BBB.
Stimulation of dopamine receptors present in either the CNS or the periphery can result in a diverse spectrum of effects depending on the location of the receptor making this system an ideal model for the proposed strategy. For example, in the brain, dopamine plays a role in the control of movement and behaviour,41–44 whereas, in the periphery, dopamine can affect the control of cardiovascular and renal function and hormone secretion.21,22,24,45 The ability to de-couple peripheral from central effects, through the proposed size exclusion mediated system, is certainly a powerful tool for investigative purposes. In addition, a conjugate able to elicit selective central or peripheral responses could potentially be used in in vivo models to mimic pathological conditions characterised by a selectively increased or decreased dopaminergic tone.
3. Conclusions
In this paper we present for the first time the synthesis and in vitro biological investigation of a PEG–haloperidol conjugate for use as an agent per se rather than in the conventional pro-drug fashion. The challenge of this study was to generate a stable derivative that would be expected to be BBB non-permeable whilst retaining some affinity for the target receptor.A PEG–haloperidol conjugate was successfully synthesised and characterised. The conjugate displayed excellent stability, upon challenge with simulated physiological conditions. Radioligand binding studies showed that PEG–haloperidol was still able to bind to the D2 receptor albeit with a lower affinity compared to the free ligand. Theoretical modelling suggested that the conjugate would dramatically restrict the passage of haloperidol across the BBB. Taken together these results indicate that the PEG–haloperidol conjugate warrants future in vivo investigation, in the context of developing a system able to selectively (peripherally or centrally) modulate the dopaminergic system.
4. Experimental section
4.1. Materials
Alpha, omega-bis-carboxy PEG (≈6000 Da) was purchased from Iris Biotech GmbH, Germany. All other chemicals and anhydrous solvents were purchased from Sigma Aldrich, apart from extra dry chloroform and sodium trifluoroacetate for MALDI-TOF-MS analysis which were purchased from Acros Organics. Other solvents were supplied by Fisher Scientific and deuterated solvents for NMR analysis were from Cambridge Isotope Laboratories. Rat plasma was obtained from whole blood taken from male Wistar rats, P > 21 (Harlan, UK), which was subsequently spun at 3, 500 rpm for 10 min and the plasma removed. Animals were humanely dispatched by a Schedule 1 (Animals Scientific Procedures Act, 1986) method prior to blood collection.4.2. Instrumentation and software
1H NMR and 13C NMR spectra were obtained using Bruker spectrometers operating at either 700 MHz or 400 MHz and processed using Topspin software. Spectra were analysed using ACD/SpecManager software Version 12.01. HPLC analysis was performed on a Hewlett Packard series 1100 instrument equipped with an ACE C18 reverse phase column (300 μm, 250 × 4.6 mm) (Hichrom, UK). MALDI-TOF spectra were obtained on a Bruker Ultraflex MALDI-TOF/TOF mass spectrometer. The MALDI data were processed using Bruker Daltonics, FlexAnalysis version 3.0 software. Radioligand competition binding assay results were analysed using GraphPad Prism software, version 5.04.4.3. Modification of haloperidol to contain an amine handle
