
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the properties of pH responsive nanoparticles to control cellular interactions in vitro and ex vivo†
S. K.
Mann
abc,
A.
Dufour
a,
J. J.
Glass
 de,
R.
De Rose
de,
S. J.
Kent
def,
G. K.
Such
*b and
A. P. R.
Johnston
*ac
de,
R.
De Rose
de,
S. J.
Kent
def,
G. K.
Such
*b and
A. P. R.
Johnston
*ac
aDrug Delivery, Disposition and Dynamics, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, Victoria 3052, Australia. E-mail: angus.johnston@monash.edu
bDepartment of Chemistry, The University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: gsuch@unimelb.edu.au
cARC Centre of Excellence in Convergent Bio-Nano Science and Technology, Monash University, Parkville, Victoria 3052, Australia
dDepartment of Microbiology and Immunology, The Peter Doherty Institute for Infection and Immunity, The University of Melbourne, Victoria 3000, Australia
eARC Centre of Excellence in Convergent Bio-Nano Science and Technology, The University of Melbourne, Melbourne, Victoria 3010, Australia
fMelbourne Sexual Health Centre, Alfred Hospital, Central Clinical School, Monash University, Prahran, Australia
First published on 12th September 2016
Abstract
Engineering the properties of nanoparticles (NPs) to limit non-specific cellular interactions is critical for developing effective drug delivery systems. In this study we investigate the differences in non-specific cell association between polymer NPs prepared with linear polyethylene glycol (PEG) and brush PEG both in vitro and ex vivo. Most studies to investigate the non-fouling properties of NPs have been performed using cell-line based assays. However, in this study we demonstrate a whole blood assay using fresh human blood. It is likely this assay reflects more accurately the fate of NPs when injected into human blood in vivo. Non-linear PEG analogues such as poly(poly(ethylene glycol)methacrylate) (PEGMA) are attractive alternatives to linear PEG as hydrophilic coatings for NP drug delivery systems due to their simple and versatile synthesis. We prepared NPs composed of a poly(2-diethylamino)ethyl methacrylate (PDEAEMA) core and a diblock copolymer of PDEAEMA and either linear PEG or brush PEGMA. These NPs depend on low-fouling properties of the hydrophillic PEG coating to avoid uptake by the mononuclear phagocyte system (MPS). In vitro cell association assays showed brush PEGMA NPs exhibited lower association with 3T3 fibroblast and C1R lymphoblast cells compared to linear PEG NPs. In an ex vivo whole blood assay, brush PEGMA nanoparticles showed similar low association with monocytes and granulocytes as linear PEG NPs with a similar length PEG component. Higher association with blood cells was observed for NPs containing a lower molecular weight PEGMA component, despite having the same molecular weight as the linear PEG NPs (2 kDa). The results demonstrate that trends observed in cell-lines are not always consistent with assays in more complex systems such as blood. Based on these results the reported PEGMA NPs are attractive alternatives to our previously reported linear PEG NPs.
Introduction
Polymeric nanoparticles (NPs) have generated a lot of interest in the field of drug-delivery due to their ability to encapsulate drugs, release the drug in a controlled fashion and their potential for cellular targeting.1–5 For this field to evolve, a greater understanding of the interactions between nanomaterial surfaces and biological systems is required. A major barrier to the widespread medical application of NPs is the undesirable interaction with blood. Primarily, non-specific binding to opsonins (blood-based proteins) has resulted in a low half-life in biological systems, due to uptake by the mononuclear phagocyte system (MPS).6 This removes NPs from the circulation and promotes accumulation in the liver and spleen.Imparting a hydrophilic shielding on the surface of these drug-delivery systems can reduce uptake by the MPS. The most widely used hydrophilic polymer is polyethylene glycol (PEG).7 The PEG chains create a protective barrier, reducing charge-based interactions with proteins and preventing NP–NP interactions.8 By avoiding opsonization, the NPs are less visible to phagocytic cells resulting in a longer blood circulation half-life and increased chance of the NP reaching its therapeutic site of action. Many studies have been performed on the effect of PEG density and alternative hydrophilic coatings for NPs to gain greater understanding on the best anti-fouling properties. Most commonly, cell-line based assays have been used to study NP biological interactions and compare levels of NP association as they are convenient and reproducible, however there is huge variability between cell-line based assays reported in the literature (e.g. species, cell-type, assay conditions etc.). Furthermore, the biological interactions of cells may differ between cell-line assays and more complex environments such as human blood. For nanomedicines to be administered intravenously, an ex vivo human whole blood assay is most representative of the NP's environment after injection.
We have previously reported a ‘pHlexi’ nanoparticle system composed of a 2-(Diethylamino)ethyl methacrylate (DEAEMA) homopolymer core and a PEG-b-PDEAEMA copolymer (Fig. 1).9 We hypothesize that above their pKa, the PDEAEMA components drive the assembly of a hydrophobic core, whilst the PEG extends into the solution to form a stabilizing hydrophilic shell. These NPs were engineered to degrade in the early endosomal compartments of the cell, and we showed they were able to induce endosomal escape of endocytosed molecules. While linear PEG is commonly used to engineer low-fouling surfaces, another attractive alternative is brush PEG, composed of a methacrylate backbone and multiple PEG side-chains. A significant advantage of brush PEG is that the length of the PEG side-chains can be tuned as can the length of repeat units by employing controlled polymerization techniques including reversible addition fragmentation chain transfer (RAFT)10 and atom transfer radical polymerization (ATRP).11 These approaches also allow synthesis of controlled PEG-based polymers such as amphiphilic block copolymers,12 random copolymers,13,14 biopolymer hybrids15,16 and modified organic or inorganic surfaces.17 In comparison, the preparation of linear PEG by the anionic polymerization of ethylene oxide is more synthetically challenging. Furthermore, non-linear PEG analogues synthesized by controlled polymerization approaches enable the possibility of post-polymerization modification of reactive end groups.18,19 Additionally, non-linear architectures can be used to increase the number of functional groups present on a single polymer chain by modification of the brush PEG side chains. Poly(ethylene glycol methacrylate) (PEGMA) surfaces have been shown to display non-fouling properties and the Chilkoti group has demonstrated that PEGMA brushes are resistant to protein absorption.20,21 PEGMA NPs are therefore particularly advantageous for the incorporation of targeting groups that we plan to include in future generation pHlexi NPs. While there are many advantages to the use of brush PEGMA NPs, it is important that we understand the impact of this different architecture on the association of the particles with cells.
 | ||
| Fig. 1 Structures of (a) PDEAEMA homopolymer, (b) linear PEG-block-PDEAEMA and (c) brush PEGMA-block-PDEAEMA. Schematic representations of PEG NP surfaces with (d) linear PEG and (e) brush PEGMA. | ||
In this study, we compare the properties of the linear PEG particle to similar particles synthesized containing brush PEGMA (Fig. 1). We investigate the effect of PEG architecture on NP properties, cell line association and phagocytic blood cell association. We demonstrate that both linear PEG and brush PEGMA are suitable for the formation of self-assembling core–shell NPs, having similar size, pH responsiveness, and cytotoxic behavior compared to linear PEG NPs. In vitro association assays demonstrate brush PEGMA NPs exhibit lower non-specific association with 3T3 fibroblast and C1R lymphoblast cells. In blood interaction assays, PEG and PEGMA of a similar length (∼45 monomer units) showed low non-specific binding to monocytes and granulocytes, whilst PEGMA NPs with similar molecular weight to the linear PEG (2 kDa), resulted in higher non-specific association with blood cells. The differences between the two assays demonstrate that we need better models to study NP biological interactions than typical cell line based assays. Furthermore, the blood assay was used to study differences between multiple donors and illustrated key differences depending on the donor patient. These types of studies cannot be done using typical cell line analyses. This study gives us greater understanding about the NP's interaction with the ex vivo environment and thus provides a valuable tool in the design of more effective delivery systems.