Haloperidol (150 mg, 0.4 mmol) was dissolved in anhydrous CH2Cl2 (10 mL). To the solution, p-nitrophenyl chloroformate (160 mg, 0.8 mmol) and triethylamine (139 μL, 1.0 mmol) were added. The reaction was stirred for one hour at room temperature and then added drop wise to a solution of ethylenediamine (267 μL, 4 mmol) in DMF (20 mL). The reaction was stirred overnight and monitored by RP-HPLC. The haloperidol-amino ethyl carbamate (haloperidol-AEC) product was purified by washing with 0.1 M sodium hydrocarbonate (3 × 200 mL), followed by chromatography on a SiO2 column (30 × 2.5 cm) eluted with a chloroform–methanol mixture (90:10 to only methanol) and determined by TLC analysis (yield 49%). Successful synthesis was confirmed using HPLC and 1H and 13C NMR spectroscopic analysis. The introduction of free amine groups was verified using the 2,4,6-trintirobenzenesulfonic acid assay.46
1H NMR (CDCl3, 400 MHz): 8.04 (1H, s, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(NH)CH2), 7.93 (2H, dd, Jab = 9.0 Hz, JaF = 5.5 Hz, Ar–F), 7.22 (4H, dd, J = 9.5 Hz, J = 9.0 Hz, Ar–Cl), 7.05–7.03 (2H, m, Ar–F), 5.20–5.10 (2H, m, NH2), 3.34–3.26 (2H, m, OCO(NH)CH2CH2NH2), 3.22–3.14 (2H, m, OCO(NH)CH2CH2NH2), 2.94 (2H, t, J = 7.5 Hz, –CO(CH2)CH2CH2–), 2.84–2.75 (2H, m, –N < (CHHCH2)2 > C), 2.5–2.43 (2H, m, CH2–N < (CH2CH2)2 > C), 2.41–2.31 (2H, m, –N < (CHHCH2)2 > C), 2.01–1.87 (4H, m, –N < (CH2CH2)2 > C). 13C NMR (CDCl3, 100 MHz) 198.38 (F–Ar–COCH2–), 165.64 (d, JFC = 250 Hz, Ar–F, (ipso-C)) 154.97 (O–CO(NH)–), 143.8 (Ar–Cl, (para-C)), 133.67 (d, JFC = 3 Hz Ar–F (para-C)), 132.92 (Ar–Cl, (ipso-C)), 130.65 (d, JFC = 10.5 Hz, Ar–F (meta-C)), 128.5 (Ar–Cl (meta-C)), 125.99 (Ar–Cl (ortho-C)), 115.62 (d, JFC = 22.0 Hz, Ar–F (ortho-C)), 78.84 ((CH2)2 > C < (O–)(Ar–Cl)), 57.67 (CH2CH2N<), 49.27 (N < (CH2)2 (CH2)2), 43.33 (CH2CH2NH2), 41.75 (NHCH2CH2), 36.20 (CO(CH2)CH2), 35.92 ((CH2)2 (CH2)2 > C<), 21.9 (CH2CH2CH2).
O(NH)CH2), 7.93 (2H, dd, Jab = 9.0 Hz, JaF = 5.5 Hz, Ar–F), 7.22 (4H, dd, J = 9.5 Hz, J = 9.0 Hz, Ar–Cl), 7.05–7.03 (2H, m, Ar–F), 5.20–5.10 (2H, m, NH2), 3.34–3.26 (2H, m, OCO(NH)CH2CH2NH2), 3.22–3.14 (2H, m, OCO(NH)CH2CH2NH2), 2.94 (2H, t, J = 7.5 Hz, –CO(CH2)CH2CH2–), 2.84–2.75 (2H, m, –N < (CHHCH2)2 > C), 2.5–2.43 (2H, m, CH2–N < (CH2CH2)2 > C), 2.41–2.31 (2H, m, –N < (CHHCH2)2 > C), 2.01–1.87 (4H, m, –N < (CH2CH2)2 > C). 13C NMR (CDCl3, 100 MHz) 198.38 (F–Ar–COCH2–), 165.64 (d, JFC = 250 Hz, Ar–F, (ipso-C)) 154.97 (O–CO(NH)–), 143.8 (Ar–Cl, (para-C)), 133.67 (d, JFC = 3 Hz Ar–F (para-C)), 132.92 (Ar–Cl, (ipso-C)), 130.65 (d, JFC = 10.5 Hz, Ar–F (meta-C)), 128.5 (Ar–Cl (meta-C)), 125.99 (Ar–Cl (ortho-C)), 115.62 (d, JFC = 22.0 Hz, Ar–F (ortho-C)), 78.84 ((CH2)2 > C < (O–)(Ar–Cl)), 57.67 (CH2CH2N<), 49.27 (N < (CH2)2 (CH2)2), 43.33 (CH2CH2NH2), 41.75 (NHCH2CH2), 36.20 (CO(CH2)CH2), 35.92 ((CH2)2 (CH2)2 > C<), 21.9 (CH2CH2CH2).