Experimental
Materials
2-(Diethylamino)ethyl methacrylate (DEAEMA) (Sigma-Aldrich, 99%) and poly(ethylene glycol methacrylate) (PEGMA) (Sigma-Aldrich, average Mn 300 Da) were passed over aluminium oxide (activated, basic; Sigma-Aldrich) to remove inhibitors prior to use. 4-Cyano-4-[(dodecylsulfanyl thiocarbonyl)sulfanyl]pentanoic acid (Sigma-Aldrich, 97%), poly(ethylene glycol) 4-cyano-4-(phenyl carbonothioylthio)pentanoate (Sigma-Aldrich, 98%, average Mn 2000 Da) and all solvents were used as received. Pentafluorophenyl methacrylate (PFPMA) was synthesized according to the method given in the ESI.† Cyanine-5 amine was purchased from Lumiprobe.Cell lines
3T3 mouse embryonic fibroblast wild type cells (3T3 MEFs WT, ATCC CRL-2752) and human C1R cells (ATCC CRL-1993) were cultured in DMEM medium supplemented with 10% fetal bovine serum, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin.Polymer synthesis
All polymers were synthesized by RAFT polymerization. The monomer: RAFT agent: initiator ratios and polymerization times are outlined in Table S1, ESI.† Poly(ethylene glycol)-b-poly(2-(diethylamino)ethyl methacrylate) (PEG-b-PDEAEMA) was prepared from poly(ethylene glycol) 4-cyano-4-(phenylcarbonothioylthio)pentanoate (average Mn 2000 Da) as previously described.9 The molecular weight of the PDEAEMA component was determined by 1H NMR analysis to be approximately 17 kDa. Poly(2-(diethylamino)ethyl methacrylate) (PDEAEMA) was prepared from 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid as previously described.9 The molecular weight was determined by 1H NMR analysis to be approximately 36 kDa.Example synthesis of poly(ethylene glycol methacrylate) (PEGMA) macro RAFT agent
Poly(ethylene glycol methacrylate) (PEGMA; average Mn 300 Da) (2.71 g, 9.03 mmol), azobisisobutyronitrile (AIBN) (0.93 mg, 6.0 μmol) and 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid (20.7 mg, 51.3 μmol) were dissolved in 1,4-dioxane (7.4 g) and placed into a Schlenk flask equipped with a magnetic stirrer bar. The flask was degassed over three freeze–pump–thaw cycles. The reaction mixture was stirred in an oil bath at 60 °C for 18 h. The reaction was terminated by exposure to air. The polymer was purified by precipitation from cold hexane twice and dried under reduced pressure. 1H NMR (400 MHz; CDCl3): δ 4.07 (–COO–CH2–CH2–), 3.65 (PEG backbone), 3.54 (–COO–CH2–CH2–), 3.37 (–O–CH3), 1.90–1.75 (backbone –CH2–C–CH3), 1.24 (CH3–C8H16–CH2–), 1.05–0.82 (backbone –CH2–C–CH3).Example synthesis of poly(ethylene glycol methacrylate)-b-poly(2-(diethylamino)ethyl methacrylate) (PEGMA-b-PDEAEMA)
DEAEMA (783 mg, 4.23 mmol), AIBN (0.63 mg, 3.9 μmol) and PEGMA8 macro-RAFT agent (100 mg, 38.5 μmol) were dissolved in 1,4-dioxane (1.6 g) and placed into a Schlenk flask equipped with a magnetic stirrer bar. The reaction mixture was stirred in an oil bath at 60 °C for 16 h. The reaction was terminated by exposure to air. The polymer was purified by dialysis in PBS pH 6, followed by MQ-water (12 kDa MWCO membrane) and the product was lyophilized. 1H NMR (400 MHz; D2O): ∂/ppm: 4.40 (PDEAEMA –COO–CH2–CH2–), 4.18 (PEGMA –COO–CH2–CH2–), 3.79 (PEG), 3.64 (PEGMA –COO–CH2–CH2–), 3.55 (–CH2–CH2–N–), 3.40 (–O–CH3), 3.32 (–N–CH2–CH3), 1.98 (backbone –CH2–C–CH3), 1.33 (–N–CH2–CH3), 1.08 (backbone –CH2–C–CH3).Synthesis of poly(2-(diethylamino)ethyl methacrylate)-r-poly(pentalfuorophenyl methacrylate) (PDEAEMA-r-PPFPMA)
PDEAEMA was synthesized as described for the non-functionalized polymer with 1% pentafluorophenyl methacrylate (PFPMA). The conversion of PFPMA was determined to be 80% (approximately 4 PFP esters per chain) from the integrals of the polymer to monomer peaks in the crude fluorine NMR. The polymer was precipitated in water, dissolved in dichloromethane (DCM) and dried over magnesium sulphate. The polymer was further purified by precipitation in cold n-hexane. The precipitate was dried in vacuo to give PDEAEMA-r-PPFPMA as a tacky solid. The molecular weight of the polymer was determined by 1H (DEAEMA) and 19F (PPFPMA) NMR analysis to be approximately 49 kDa with 4 PFP esters per chain. 1H NMR (400 MHz; CDCl3): ∂/ppm: 3.99 (–COO–CH2–CH2–), 2.69 (–CH2–CH2–N–), 2.56 (–N–CH2–CH3), 1.90–1.78 (backbone –CH2–C–CH3–), 1.26 (CH3–C8H16–CH2–), 1.03 (–N–CH2–CH3), 0.88 (backbone –CH2–C–CH3–); 19F NMR (376 MHz; CDCl3): ∂/ppm: −149.27 (1F, ortho), −151.35 (1F, ortho′), −157.83 (1F, para), −162.11 (2F, meta).Cy5 functionalization of PDEAEMA-r-PPFPMA
PDEAEMA-co-PPFPMA (20 mg, 1.63 μmol PFP ester groups) was dissolved in 1 ml of acetonitrile. Cy5 amine (0.5 mg, 0.82 μmol) and triethylamine (1 μl, 6.6 μmol) were added. The reaction was stirred at 50 °C for 48 h. The polymer was purified by dialysis in PBS pH 6, followed by MQ-water (12k membrane) and the product was lyophilized.Synthesis of nanoparticles
PDEAEMA and PEG-b-PDEAEMA or PEGMA-b-PDEAEMA were co-dissolved into 3 ml PBS (pH 6) to a total mass of 3 mg. Cy5-labelled PDEAEMA was used in the particles for the cell line and blood association experiments. The polymer mixture was dialyzed overnight against PBS pH 8 in 3.5 kDa MWCO dialysis devices to gradually increase the pH. The resulting particles were purified by further dialysis against PBS pH 8 in 100 kDa MWCO dialysis devices. The buffer was changed six times over a 24 hour period. The NP solution was removed from dialysis and left to sit for 24 hours. The particles were filtered with a 0.45 μm polyethersulfone (PES) filter immediately prior to any experiments.pH-Dependent disassembly of nanoparticles analysis by DLS
NPs were prepared as described above. 15 μl of filtered particles was added to 100 μl of PBS adjusted to various pHs (ranging from pH 8.0 to pH 6.8 in 0.2 pH increments) for analysis by dynamic light scattering. All measurements were performed in triplicate.Alamar blue viability assay in 3T3 fibroblast and C1R cells
3T3 MEFs WT cells were seeded at 1.0 × 104 cells per well and incubated overnight (37 °C and 5% CO2). C1R cells were seeded at 1.5 × 104 cells per well. Particles were added and incubated for 4 hours. The media was replaced with fresh media containing 10 μl Alamar Blue reagent and incubated for a further 4 hours. Fluorescence was measured on a FLUOstar OPTIMA microplate reader.Assessing association of nanoparticles with 3T3 fibroblast and C1R cells
3T3 MEFs WT cells were seeded at 6.0 × 104 cells per well in 24 well plates and incubated overnight (37 °C and 5% CO2). C1R cells were seeded at 9.0 × 104 cells per well in 24 well plates. Cy5-labelled particles were added at various concentrations and incubated for 4 hours. 3T3 cells were washed twice in PBS, lifted with trypsin, centrifuged and re-suspended in PBS plus propidium iodide (PI). C1R cells were washed twice in PBS and re-suspended in PBS plus PI for analysis by flow cytometry.Assessing the localization of nanoparticles in vitro
3T3 MEFs WT cells were seeded at 1.5 × 104 cells per well in an 8 well chamber slide and late endosomes/lysosomes were labelled using CellLight Lysosomes-GFP and BacMam enhancer (Life Technologies) according to the manufacturer's instructions. NPs were added to the cells at a concentration of 5 μg mL−1 and allowed to incubate for 4 h. The cells were washed 3 times in PBS, and FluoroBrite imaging media (Thermo Fisher Scientific) containing Hoeschst stain (Thermo Fisher Scientific, 2.5 μg mL−1) was added and incubated for 5 minutes. The cells were imaged by live cell fluorescence microscopy.Nuclear magnetic resonance (NMR) spectroscopy
NMR spectra were recorded on a 400 MHz Bruker NMR using the residual proton resonance of the solvent (CDCl3 or D2O) as the internal standard.Gel permeation chromatography (GPC)
Polymer molecular weights and polydispersity were measured by gel permeation chromatography (GPC) on a Shimadzu liquid chromatography system in dimethylacetamide (containing 4.3 g L−1 lithium bromide) relative to polymethyl methacrylate standards at 80 °C at a flow rate of 1 mL min−1.Dynamic light scattering (DLS)
NP size, polydispersity, and pH-dependent degradation measurements were performed on a Horiba nanopartica SZ-100 (Horiba Scientific, Japan) operating at a fixed scattering angle of 90°. All DLS measurements were performed in triplicate.Zeta potential
NP samples were prepared by the addition of 10 μl of filtered particles to 1 ml phosphate buffer (pH 8). The zeta potential was determined using Malvern Zetasizer (Malvern Instruments, Worcestershire, UK). Zeta potential was measured in triplicate.Cryo electron microscopy (cryo-EM)
Small drops of particle solution were deposited onto copper TEM grids. TEM images were taken using a Tecnai F30 TEM with cryo tomography (FEI), and ran at 300 kV acceleration voltage.Research donor blood
Blood was collected from healthy human donors after obtaining informed consent in accordance with University of Melbourne Human ethics approval 1443420 and the Australian National Health and Medical Research Council Statement on Ethical Conduct in Human Research. Blood was drawn by venipuncture into Vacuette® (Greiner Bio-One) collection tubes containing heparin and inverted 5 times. Blood was studied within 1 h of collection.Ex vivo human blood assay
Blood (100 μl) was added to 5 ml polystyrene round-bottom tubes (Falcon), capped, and incubated at 37 °C for 15 min. NPs (10 μg mL−1) or PBS control were added to blood, mixed briefly by vortex mixer and returned to 37 °C for 90 min. Following incubation, erythrocytes were lysed with Pharm Lyse™ buffer (BD Biosciences). Samples were washed thrice with PBS, and cells phenotyped using titrated concentrations of 3 fluorescent antibodies (CD3 AF700, SP34-2; CD14 APC-H7, MΦP9; CD45 V500, HI30) for 30 min, room temperature. Antibodies were purchased from BD Biosciences. Samples were washed twice with FACS Wash Buffer [1× PBS containing 0.5% w/v bovine serum albumin (Sigma Aldrich) and 2 mM EDTA pH 8 (Ambion)] and fixed with 1% formaldehyde.Flow cytometry
NP association with 3T3 fibroblast and C1R cells was studied using a S100EXi flow cytometer (Stratedigm). Association with human blood cells was analyzed using a BD LSRFortessa (BD Biosciences). Fluorescence compensation was performed with VersaComp Antibody Capture Beads (Beckman Coulter). Gated populations of single cells were analyzed for association with Cy5-NPs (FlowJo v10) and relative fluorescence intensity (RFI) summarized using GraphPad Prism 6 (GraphPad Software).Live cell fluorescence microscopy
Live cell imaging was performed using an Olympus IX83 microscope with a 60× 1.3 NA silicone objective. The cells were imaged in an incubation chamber with 10% CO2 maintained at 37 °C.Results and discussion
Design, synthesis and characterization of nanoparticles
PEGMA-b-PDEAEMA diblock copolymers were synthesised by RAFT polymerization of PEGMA with an average degree of polymerization of 5 (Mn = 300). Polymer molecular weights were determined by NMR and GPC (Table S2†). We compared a 2 kDa linear PEG (PEG45) with a brush PEGMA of the same molecular weight (PEGMA8), and a brush PEGMA of the same length (PEGMA37). The brush PEGMA diblock polymer with the same molecular weight contains approximately the same number of ethylene glycol units as the linear PEG, however it will be shorter in length (due to the branching), leading to a thinner hydrophilic layer. We therefore also synthesised a longer brush PEGMA with the same number of monomer units (approximately 37) as a further comparison between linear and brush PEGs. The NPs are subsequently referred to by the number of repeating PEG/PEGMA units: PEG45, PEGMA8 and PEGMA37. The PDEAEMA component of the diblock polymers was approximately 17 kDa for all NPs (Table S2†). Previously our group reported a method to form NPs by a nano-precipitation technique.9 We have developed this method further to synthesise NPs in an organic-solvent free assembly by changing the pH of the assembly solution. This aqueous nanoprecipitation is ideal for encapsulating water-soluble cargo such as DNA and proteins, whereas the ethanol precipitation is better suited to hydrophobic cargo. In aqueous method, the diblock copolymer and pH responsive PDEAEMA core polymer are co-dissolved into PBS (pH 6) and dialyzed against PBS at pH 8 to induce particle formation. The ratio of the two polymer components was varied in order to keep the PEG content of the NPs constant at 5% (w/w).The NPs were characterized by dynamic light scattering (DLS; Table 1). The linear PEG and brush PEGMA NPs were all similar in size, ranging from 110–150 nm. The dispersity of all particles was low (<0.18). NPs showed slightly negative zeta potential (∼−5 mV) with no significant difference between zeta potential values of the NPs of different PEG surfaces. Cryo-EM images of the NPs show similar sizes to the DLS analysis (Fig. 2). A minor second population of smaller NPs (∼20–50 nm) can be seen in cryo-EM images for the two PEGMA samples that were not detected by DLS analysis. These are thought to be single component micelles from the PEGMA block copolymers. As only the PDEAEMA component of the NPs is fluorescently labelled, binding of micelles to cells will not be detected in the cell association assay.