4.4. Activation of bis-carboxy PEG with N-hydroxysuccinimide
Bis-carboxy-PEG (Mw ∼ 6000, 1 g, 167 μmol) was dissolved in anhydrous CH2Cl2 (∼20 mL). To this was added, N-hydroxysuccinimide (NHS) (3 equiv., 64 mg, 555 μmol), followed by N,N′-dicyclohexylcarbodiimide (DCC) (6.45 equiv., 0.222 g, 1.1 mmol). The reaction was left stirring for 5 hours. Within 30 minutes the reaction had turned cloudy. The reaction mixture was filtered through celite and the filtrate directly added into cold diethyl ether (500 mL) with vigorous stirring. This precipitated material was left on ice for 1 h, before filtering to recover the white precipitate. The recovered solid was dried under vacuum. The degree of end group activation of the polymer was determined indirectly by measuring the amount of free amine groups remaining after reaction of the activated PEG with an equimolar quantity of the di-peptide gly–gly in borate buffer at pH 8, using the colorimetric 2,4,6-trinitrobenzene sulfonic acid assay.464.5. Synthesis of PEG–haloperidol conjugate
Haloperidol-AEC (7 equiv., 64 mg, 139 μmol) was dissolved in anhydrous CHCl3 (5 mL) and anhydrous N,N-diisopropylethylamine (DIPEA) (25 μL, 139 μmol, 7 equiv.) was added. NHS-activated PEG (Mw 6308 equiv., 112 mg, 17 μmol) was added portion-wise and the reaction then stirred under a nitrogen atmosphere, protected from light for 3 days. The reaction was monitored in the early stages using RP-HPLC. The solvent was then evaporated in vacuo, to leave ∼3 mL, before adding this dropwise to ice cold diethyl ether (200 mL). The solution was kept on ice for one hour before filtering to recover the fine white precipitate. The solid was then dried in vacuo before dissolving in water (∼10 mL). This was dialysed against distilled water, using a regenerated cellulose membrane of MWCO 1000 Da, with 3 changes of water over a 24 hour period. The product was freeze dried and subjected to RP-HPLC to determine purity. A small amount of free haloperidol-AEC was still present, which resulted in the product needing further purification. PEG–haloperidol-AEC was re-dissolved in around 4 mL of methanol and loaded on to a LH-20 sephadex column. The column was run using 100% methanol and fractions containing UV active material (as determined via TLC analysis) were analysed by HPLC. Fractions containing the conjugate were combined, and the methanol removed in vacuo. The residue was dissolved in distilled water and freeze dried once again (yield, 24%). The conjugate was characterised by 1H and 13C-NMR spectroscopic analysis, MALDI-TOF mass spectrometry, RP-HPLC and UV absorbance spectrophotometry. The amount of haloperidol bound to the polymer and free drug was determined using 1H NMR spectroscopic analysis and RP-HPLC.
1H NMR (CDCl3, 500 MHz): 8.03 (dd, haloperidol-AEC, Jab = 7.5 Hz, JaF = 4.5 Hz, Ar–F), 7.33–7.29 (m, haloperidol, Ar–Cl), 7.15–7.12 (m, haloperidol, Ar–F), 3.8–3.4 (m, PEG, CH2CH2O), 3.33–3.29 (m, haloperidol-AEC NHCH2CH2NH), 3.21–3.15 (m, haloperidol-AEC), 2.60–2.50 (m, PEG and haloperidol-AEC), 2.25–2.00 (m, haloperidol-AEC). 13C NMR (CDCl3, 125 MHz) 173.16 (PEG, –CH2CO(OH)), 172.93 (PEG, NHCO(CH2)–), 165.82 (d, JFC = 253.0 Hz, haloperidol-AEC, Ar–F, (ipso-C)), 154.92 (haloperidol-AEC, –OCO(NH)–), 133.25 (haloperidol-AEC, Ar–Cl, (ipso-C)), 130.76 (d, JFC = 9.5 Hz, haloperidol-AEC, Ar–F (meta-C)), 128.64 (haloperidol-AEC, Ar–Cl (meta-C)), 126.04 (haloperidol-AEC, Ar–Cl (ortho-C)), 115.75 (haloperidol-AEC, d, JFC = 20.5 Hz, Ar–F (ortho-C)), 72.57 (haloperidol-AEC, (CH2)2 > C < (O)(Ar–Cl)), 70.55 (PEG, –(OCH2CH2)n), 56.83 (haloperidol-AEC, (CH2CH2N<)), 48.62 (haloperidol-AEC, N < (CH2)2 (CH2)2), 40.23 (PEG, NHCH2CH2 (OCH2CH2)n), 39.6 (haloperidol-AEC, (CH2)2(CH2)2 > C<), 39.24 (haloperidol-AEC,-NHCH2CH2NH–), 39.24 (haloperidol-AEC, –NHCH2CH2NH–), 36.09 (haloperidol-AEC, CO(CH2)CH2), 32.13 (PEG, COCH2CH2CO), 31.95 (PEG, COCH2CH2CO(OH)), 29.69 (PEG, –CH2CH2CO(OH)), 28.13 (haloperidol-AEC, CH2CH2CH2).