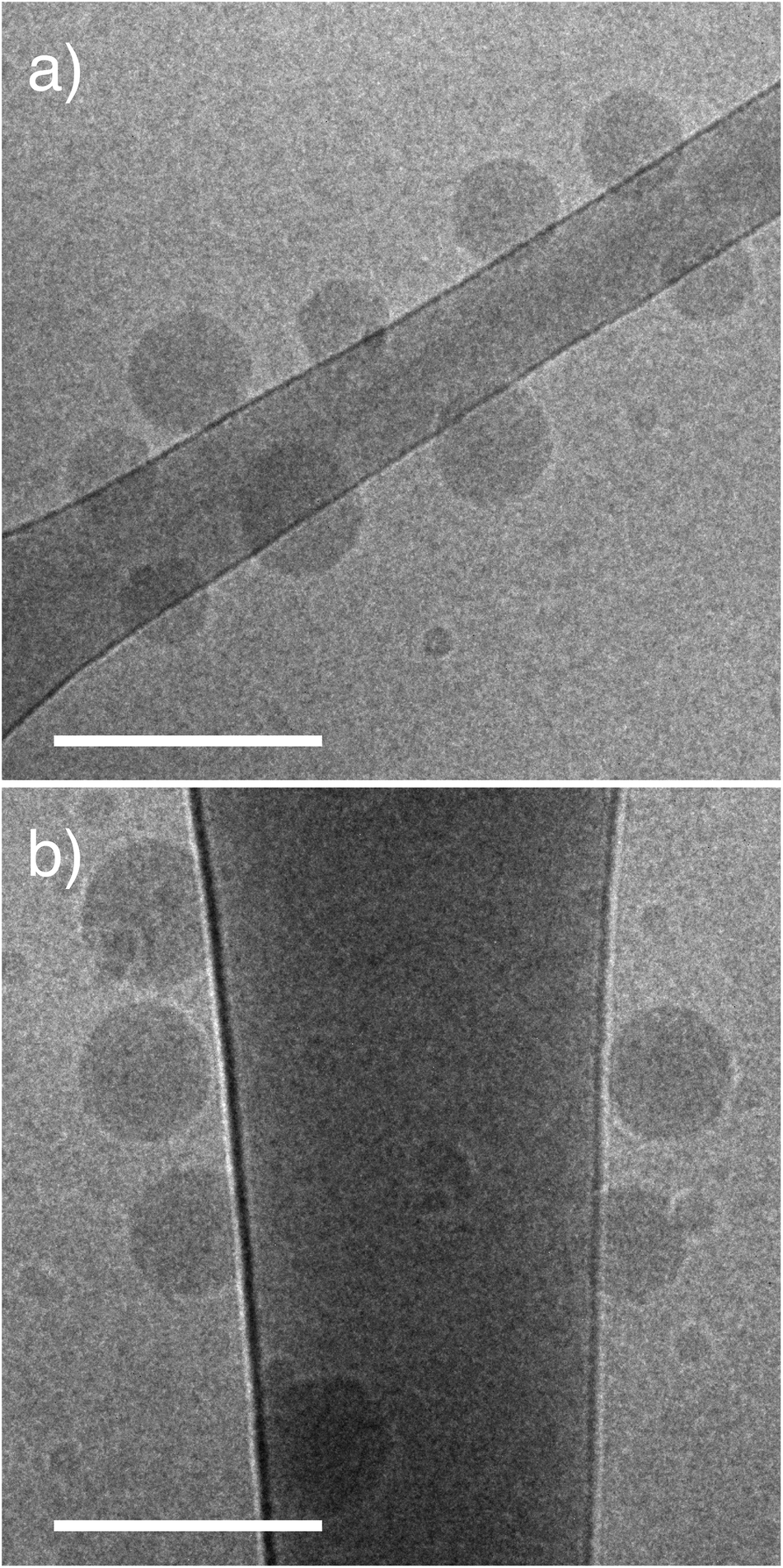 | ||
| Fig. 2 Brush PEGMA pHlexi particles form spherical structures. Representative cryo-TEM images of brush PEGMA NPs. (a) PEGMA8 and (b) PEGMA37. Scale bar represents 200 nm. | ||
| Sample | Sizea (nm) | PDIa | ζb (mV) |
|---|---|---|---|
| a NP size and polydispersity index (PDI) were measured in PBS pH 8 at 37 °C. b Zeta potential (ζ) was measured in phosphate buffer pH 8 at 25 °C. (n = 3, reported as mean ± standard deviation). | |||
| Linear PEG45 | 135 ± 8 | 0.16 | −3 ± 1 |
| Brush PEGMA8 | 110 ± 3 | 0.18 | −5 ± 1 |
| Brush PEGMA37 | 150 ± 6 | 0.08 | −3 ± 1 |
Cellular behavior of nanoparticles
In order for NPs to be useful for drug delivery applications they must be stable at physiological pH (7.4) and ideally dissociate rapidly at the pH of the endosomes/lysosomes. To investigate this, the size of the NPs was measured by DLS in the pH range 6.8 to 8.0 at 37 °C. As the pH is decreased the PDEAEMA core becomes protonated causing the charged amine groups to repel each other and induce disassembly of the NPs. All of the NPs completely disassembled at pH 7 (Fig. 3 and S9†). As we have previously reported, the linear PEG NPs appear to have an intermediate stage of instability between pH 7.0–7.2.9 At this pH, the diameter via DLS increases to ∼250 nm, indicating partial aggregation before disassembly at lower pH (Fig. 3a). This behavior was not observed for the brush PEGMA8 NPs, where complete NP disassembly occurred at pH 7 (Fig. 3b). PEGMA37 NPs showed a small increase in particle diameter down to pH 7.2, indicating slight swelling or aggregation of the NPs, before degrading at pH 7.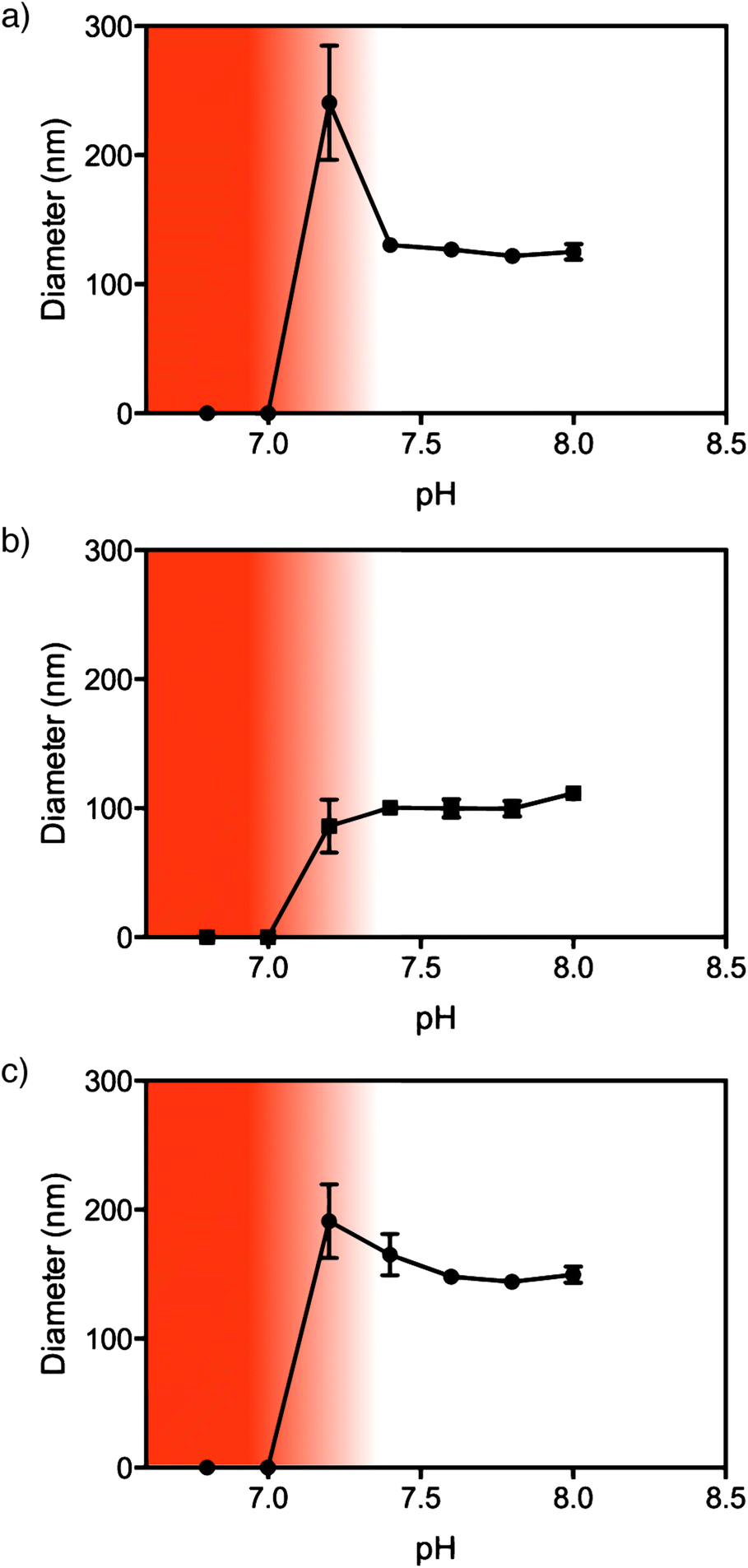 | ||
| Fig. 3 pHlexi particles disassemble rapidly below pH 7.2. pH response of NPs measured by DLS at 37 °C. (a) PEG45, (b) PEGMA8 and (c) PEGMA37. The white region indicates stable NPs. The dark shaded region indicates disassembled particles whereas the gradient indicates a region of NP instability. | ||
The assembly of PDEAEMA NPs using PEGMA rather than linear PEG showed similar cellular cytotoxicity as measured by Alamar Blue viability assay (Fig. S11†). In 3T3 fibroblast cells, PEGMA8, PEGMA37 and linear PEG similarly exhibited >80% viability at concentrations below 30 μg ml−1. The toxicity of all three NPs was significantly higher in C1R lymphoblast cells compared to 3T3s however linear PEG and brush PEGMA NPs all exhibited comparable toxicity profiles below 30 μg ml−1 in 3T3 and below 3 μg ml−1 in C1R cell lines. The association of NPs with both cell lines was similar when the same concentration of NPs was incubated with the cells (Fig. 4). This suggests the different toxicity observed is not a result of nanoparticle association, but differences in how the cells process the NPs. The analogous toxicity of brush PEGMA and linear PEG is consistent with previous findings.22
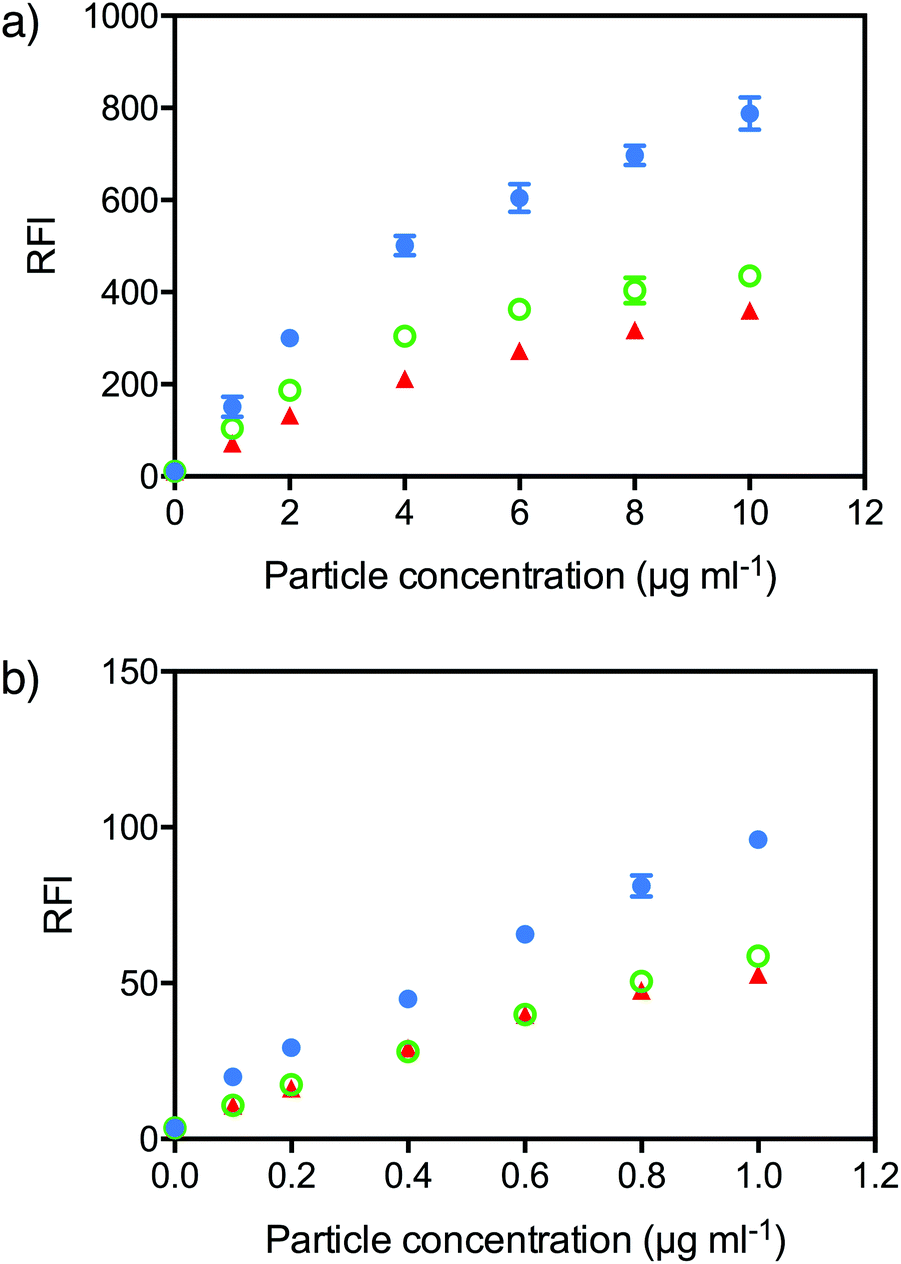 | ||