4.6. RP-HPLC analysis of PEG–haloperidol
PEG–haloperidol was analysed using RP-HPLC with a C18 (300 μm, 250 × 4.6 mm) column at a flow rate of 1 mL min−1. The eluent was: A; acetonitrile and B; water containing 0.25% acetic acid. The protocol for analysis of PEG–haloperidol comprised UV absorbance detection at 254 nm, using a gradient of 10% A increasing to 70% A over 25 minutes, returning to 10% A in the following 10 minutes with a total run time of 40 minutes.4.7. MALDI-TOF
For MALDI-TOF analysis sample solutions at a concentration of 5 mg mL−1 at a 1:1:1 ratio with dithranol (40 mg mL−1, THF) and sodium trifluoroacetate (10 mg mL−1, THF) as matrix were employed. Linear positive mode was used on a Bruker Ultraflex MALDI-TOF/TOF mass spectrometer with an acceleration voltage of 25 kV.
4.8. Stability of PEG–haloperidol under physiological conditions
Stability studies of PEG–haloperidol were carried out in rat plasma. Briefly, 22 μL of either free haloperidol or PEG–haloperidol conjugate solution was added to 200 μL of rat plasma, before vortexing for 1 min to ensure complete mixing.200 μl of rat plasma was aliquoted out, and to this 22 μl, of either, free haloperidol or PEG–haloperidol conjugate, solution was added. This was vortexed for 1 min to ensure complete mixing.
Mixtures were then divided into five equal aliquots for analysis via HPLC at 0 h, 48 h, 24 h, 72 h and 168 h. The samples were incubated at 37 °C to mimic physiological temperatures. At the appropriate time point samples were removed from incubation and quenched with 160 μl of, 1:1, acetonitrile:methanol. This was then vortexed for 1 min, followed by centrifugation at 3500 rpm for 2 min. This supernatant was removed and filtered through a 0.45 μm syringe filter. The filtered supernatant was then analysed via HPLC, using the elution programme outlined in section 4.6.
4.9. Radioligand binding studies
Cell membranes (25 μg), of cells from CHO expressing the D2 receptor, were incubated with [3H]spiperone (0.25 nM) and competing drugs in HEPES buffer (20 mM HEPES, 1 mM EGTA, 1 mM EDTA, 10 mM MgCl2, pH 7.4 (using HCl) containing 0.1 mM dithiothreitol) in a final volume of 1 ml for 3 h at 25 °C. The assay was terminated by rapid filtration (through Whatman GF/C filters) using a Brandel cell harvester, followed by four washes with 4 ml ice-cold phosphate-buffered saline (0.14 M NaCl, 3 mM KCl, 1.5 mM KH2PO4, 5 mM Na2HPO4; pH 7.4) to remove unbound radioactivity. Filters were soaked in 2 ml of scintillation fluid for at least 5 h and bound radioactivity was determined by liquid scintillation counting. Non-specific binding of [3H] spiperone was determined in the presence of 3 μM (+)-butaclamol. The data was analysed using GraphPad Prism software, version 5.04 and fitted to equations describing a one-site binding model. pKi values for the data were obtained from non-linear regression.4.10. Theoretical modelling to assess the ability of PEG conjugation to restrict passage haloperidol across the BBB
The following equation proposed by Fu et al.36 was used to predict the BBB penetration of the free ligands and PEG-ligand conjugates.| logBB = −9.880 × 10−6Mw2 + 7.339 × 10−3Mw − 0.2268npol − 0.1143 | (1) |
The components of the model are defined as follows: logBB = logarithm of the ratio of the steady state concentration of the compound in the brain to the concentration of compound in the blood, Mw = molecular weight, npol = the number of polar atoms (oxygen, nitrogen and attached hydrogen).
The model was developed using 86 compounds, of which 8 were removed as outliers. The correlation coefficient for the 78 remaining compounds was, r2 = 0.74.36
Acknowledgements
We would like to thank the Engineering and Physical Sciences Research Council (EPSRC Grant EP/H021477/1) for financial support. The authors also thank Peter Heath for help with the acquisition of NMR spectra, Dr Olga Khutoryanskaya for assistance with the conjugate stability studies and Dr Davinia Mills for support with the MALDI-TOF analysis. We would also like to thank the University of Reading for use of the Chemical Analysis Facility.References
- J. A. Borovac, Yale J. Biol. Med., 2016, 89, 37–47 Search PubMed.
- P. Dietrichson, J. Presthus and R. Holmsen, Eur. Neurol., 1975, 13, 339–349 CrossRef CAS PubMed.
- F. E. R. Simons, Clin. Exp. Allergy, 1999, 29, 125–132 CrossRef CAS PubMed.
- I. Hindmarch, Z. Shamsi and S. Kimber, Clin. Exp. Allergy, 2002, 32, 133–139 CrossRef CAS PubMed.
- N. J. Abbott, J. Inherited Metab. Dis., 2013, 36, 437–449 CrossRef CAS PubMed.
- W. M. Pardridge and L. J. Mietus, J. Clin. Invest., 1979, 64, 145–154 CrossRef CAS PubMed.
- W. M. Pardridge, Drug Discovery Today, 2007, 12, 54–61 CrossRef CAS PubMed.
- R. Satchi-Fainaro, M. Puder, J. W. Davies, H. T. Tran, D. A. Sampson, A. K. Greene, G. Corfas and J. Folkman, Nat. Med., 2004, 10, 255–261 CrossRef CAS PubMed.
- JAMA, J. Am. Med. Assoc., 2016, 315, 194–195 Search PubMed.
- J. Yang and J. Kopecek, J. Controlled Release, 2014, 190, 288–303 CrossRef CAS PubMed.
- T. W. Chu and J. Kopecek, Biomater. Sci., 2015, 3, 908–922 RSC.
- W. A. Banks, BMC Neurol., 2009, 9, S3 CrossRef PubMed.
- R. A. Williamson, S. Worrall, P. L. Chazot and P. G. Strange, EMBO J., 1988, 7, 4129–4133 CAS.
- T. Etrych, J. Strohalm, M. Šírová, B. Tomalová, P. Rossmann, B. Říhová, K. Ulbrich and M. Kovář, Polym. Chem., 2015, 6, 160–170 RSC.
- F. M. Veronese, O. Schiavon, G. Pasut, R. Mendichi, L. Andersson, A. Tsirk, J. Ford, G. Wu, S. Kneller, J. Davies and R. Duncan, Bioconjugate Chem., 2005, 16, 775–784 CrossRef CAS PubMed.
- F. Greco and M. J. Vincent, Front. Biosci., 2008, 13, 2744–2756 CrossRef CAS.
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688–701 CrossRef CAS PubMed.
- A. S. Mikhail and C. Allen, Biomacromolecules, 2010, 11, 1273–1280 CrossRef CAS PubMed.
- H. Hall, G. Sedvall, O. Magnusson, J. Kopp, C. Halldin and L. Farde, Neuropsychopharmacol., 1994, 11, 245–256 CrossRef CAS PubMed.
- N.-E. Anden, A. Carlsson, A. Dahlstrom, K. Fuxe, N.-A. Hillarp and K. Larsson, Life Sci., 1964, 3, 523–530 CrossRef CAS PubMed.
- J.-M. Beaulieu and R. R. Gainetdinov, Pharmacol. Rev., 2011, 63, 182–217 CrossRef CAS PubMed.
- C. C. Felder, A. M. Mckelvey, M. S. Gitler, G. M. Eisner and P. A. Jose, Kidney Int., 1989, 36, 183–193 CrossRef CAS PubMed.