| Fig. 4 Brush PEG pHlexi NPs show lower non-specific binding to cells in vitro. NP association with (a) 3T3 fibroblast cells and (b) C1R cells; PEG45 (blue circles), PEGMA8 (green open circles) and PEGMA37 (red triangles). | ||
Association and trafficking of NPs within the cell is of interest for improving NPs as efficient therapeutic delivery systems. To investigate the stealth properties of the NPs, we first investigated the association of NPs with 3T3 and C1R cells by flow cytometry. Fluorescently labelled NPs were prepared using a random polymer of PDEAEMA containing 1% pentafluorophenyl methacrylate (PFPMA), which was reacted with cyanine-5 amine. The concentration of Cy5-labelled NPs after filtration was determined by UV-vis spectroscopy using the absorbance of the Cy5 (649 nm) (Fig. S10†)).
To measure cell association, Cy5-labelled NPs were incubated with 3T3 or C1R cells at different concentrations for 4 hours and the fluorescence intensity of the cells measured by flow cytometry. In both cell lines, both PEGMA8 and PEGMA37 NPs exhibited lower binding to 3T3 and C1R cells in vitro compared to linear PEG NPs (Fig. 4). Each NP formulation had a slightly different concentration of fluorescent label, so to directly compare association, the mean fluorescence intensity (MFI) of the cells was divided by the degree of fluorescent labeling of the particles to generate the relative fluorescence intensity (RFI). In 3T3 fibroblast cells, the RFI of cells incubated with linear PEG NPs at a concentration of 10 μg ml−1 was 788 a.u., approximately twice the RFI observed for both PEGMA NPs (∼400 a.u.). Likewise in C1R cells a RFI of 96 a.u. was observed for linear PEG NPs at a concentration of 1 μg ml−1 compared to 59 a.u. and 53 a.u. for PEGMA8 and PEGMA37 NPs respectively. A lower concentration of NPs was used with C1R cells due to the higher toxicity observed in C1R cells compared with 3T3 cells. The decreased non-specific cell association of PEGMA NPs is potentially due to the different structure of the hydrophilic shell provided by the brush PEGMA compared to the linear PEG.
We have shown previously that these NPs are internalized rapidly into 3T3 cells, with more than 75% of the NPs that bound to cells internalized within 2 hours.23 To assess cellular localization of the endocytosed NPs, Cy5-labelled NPs were incubated with 3T3 cells expressing GFP-tagged lysosomal associated membrane protein 1 (LAMP1). The PEG and PEGMA NPs were all co-localized with LAMP1, suggesting that the NPs are endocytosed and trafficked to the lysosomes (Fig. 5). This is consistent with our previous observations that while the PDEAEMA NPs may be able to induce endosomal escape of small molecules, the NP remains largely confined to the lysosome.9
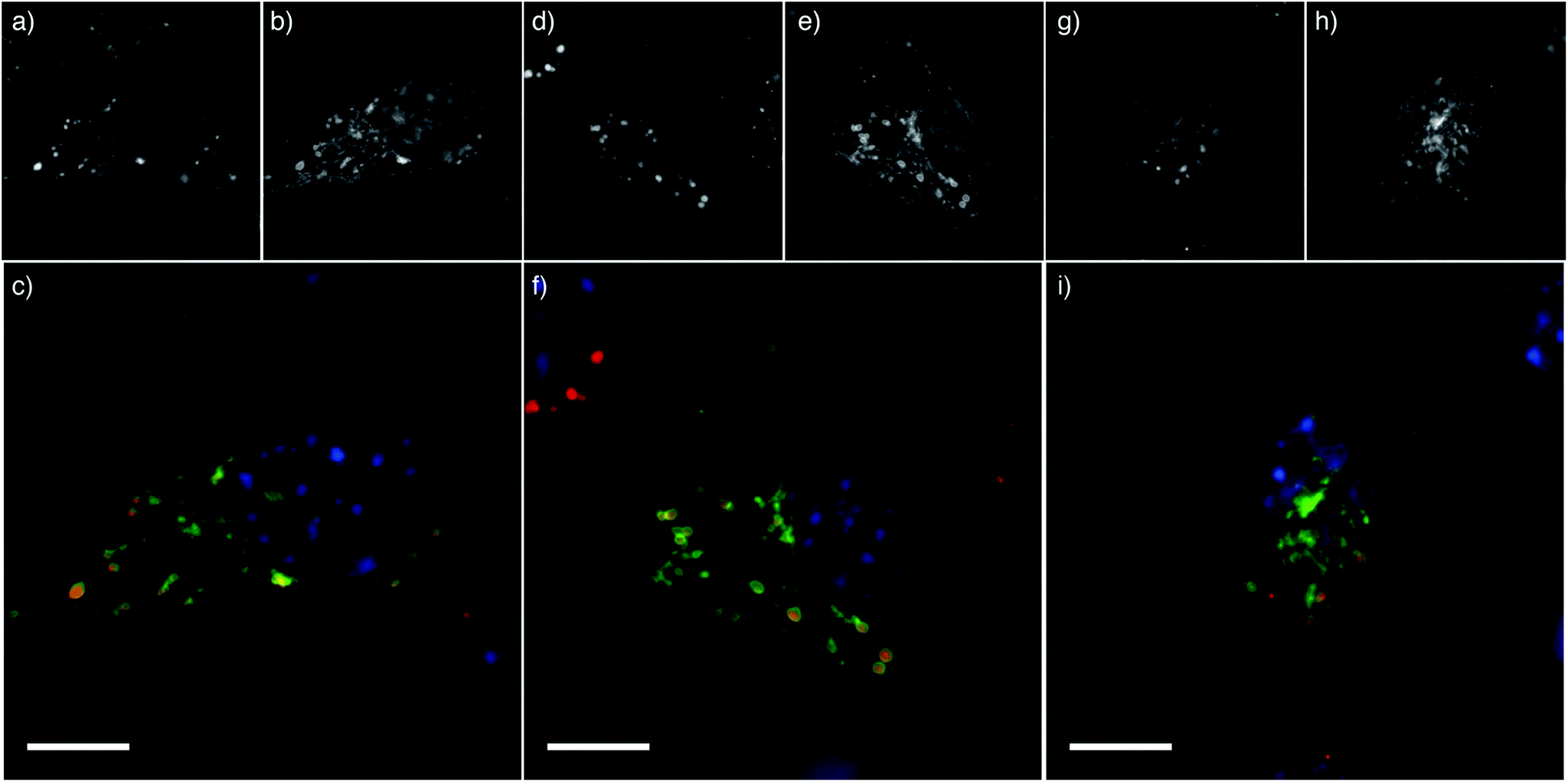 | ||
| Fig. 5 pHlexi particles are internalized into lysosomes. Multichannel cell images showing 3T3 cells incubated with NPs; (a–c) PEG45, (d–f) PEGMA8 and (g–i) PEGMA37. (a, d, g) Cy5-labelled PDEAEMA NP fluorescence, (b, e, h) lysosomal associated membrane protein (LAMP1) staining and (c, f, i) an overlay of NP fluorescence (red) and LAMP1 late endosomal/lysosomal staining (green). The cell nucleus is stained blue with Hoescht DNA stain. Scale bar represents 10 μm. | ||
Ex vivo whole blood assay
We have previously reported an ex vivo human blood assay able to predict in vivo circulation behavior of different sized cross-linked PEG particles more accurately than traditional cell-line based assays.24,25 The presence of autologous opsonins in this model likely better reflects what the MPS encounters upon human intravenous NP administration in vivo. We therefore incubated the linear and brush PEG NPs in fresh human blood at 10 μg mL−1 at 37 °C and investigated association with phagocytic (monocytes, granulocytes) and non-phagocytic cells (T cells) by flow cytometry (Fig. 6). Cells were identified after phenotyping with fluorescent antibodies (Fig. S12†). NP association with T cells, a non-phagocytic population, was negligible and not influenced by the NP surface. This indicates the NPs have a low level of nonspecific binding, as would be expected for PEGylated systems. As we have observed previously, the association of NPs to monocytes and granulocytes was significantly greater than to T cells,24 with monocyte association greater (higher RFI) than granulocyte association for each NP. Similar association patterns were observed for the different NPs to monocytes and granulocytes. For example, increasing the molecular weight of PEGMA (PEGMA37) resulted in less association (p < 0.01) with granulocytes (RFI 171 a.u.) compared with the smaller molecular weight PEGMA NPs (PEGMA8; RFI 335 a.u.). Monocytes also exhibited lower association of PEGMA37 NPs (RFI 834 a.u.) compared to PEGMA8 (RFI 1692 a.u.). Interestingly, linear PEG NPs (PEG45) displayed significantly less association with monocytes (p < 0.05) and granulocytes (p < 0.01) than PEGMA8 despite having the same molecular weight PEG. We expect this is due to the PEGMA8 NPs having a thinner hydrophilic layer due to branching. As well as the nature of the hydrophilic layer, the adsorption of proteins of the surface of the nanoparticles may also depend on the particle size, surface curvature and PEG density. Several studies have been performed on the nature of protein adsorption onto silica nanoparticles of varying sizes.26–28 In these studies, smaller nanoparticles, perhaps due to higher surface curvature, retained higher levels of specific native-like proteins (lysozyme and human carbonic anhydrase) compared to larger particles.27,28 There was no significant difference between the association of linear PEG NPs and brush PEGMA of the same monomer units (PEGMA37). Taken together, these results indicate that tuning the NP surface can minimize association with primary phagocytic cells and enhance their stealth properties in a whole blood environment. | ||
| Fig. 6 pHlexi particles with low MW brush PEG exhibit higher association with monocytes and granulocytes than linear PEG and high MW brush PEG. NPs were incubated with whole human blood at 37 °C and examined for their association with phagocytic (monocytes, granulocytes) and non-phagocytic cells (T cells) by flow cytometry. (a) Sample histograms for two donors. (b) Association of PEG-based NPs in 3 cell populations in fresh whole human blood from 8 healthy donors. *p < 0.05, **p < 0.01, ***p < 0.001 (Friedman non-parametric test, followed by Dunn's multiple comparisons test). | ||
We performed the whole-blood association assay with 8 donors and observed significant differences in cell association between each donor. The RFI for monocyte binding varied by more than 4 fold between subjects for all NPs tested. A consistent trend was observed, where donors that exhibited high binding with one type of NP also showed high levels of binding with the other NPs. An example of histograms for a donor showing low association (Donor A) and high association (Donor B) is shown in Fig. 6a. Donors C, D and F show a bimodal distribution of NP association with monocytes (Fig. S13†). The variation in NP association across the different donors is not surprising given the different environmental conditions each donor has been exposed to, and differences in age, sex and ethnicity. To probe these factors further, larger cohorts would need to be included in the studies. The high degree of variation between individuals highlights the importance of studies on multiple human donors.
NP association with cells was found to differ between assays employing ex vivo whole blood and cultured cell lines. Lower molecular weight PEGMA NPs (PEGMA8) showed higher cell association in whole blood compared to PEGMA37 NPs, despite similarly low association in 3T3 and C1R cell lines. Many studies employ only traditional cell-line based assays to test the stealth properties of particles; although cell lines are a convenient and accessible strategy to obtain preliminary NP association and toxicity data, observations in this study suggest that the use of ex vivo whole blood is likely to better inform the design of polymeric NP delivery systems. Cell-line based assays also give no insight into the variation that is observed between individuals. The major causes of discrepancy between these two assay systems are likely to include phenotypical differences between the culture-adapted cell lines and the complex distribution of cell types present in circulation in vivo, as well as the differing composition of the milieu of proteins NPs become exposed to (i.e. culture medium containing bovine serum compared to human serum). The latter is likely to modify the protein corona established upon NPs following exposure to serum proteins, which may alter the surface interactions and behavior of the NP.29,30 Understanding the specific nature of the protein corona will be the focus of our future work.
Conclusions
In summary, we have demonstrated that both linear PEG and brush PEGMA are suitable for the formation of self-assembling pHlexi NPs. We investigated the difference in cellular association of linear and brush PEG NPs using a basic cell-line association assay and an ex vivo human blood association assay. Brush PEGMA NPs exhibited lower non-specific association with 3T3 fibroblast and C1R lymphoblast cells, however PEGMA8 NPs showed significantly higher association to monocytes and granulocytes compared to linear PEG45 and PEGMA37 in the ex vivo blood assay. The differences between the two assays underscore the need for better models to study NP biological interactions beyond cell-line based assays. The blood assay also highlights the donor variation in binding of NPs to blood cells. We believe improved understanding of NPs/cellular interactions is fundamental for driving the design of next generation delivery systems.Acknowledgements
This work was supported by the Australian Research Council through the Future Fellowship Scheme (FT120100564 – GKS and FT110100265 – APRJ) and Centre of Excellence in Convergent Bio-Nano Science and Technology (APRJ and SJK). APRJ is also supported through the Monash University Larkin's Fellowship Scheme.Notes and references
- L. Brannon-Peppas, Int. J. Pharm., 1995, 116, 1–9 CrossRef CAS.
- J. Moraes, K. Ohno, T. Maschmeyer and S. Perrier, Chem. Mater., 2013, 25, 3522–3527 CrossRef CAS.
- B. G. De Geest, M. A. Willart, H. Hammad, B. N. Lambrecht, C. Pollard, P. Bogaert, M. De Filette, X. Saelens, C. Vervaet, J. P. Remon, J. Grooten and S. De Koker, ACS Nano, 2012, 6, 2136–2149 CrossRef CAS PubMed.
- S. R. Abulateefeh, S. G. Spain, K. J. Thurecht, J. W. Aylott, W. C. Chan, M. C. Garnett and C. Alexander, Biomater. Sci., 2013, 1, 434–442 RSC.
- S. K. Hamilton, A. L. Sims, J. Donavan and E. Harth, Polym. Chem., 2011, 2, 441–446 RSC.
- D. E. Owens Iii and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef PubMed.
- R. Gref, Y. Minamitake, M. T. Peracchia, V. Trebetskoy, V. Torchilin and R. Langer, Science, 1994, 263, 1600 CAS.
- L. van Vlerken, T. Vyas and M. Amiji, Pharm. Res., 2007, 24, 1405–1414 CrossRef CAS PubMed.
- A. S. M. Wong, S. K. Mann, E. Czuba, A. Sahut, H. Liu, T. C. Suekama, T. Bickerton, A. P. R. Johnston and G. K. Such, Soft Matter, 2015, 11, 2993–3002 RSC.
- M. Mertoglu, S. Garnier, A. Laschewsky, K. Skrabania and J. Storsberg, Polymer, 2005, 46, 7726–7740 CrossRef CAS.
- J.-F. Lutz, K. Weichenhan, Ö. Akdemir and A. Hoth, Macromolecules, 2007, 40, 2503–2508 CrossRef CAS.
- S. J. Holder, N. A. A. Rossi, C.-T. Yeoh, G. G. Durand, M. J. Boerakker and N. A. J. M. Sommerdijk, J. Mater. Chem., 2003, 13, 2771–2778 RSC.
- J.-F. Lutz and A. Hoth, Macromolecules, 2006, 39, 893–896 CrossRef CAS.
- M. M. Ali and H. D. H. Stöver, Macromolecules, 2004, 37, 5219–5227 CrossRef CAS.
- B. S. Lele, H. Murata, K. Matyjaszewski and A. J. Russell, Biomacromolecules, 2005, 6, 3380–3387 CrossRef CAS PubMed.
- J. Nicolas, V. S. Miguel, G. Mantovani and D. M. Haddleton, Chem. Commun., 2006, 4697–4699, 10.1039/B609935A.
- J. Pyun, T. Kowalewski and K. Matyjaszewski, Macromol. Rapid Commun., 2003, 24, 1043–1059 CrossRef CAS.
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337–377 CrossRef CAS.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- H. Ma, J. Hyun, P. Stiller and A. Chilkoti, Adv. Mater., 2004, 16, 338–341 CrossRef CAS.
- H. Ma, M. Wells, T. P. Beebe and A. Chilkoti, Adv. Funct. Mater., 2006, 16, 640–648 CrossRef CAS.
- J.-F. Lutz, J. Andrieu, S. Üzgün, C. Rudolph and S. Agarwal, Macromolecules, 2007, 40, 8540–8543 CrossRef CAS.
- S. K. Mann, E. Czuba, L. I. Selby, G. K. Such and A. P. R. Johnston, Pharm. Res., 2016, 1–12, DOI:10.1007/s11095-016-1984-3.
- J. Cui, R. De Rose, K. Alt, S. Alcantara, B. M. Paterson, K. Liang, M. Hu, J. J. Richardson, Y. Yan, C. M. Jeffery, R. I. Price, K. Peter, C. E. Hagemeyer, P. S. Donnelly, S. J. Kent and F. Caruso, ACS Nano, 2015, 9, 1571–1580 CrossRef CAS PubMed.
- R. De Rose, A. N. Zelikin, A. P. R. Johnston, A. Sexton, S.-F. Chong, C. Cortez, W. Mulholland, F. Caruso and S. J. Kent, Adv. Mater., 2008, 20, 4698–4703 CrossRef CAS.
- P. Asuri, S. S. Bale, S. S. Karajanagi and R. S. Kane, Curr. Opin. Biotechnol., 2006, 17, 562–568 CrossRef CAS PubMed.
- M. Lundqvist, I. Sethson and B.-H. Jonsson, Langmuir, 2004, 20, 10639–10647 CrossRef CAS PubMed.
- A. A. Vertegel, R. W. Siegel and J. S. Dordick, Langmuir, 2004, 20, 6800–6807 CrossRef CAS PubMed.
- S. R. Saptarshi, A. Duschl and A. L. Lopata, J. Nanobiotechnol., 2013, 11, 26–26 CrossRef CAS PubMed.
- P. d. Pino, B. Pelaz, Q. Zhang, P. Maffre, G. U. Nienhaus and W. J. Parak, Mater. Horiz., 2014, 1, 301–313 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of pentafluorophenyl methacrylate (PFPMA) monomer, characterization of polymers, Cy5-PDEAEMA absorbance standard curve, NP viability, gating strategy for whole blood assay and flow cytometry histograms for whole blood assay. See DOI: 10.1039/c6py01332e |
| This journal is © The Royal Society of Chemistry 2016 |