- A. Eliassi, F. Alealli and T. Ghasemi, Clin. Exp. Pharmacol. Physiol., 2008, 35, 1065–1070 CrossRef CAS PubMed.
- C. Missale, S. Russell Nash, W. W. Robinson, M. Jaber and M. G. Caron, Physiol. Rev., 1998, 78, 189–225 CAS.
- W. B. Liechty, D. R. Kryscio, B. V. Slaughter and N. A. Peppas, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 149–173 CrossRef CAS PubMed.
- G. Pasut and F. M. Veronese, Prog. Polym. Sci., 2007, 32, 933–961 CrossRef CAS.
- M. Hans, K. Shimoni, D. Danino, S. J. Siegel and A. Lowman, Biomacromolecules, 2005, 6, 2708–2717 CrossRef CAS PubMed.
- M. L. Hans, C. Maxwell, R. S. Ehrlichman, K. Metzger, Y. Liang, S. J. Siegel and A. M. Lowman, J. Biomed. Mater. Res., Part B, 2007, 83B, 422–430 CrossRef CAS PubMed.
- A. Mukherjee, T. K. Prasad, N. M. Rao and R. Banerjee, J. Biol. Chem., 2005, 280, 15619–15627 CrossRef CAS PubMed.
- K. Pal, S. K. Pore, S. Sinha, R. Janardhanan, D. Mukhopadhyay and R. Banerjee, J. Med. Chem., 2011, 54, 2378–2390 CrossRef CAS PubMed.
- F. Bettio, M. Canevari, C. Marzano, F. Bordin, A. Guiotto, F. Greco, R. Duncan and F. M. Veronese, Biomacromolecules, 2006, 7, 3534–3541 CrossRef CAS PubMed.
- F. M. H. de Groot, L. W. A. Van Berkom and H. W. Sheeren, J. Med. Chem., 2000, 43, 3093–3102 CrossRef CAS PubMed.
- V. A. Levin, J. Med. Chem., 1980, 23, 682–684 CrossRef CAS PubMed.
- W. M. Pardridge, Adv. Drug Delivery Rev., 1995, 15, 5–36 CrossRef CAS.
- M. M. Almutairi, C. Gong, Y. G. Xu, Y. Chang and H. Shi, Cell. Mol. Life Sci., 2016, 73, 57–77 CrossRef CAS PubMed.
- X.-C. Fu, G.-P. Wang, H.-L. Shan, W.-Q. Liang and J.-Q. Gao, Eur. J. Pharm. Biopharm., 2008, 70, 462–466 CrossRef CAS PubMed.
- F. Lopez-Munoz and C. Alamo, Brain Res. Bull., 2009, 79, 130–141 CrossRef CAS PubMed.
- K. Iwaki, T. Sakaeda, M. Kakumoto, T. Nakamura, C. Komoto and N. Okamura, et al. , J. Pharm. Pharmacol., 2006, 58, 1617–1622 CrossRef CAS PubMed.
- J. S. Wang, H. J. Zhu, J. S. Markowitz, J. L. Donovan, H. J. Yuan and C. L. DeVane, Basic Clin. Pharmacol. Toxicol., 2008, 103, 336–341 CrossRef CAS PubMed.
- Q. Shena, Y. Lina, T. Handaa, M. Doia and M. Sugiea, et al. , Int. J. Pharm., 2006, 313, 49–56 CrossRef PubMed.
- M. W. Howe and D. A. Dombeck, Nature, 2016, 535, 505–510 CrossRef CAS PubMed.
- D. R. Sibley, Annu. Rev. Pharmacol. Toxicol., 1999, 39, 313–341 CrossRef CAS PubMed.
- S. D. Iversen and L. L. Iversen, Trends Neurosci., 2007, 30, 188–193 CrossRef CAS PubMed.
- R. A. Bressen and J. A. Crippa, Acta. Pyschiatr. Scand., 2005, 111, 14–21 CrossRef PubMed.
- J. J. Gildea, I. T. Shah, R. E. Van Sciver, J. A. Israel, C. Enzensperger, H. E. McGrath, P. A. Jose and R. A. Felder, Kidney Int., 2014, 86, 118–126 CrossRef CAS PubMed.
- S. L. Snyder and P. Z. Sobocinski, Anal. Biochem., 1975, 64, 284–288 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |