
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biodegradable hyperbranched polyether-lipids with in-chain pH-sensitive linkages
S. S.
Müller
ab,
T.
Fritz
c,
M.
Gimnich
a,
M.
Worm
a,
M.
Helm
c and
H.
Frey
*a
aInstitute of Organic Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10-14, 55128 Mainz, Germany. E-mail: hfrey@uni-mainz.de
bGraduate School Materials Science in Mainz (MAINZ), Staudingerweg 9, 55128 Mainz, Germany
cInstitute of Pharmacy and Biochemistry, Johannes Gutenberg University Mainz, Staudingerweg 5, 55128 Mainz, Germany
First published on 19th September 2016
Abstract
Hyperbranched polyether-based lipids with cleavable acetal units were obtained via copolymerization of the epoxide inimer 1-(glycidyloxy)ethyl ethylene glycol ether (GEGE) and glycidol, using anionic ring-opening polymerization. Cholesterol-linear polyglycerol (Ch-linPG) was used as a macroinitiator, resulting in branched polyethers with an adjustable amount of acid-cleavable units. Random copolymerization led to Ch-P(GEGEx-co-Gy) copolymers, whereas sequential copolymerization provided access to Ch-P(GEGEx-b-Gy) amphiphiles. The amount of GEGE was varied between 8–49 mol% of the total amount of monomer units. In addition, hyperbranched polyethers with a single acetal unit were prepared using glycol-1-(cholesteryloxy)ethylether as an initiator for the polymerization of allyl glycidyl ether (AGE) in bulk. Subsequent thiol–ene coupling of mercaptoethanol resulted in the hydroxyl functional macroinitiator used for the polymerization of glycidol. The novel polyether-based lipids were characterized in detail by 1H NMR spectroscopy and size exclusion chromatography, revealing narrow to moderate molecular weight distributions. Degradation was achieved at pH 2 in a proof-of-principle experiment. Acid-triggered shedding of liposomes was proven using the linear analogue α-(1-(cholesteryloxy)ethoxy)-ω-hydro-PEG-CH2-C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH with one cleavable group and a fluorescence label, Atto 488 azide. Investigation of the acetal-cleavage under neutral and acidic pH (7.4–2.0) via fluorescence spectroscopy was carried out.
CH with one cleavable group and a fluorescence label, Atto 488 azide. Investigation of the acetal-cleavage under neutral and acidic pH (7.4–2.0) via fluorescence spectroscopy was carried out.
Introduction
Drug delivery vehicles, such as liposomes, are well known carriers, especially in anti-cancer treatment.1,2 Liposomes consist of amphiphilic phospholipids that form spherical vesicles in aqueous solution. Effective delivery systems enable an important step towards minimizing undesired effects of extremely toxic drugs, such as doxorubicin, on healthy tissue. Disadvantages like cardiotoxicity of doxorubicin and short circulation times can be avoided. The use of poly(ethylene glycol) (PEG) as a polymer shell at the liposome surface has become a popular approach to create long-circulating liposomes.3,4 These systems are also called sterically stabilized or “stealth” liposomes, since the PEGylated liposomes obtain extraordinary properties due to the water-soluble and flexible polymer chains. The presence of PEG results in prolonged blood circulation times,5,6 reduced mononuclear phagocyte system (MPS) uptake,7 reduced aggregation of the carriers and increasing colloidal stability.8 Nevertheless, the gold standard PEG possesses drawbacks that need to be overcome in advanced drug delivery systems. Having reached the target tissue and cells, the PEG shell can prevent the liposome from penetrating into cells or escaping from the endosome after endocytosis (PEG dilemma).9,10 Currently established drug delivery systems that are based on such liposomes fail to release the drug actively and rely on passive diffusion or slow and non-specific liposome degradation. Ideally, PEGylated liposomes profit from the enhanced permeability and retention (EPR) effect,11 and are stable under physiological pH (pH 7.4). Preferentially, the polymer coating would be detached under local pathological conditions such as low pH, leading to destabilization and release of the cargo. The slightly acidic environment in endosomes, tumor tissues, or inflammatory tissue is one of the main differences to healthy tissue. Endocytic pathways lead to a drop from a neutral pH to pH 6 in late endosomes and pH 4 in lysosomes.12,13 Cancer and inflammatory tissue also exhibit acidic pH around pH 6.5.14 A perfectly tuned pH-sensitive nanoparticle would shed the polymer coating and thus enable membrane–membrane fusion and internalization. Fusion with the endosomal membrane is especially important for the release of a cargo into the cytoplasm.15,16 This escape is crucial for acid-degradable cargo such as small interfering RNA (siRNA), proteins or acid-labile drugs.17 The need for “stealth” liposomes that are stable under physiological conditions, but release their content at decreased pH at the site of action is proposed in an increasing number of publications in this field. Several examples have been reported for controlled release including acetals,18,19 vinyl ethers,20–22 (di-)ortho esters,23–25 hydrazones,26–28 and esters (e.g. succinates).29,30 The second drawback of PEG is its lack of functional groups. Methoxypoly(ethylene glycol) (mPEG), that is commonly used for the linkage to phospholipids or cholesterol, does not possess additional functional groups for derivatization. However, functional groups are important for the attachment of markers, antibodies or proteins for “active” tumor targeting.31 Our group has introduced multifunctional polyether-based lipids synthesized from an epoxide “construction kit”.32–34 By using cholesterol directly as the initiator for the oxyanionic ring-opening polymerization (ROP) of various epoxides (ethylene oxide (EO), ethoxyethyl glycidyl ether (EEGE), isopropylidene glycidyl glyceryl ether (IGG), and glycidol), a variety of tailor-made architectures like linear, hyperbranched or linear-hyperbranched polyether amphiphiles can be obtained. The hyperbranched structures can be realized by using glycidol in the ring-opening multibranching polymerization technique via slow monomer addition (SMA) of the AB2 monomer to a multihydroxy-precursor. With this method reproducible and narrowly distributed hyperbranched polyglycerol (hbPG) derivatives can be obtained.35 Furthermore, the number of hydroxyl groups can be accurately tuned by the amount of glycidol utilized. HbPG has been extensively studied in drug delivery applications and as a biorepellent material due to its extraordinary biocompatibility,36–38 multifunctionality and water-solubility.39 Similar to the abovementioned PEG-lipids, its non-biodegradability can be an issue in special applications. To overcome this drawback, pH-sensitive polyether polyketals with tunable degradation in solution were synthesized by Kizhakkedathu and coworkers.40–42 Hyperbranched acetal-containing polyethers were introduced by our group using the epoxide inimer 1-(glycidyloxy)ethyl ethylene glycol ether (GEGE) in a copolymerization reaction with glycidol.43In the present study, we combine the benefits of multifunctional polyether-based lipids with the degradability of pH-sensitive acetal-containing polyglycerol. In all cases, cholesterol moieties were employed as the hydrophobic part of the amphiphilic structure, ensuring strong interaction with the phospholipid bilayer of liposomes. In the first strategy, we show the synthesis of hyperbranched acetal-containing polyglycerol using GEGE and glycidol in a random copolymerization or a sequential polymerization (Fig. 1a and Scheme 1). In the second approach, we expand the concept of glycol-1-(cholesteryloxy)ethylether recently presented by our group.18 This initiator is used for the polymerization of AGE, and subsequent thiol–ene coupling with 2-mercaptoethanol followed by the polymerization of glycidol. This pathway results in either linear multihydroxy-functional polyethers or hyperbranched polyethers having exactly one acetal group between the cholesterol anchor and the polymer chain (Fig. 1b and Scheme 2). In the third case, the scope of α-(1-(cholesteryloxy)ethoxy)-ω-hydro-PEG is extended by the functionalization with propargyl bromide followed by copper-catalyzed cycloaddition with Atto 488 azide. Acidic cleavage of the acetal group in functionalized liposomes was investigated via fluorescence spectroscopy. The shedding process could be monitored by fluorescence emission of the dye Atto 488 (Fig. 1c and 8).
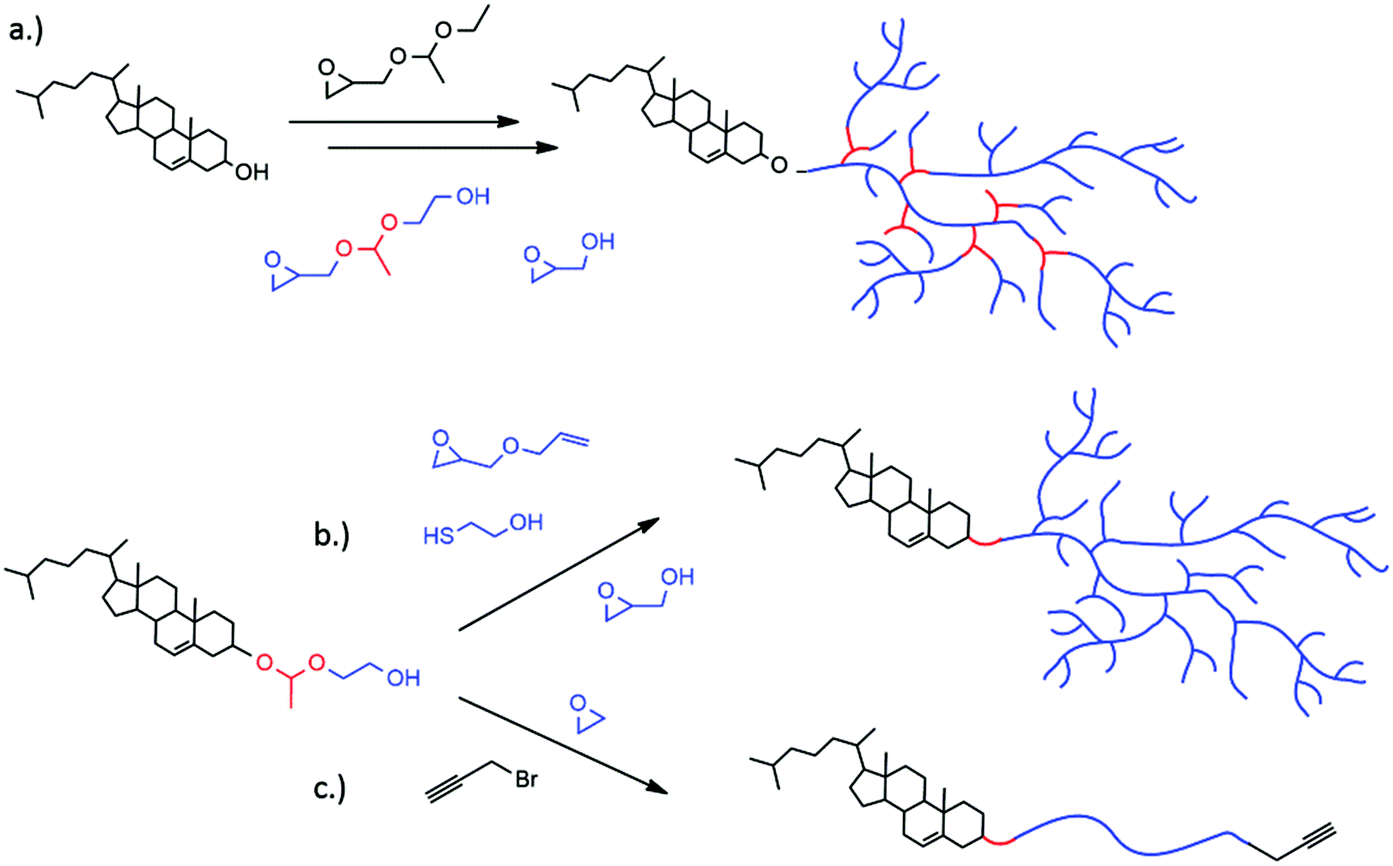 | ||
| Fig. 1 Synthesis of (a) hyperbranched polyglycerol lipids containing multiple acetal groups by using GEGE and glycidol in a random copolymerization or a sequential polymerization (b) hyperbranched polyether lipids with exactly one acetal group at the block junctions utilizing glycol-1-(cholesteryloxy)ethylether as an initiator (c) linear PEG lipids with one cleavable group functionalized with the fluorescent dye Atto 448. | ||
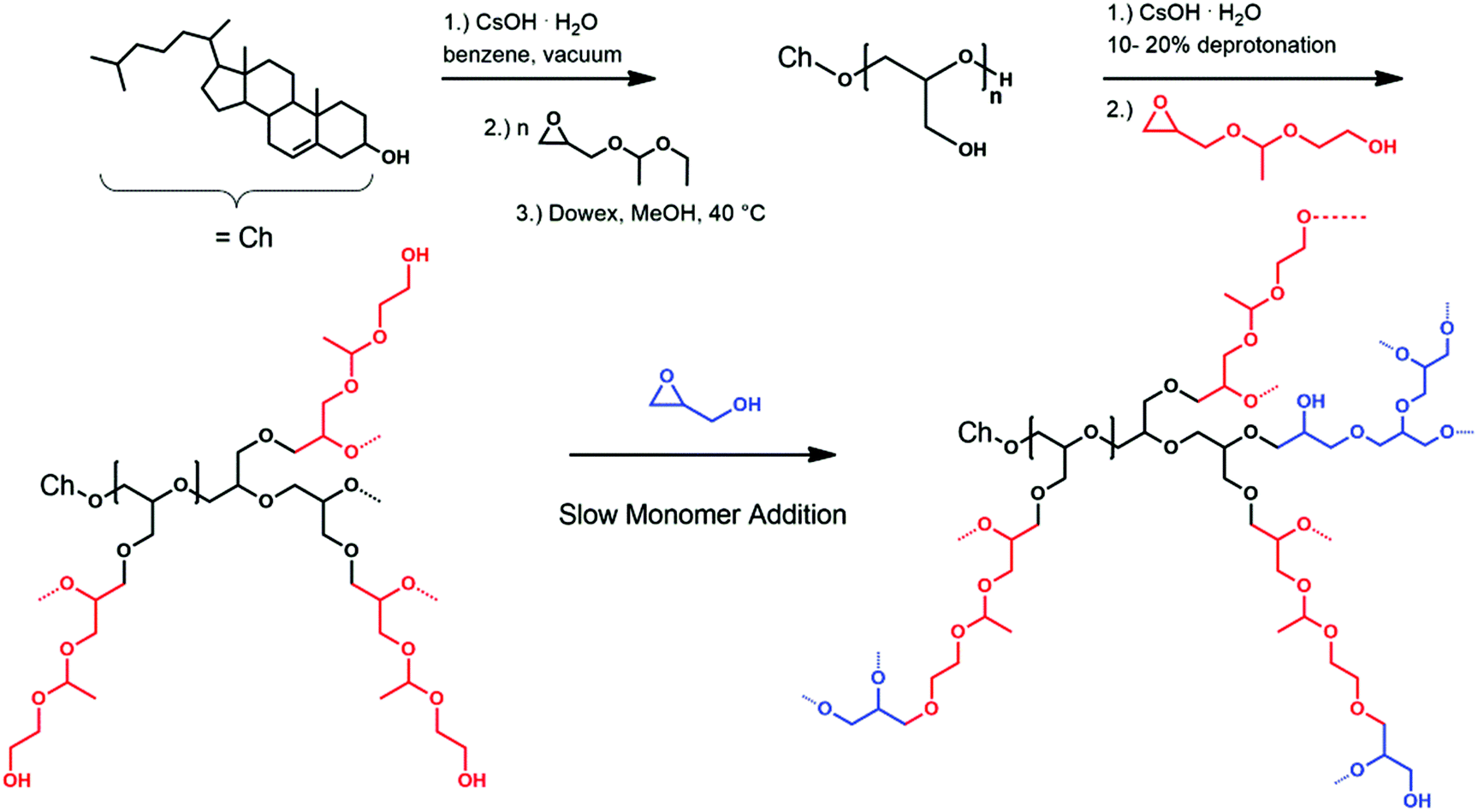 | ||
| Scheme 1 Reaction scheme for the synthesis of the macroinitiator Ch-linPGn, followed by the slow monomer addition of GEGE and glycidol, which can be carried out simultaneously or sequentially as depicted here. | ||
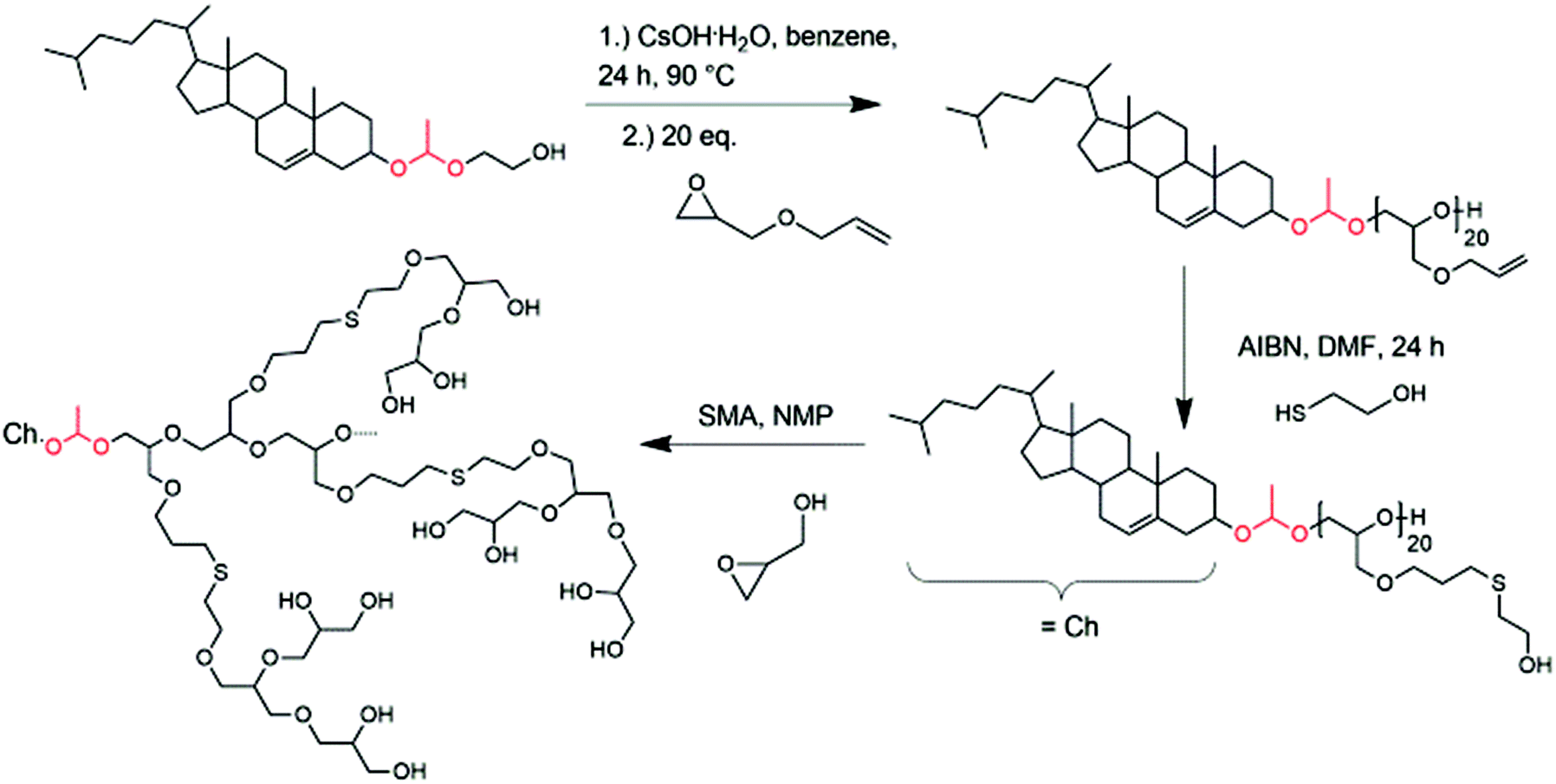 | ||
| Scheme 2 Synthesis of hyperbranched cholesterol-lipids with exactly one acetal group starting from glycol-1-(cholesteryloxy)ethylether (Ch-acetal); (SMA = slow monomer addition). | ||
With these different polymer systems comprising multiple or single acetal groups in hand, we introduce a platform of lipids that combine desired lipid pH-responsiveness with multifunctionality for drug delivery applications.
Experimental
Methods
1H NMR spectra (300 MHz and 400 MHz) were recorded using a Bruker AC300 or a Bruker AMX400, employing CD2Cl2, MeOD, or THF-d8 as solvent. All spectra were referenced internally to residual proton signals of the deuterated solvent. Size exclusion chromatography (SEC) measurements were carried out in dimethylformamide (DMF) with 0.25 g L−1 LiBr on PSS HEMA columns (300/100/40). For SEC measurements UV (275 nm) and RI detectors were used. Calibration was carried out using poly(ethylene glycol) (PEG) standards provided by Polymer Standards Service (PSS). Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-ToF MS) measurements were performed on a Shimadzu Axima CFR MALDI-ToF mass spectrometer equipped with a nitrogen laser delivering 3 ns laser pulses at 337 nm. Dithranol was used as a matrix. The sample was prepared by dissolving the polymer in methanol at a concentration of 10 g L−1. A 10 μL aliquot of this solution was added to 10 μL of a 10 g L−1 solution of the matrix and 1 μL of a solution of potassium trifluoroacetate (KTFA) (0.1 M in methanol as a cationization agent). A 1 μL aliquot of the mixture was applied to a multistage target, methanol evaporated and a thin matrix/analyte film created.Liposome preparation
Liposomes bearing acetal-containing cholesterol-based amphiphiles were prepared as described previously.44,45 Briefly, 32.5 μl of phosphate buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) was added to 17.5 mg of a dried lipid film consisting of cholesterol (Carl Roth), egg phosphatidyl choline (EPC, kindly provided by Lipoid) and α-(1-(cholesteryloxy)ethoxy)-ω-Hydro-PEG46-CH2-CCH (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :55:5 molar ratio) and 250 mg ceramic beads (SiLiBeads ZY 0.6–0.8 mm, kindly provided by Sigmund Lindner, Warmensteinach, Germany). Non-cleavable liposomes were prepared with an equivalent cholesterol-PEG-CH2-CCH compound. After dual centrifugation in a Rotanta 400 centrifuge (customized with a prototype DC-rotor, Hettich, Tuttlingen, Germany) for 20 min at 2500 RPM and dilution with 100 μl PBS, 5 μl of the liposome suspension were exposed to click-modification with 50 μM Atto 488 azide (Atto-Tec, Siegen, Germany) in Milli-Q water (to a total volume of 40 μl), phosphate buffer (5.3 mM NaH2PO4, 94.7 mM Na2HPO4, pH 8), Tris(hydroxypropyltriazolylmethyl)amine (THPTA) (0.5 mM), CuSO4 (0.1 mM) and sodium ascorbate (2.5 mM) and subsequent gel filtration through Sepharose 2B. Liposome size was obtained via dynamic light scattering (DLS) on a Malvern Zetasizer Nano ZS, using disposable poly(styrene) cuvettes (Sarstedt, Germany). 1 μl of liposome stock suspension was diluted in 1 ml PBS. After equilibration to 25 °C, three measurements were performed, using the internal measurement optimization for both attenuator and measurement position. Refractive index (RI) and viscosity of the dispersant was set to 1.330 and 0.8872 cP, the RI of the particle to 1.59. The absorption of the particle was set to 0.01 and all measurements were performed at a scattering angle of 173°. Cleavage of the polymeric liposome shell was observed via fluorescence spectroscopy on a FP-6500 spectrofluorimeter (Jasco, Tokyo, Japan) at 488 nm excitation wavelength and 523 nm emission wavelength during a time course measurement with a data interval of 1 min. 50 μl of purified liposomes were combined with 3 ml PBS in a 1 cm3 quartz glass cuvette (Hellma Analytics, Müllheim, Germany) with a magnetic stirrer. After equilibration for 12 h at 37 °C, the pH of the suspension was adjusted by addition of 50 μl 2 M hydrochloric acid (HCl) (pH 2), 20 μl 2 M HCl (pH 3) or 15 μl 2 M HCl (pH 5.5). Fluorescence intensity was normalized to the intensity at the beginning of acidification.
:55:5 molar ratio) and 250 mg ceramic beads (SiLiBeads ZY 0.6–0.8 mm, kindly provided by Sigmund Lindner, Warmensteinach, Germany). Non-cleavable liposomes were prepared with an equivalent cholesterol-PEG-CH2-CCH compound. After dual centrifugation in a Rotanta 400 centrifuge (customized with a prototype DC-rotor, Hettich, Tuttlingen, Germany) for 20 min at 2500 RPM and dilution with 100 μl PBS, 5 μl of the liposome suspension were exposed to click-modification with 50 μM Atto 488 azide (Atto-Tec, Siegen, Germany) in Milli-Q water (to a total volume of 40 μl), phosphate buffer (5.3 mM NaH2PO4, 94.7 mM Na2HPO4, pH 8), Tris(hydroxypropyltriazolylmethyl)amine (THPTA) (0.5 mM), CuSO4 (0.1 mM) and sodium ascorbate (2.5 mM) and subsequent gel filtration through Sepharose 2B. Liposome size was obtained via dynamic light scattering (DLS) on a Malvern Zetasizer Nano ZS, using disposable poly(styrene) cuvettes (Sarstedt, Germany). 1 μl of liposome stock suspension was diluted in 1 ml PBS. After equilibration to 25 °C, three measurements were performed, using the internal measurement optimization for both attenuator and measurement position. Refractive index (RI) and viscosity of the dispersant was set to 1.330 and 0.8872 cP, the RI of the particle to 1.59. The absorption of the particle was set to 0.01 and all measurements were performed at a scattering angle of 173°. Cleavage of the polymeric liposome shell was observed via fluorescence spectroscopy on a FP-6500 spectrofluorimeter (Jasco, Tokyo, Japan) at 488 nm excitation wavelength and 523 nm emission wavelength during a time course measurement with a data interval of 1 min. 50 μl of purified liposomes were combined with 3 ml PBS in a 1 cm3 quartz glass cuvette (Hellma Analytics, Müllheim, Germany) with a magnetic stirrer. After equilibration for 12 h at 37 °C, the pH of the suspension was adjusted by addition of 50 μl 2 M hydrochloric acid (HCl) (pH 2), 20 μl 2 M HCl (pH 3) or 15 μl 2 M HCl (pH 5.5). Fluorescence intensity was normalized to the intensity at the beginning of acidification.
Syntheses and reagents
All reagents and solvents were purchased from Acros and used as received, unless otherwise mentioned. Anhydrous solvents were stored over molecular sieves and were purchased from Aldrich. Deuterated solvents were purchased from Deutero GmbH, and stored over molecular sieves. Cholesterol was purchased from Acros and stored at 8 °C. Ethoxyethyl glycidyl ether (EEGE) was synthesized as described in the literature,46 dried over CaH2 and cryo-transferred prior to use. Glycidol and N-methyl-2-pyrrolidon (NMP) were purified by distillation from CaH2 directly prior to use. 1-(Glycidyloxy)ethyl ethylene glycol ether (GEGE) was synthesized according to the literature.43 To release the hydroxyl group in the final reaction step, the benzyl protecting group was removed using catalytic hydrogenation (Pd(OH)2/C, 10 wt%). The product was purified by column chromatography in ethyl acetate. Glycol-1-(cholesteryloxy)ethylether and α-(1-(cholesteryloxy)ethoxy)-ω-Hydro-PEG46 were synthesized following reported procedures.18 Propargyl bromide (0.8 M in toluene) was stored at 4 °C and used as received. Allyl glycidyl ether (AGE) was purchased from Acros Organics, dried over CaH2 and cryo-transferred prior to use. Azobisisobutyronitrile (AIBN, Acros) was recrystallized from MeOH. 2-Mercaptoethanol (Acros) was stored at 5 °C and was used as received.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH cholesterol), 3.80–3.40 (polyether backbone; CHO cholesterol), 2.40–0.82 (CH2, CH cholesterol), 0.72 (–CH3 cholesterol).
CH cholesterol), 4.79 (H3C–CHO2, GEGE acetal group), 3.80–3.40 (polyether backbone; CHO cholesterol), 2.36–0.87 (CH2, CH cholesterol), 1.32 (H3C–CHO2, GEGE), 0.72 (–CH3 cholesterol).
CH2, allyl), 5.39 (CCH cholesterol), 5.33–5.17 (–CH2CHCH2, allyl), 4.82 (H3C–CHO2, acetal group), 4.01 (–CH2CHCH2), 3.80–3.39 (polyether backbone; CHO cholesterol), 2.32–0.88 (CH2, CH cholesterol), 1.30 (H3C–CHO2), 0.72 (–CH3 cholesterol).
CH cholesterol), 4.81 (H3C–CHO2, acetal group), 4.00–3.30 (polyether backbone; CHO cholesterol), 2.72–2.40 (–CH2SCH2), 1.83 (–CH2CH2S), 2.37–0.87 (CH2, CH cholesterol), 0.72 (–CH3 cholesterol).
CH cholesterol), 3.80–3.40 (polyether backbone; CHO cholesterol), 2.40–0.82 (CH2, CH cholesterol), 0.72 (–CH3 cholesterol).
CH cholesterol), 4.79 (H3C–CHO2, GEGE acetal group), 3.80–3.40 (polyether backbone; CHO cholesterol), 2.36–0.87 (CH2, CH cholesterol), 1.32 (H3C–CHO2, GEGE), 0.72 (–CH3 cholesterol).
CH2, allyl), 5.39 (CCH cholesterol), 5.33–5.17 (–CH2CHCH2, allyl), 4.82 (H3C–CHO2, acetal group), 4.01 (–CH2CHCH2), 3.80–3.39 (polyether backbone; CHO cholesterol), 2.32–0.88 (CH2, CH cholesterol), 1.30 (H3C–CHO2), 0.72 (–CH3 cholesterol).
CH cholesterol), 4.81 (H3C–CHO2, acetal group), 4.00–3.30 (polyether backbone; CHO cholesterol), 2.72–2.40 (–CH2SCH2), 1.83 (–CH2CH2S), 2.37–0.87 (CH2, CH cholesterol), 0.72 (–CH3 cholesterol).
1H NMR (400 MHz, DMSO-d6): δ (ppm) = 5.32 (CCH cholesterol), 5.11 (–OH) 4.81 (H3C–CHO2, acetal group), 3.90–3.20 (polyether backbone; CHO cholesterol), 2.65–2.52 (–CH2SCH2), 1.74 (–CH2CH2S), 2.40–0.84 (CH2, CH cholesterol), 0.65 (–CH3 cholesterol).
CH cholesterol), 4.79 (H3C–CHO2, acetal group), 4.18 (–OCH2–CCH), 3.80–3.40 (polyether backbone; CHO cholesterol), 2.49 (–CCH), 2.27–0.82 (CH2, CH cholesterol), 0.68 (–CH3 cholesterol).
Results and discussion
Synthesis of amphiphilic hyperbranched lipids with multiple acid-cleavable moieties
Hyperbranched polyether-lipids with multiple pH-responsive acetal groups were prepared by the oxyanionic ring-opening polymerization using cholesterol as an initiator. After deprotonation with cesium hydroxide, the alkoxide was used for the polymerization of ethoxyethyl glycidyl ether (EEGE) with subsequent acidic hydrolysis of the EEGE protecting groups (Scheme 1). Molecular weights for the macroinitiators were in the range of 1300 to 1900 g mol−1 (Table 1) according to 1H NMR spectroscopy, with an expected underestimation by size exclusion chromatography (SEC). The linear polyglycerol repeating units were calculated by comparing the methyl group of cholesterol (0.72 ppm) and the polyether backbone resonances after acidic cleavage of the acetal protecting groups from the EEGE moieties. Molecular weights calculated by 1H NMR spectroscopy for both macroinitiators were higher than the theoretical values. We aimed at seven glycerol units per macroinitiator, however, 20 and 13 units were obtained, respectively. Fast proton exchange results in the protonated form of cholesterol and deprotonated adducts of initiator and the first monomer unit.47–49 This leads to different kinetics in the initiation step and the propagation step, since the ethoxylated cholesterol exhibits higher reactivity than the cholesterol alkoxide. Hence, significant amounts of free initiator are found in the reaction mixture when oligomers are targeted.50 After acidic hydrolysis of the acetal groups, the polymer was precipitated multiple times in cold diethyl ether to remove excess cholesterol. This led to reduced yields and an enhanced concentration of longer polymer chains.| Composition | M theon | M NMRn | M SECn | Đ SEC | GEGE mol% |
|---|---|---|---|---|---|
| g mol−1 | |||||
| M theon: theoretical molecular weights; MNMRn: molecular weights calculated from 1H NMR via end group analysis; MSECn: molecular weights obtained from size exclusion chromatography using PEG standards for calibration; ĐSEC = Mw/Mn: dispersity determined from SEC via PEG calibration; GEGE mol%: molar content of GEGE calculated from 1H NMR. | |||||
| Ch-linPG20 | 904 | 1870 | 830 | 1.05 | — |
| Ch-P(GEGE4-co-G48) | 4160 | 4600 | 1600 | 1.57 | 8 |
| Ch-P(GEGE20-co-G49) | 5040 | 7200 | 2100 | 1.60 | 29 |
| Ch-linPG13 | 904 | 1350 | 980 | 1.14 | — |
| Ch-P(GEGE10-b-G36) | 4220 | 4700 | 2500 | 1.24 | 22 |
| Ch-P(GEGE18-b-G36) | 4660 | 6000 | 3000 | 1.29 | 33 |
| Ch-P(GEGE17-b-G18) | 4600 | 4500 | 2200 | 1.22 | 49 |
The resulting linear polyglycerol amphiphiles (Ch-linPG20; Ch-linPG13) functioned as a macroinitiator for the slow monomer addition of 1-(glycidyloxy)ethyl ethylene glycol ether (GEGE) and glycidol. Partial deprotonation and slow monomer addition allows good control over the alkoxide concentration resulting in equal growth of the polymer chain ends. This technique narrows the molecular weight distribution and prevents undesired homopolymerization of the cyclic inimers.51 Deprotonation of the macroinitiator was adjusted to 10% (relative to the total amount of hydroxyl groups) in order to ensure solubility in N-methyl-2-pyrrolidon (NMP). Both monomers, i.e. GEGE and glycidol, can be copolymerized randomly or sequentially. The reaction route is shown in Scheme 1 (vide supra). The advantage of a sequential addition of the monomers, in which GEGE is polymerized first, derives from the proximity of the cleavable acetal group to the cholesterol anchor group. Acidic cleavage would lead to scission near the phospholipid bilayer in the final liposomes, which could enhance shedding of the polymer and drug release.
This approach enabled the synthesis of a series of copolymers bearing multiple acetal groups distributed in the hyperbranched polymer or located near the cholesterol anchor (Table 1).
The random copolymers were prepared from a macroinitiator consisting of 20 linear glycerol units, whereas for the sequentially synthesized copolymers the macroinitiator Ch-linPG13 was used. Molecular weights of the hyperbranched lipids were approx. 5000 g mol−1, again with an underestimation in SEC due to differences in the hydrodynamic radius compared to the linear PEG polymer used for calibration. On the other hand, we assume a slight overestimation of the molecular weights in 1H NMR spectroscopy, since residual solvent resonances can overlap with the polyether backbone signals. Molecular weight distributions (Đ = Mw/Mn) below 1.15 were found for the two macroinitiators, whereas for the hyperbranched copolymers Đs below 1.60 could be achieved. The last column of Table 1 summarizes the mol% of GEGE, which were successfully tuned between 8 mol% and 49 mol% (in relation to the total amount of monomer units).
Fig. 2 depicts SEC traces for a selection of copolymers that were synthesized from Ch-linPG13 (black solid line, Fig. 2). All distributions were monomodal and molecular weights for the Ch-P(GEGEx-b-Gy) copolymers were shifted to lower elution volumes compared to the macroinitiator. Due the underestimation of molecular weights in SEC, absolute molecular weights were calculated from 1H NMR integration ratios of the resonances for the methyl group of cholesterol at 0.72 ppm and the polyether signals between 4.00–3.27 ppm (Fig. 3). The number of GEGE units was calculated by comparing the integration of the initiator signals (–CH3; 0.72 ppm) with the acetal resonance at 4.79 ppm (arrow in Fig. 3). The methyl group of the acetal moieties is observed at 1.32 ppm. As an example, Fig. 3 illustrates the 1H NMR spectra of Ch-linPG13 (bottom) and Ch-P(GEGE18-b-G36) (top) in MeOD.
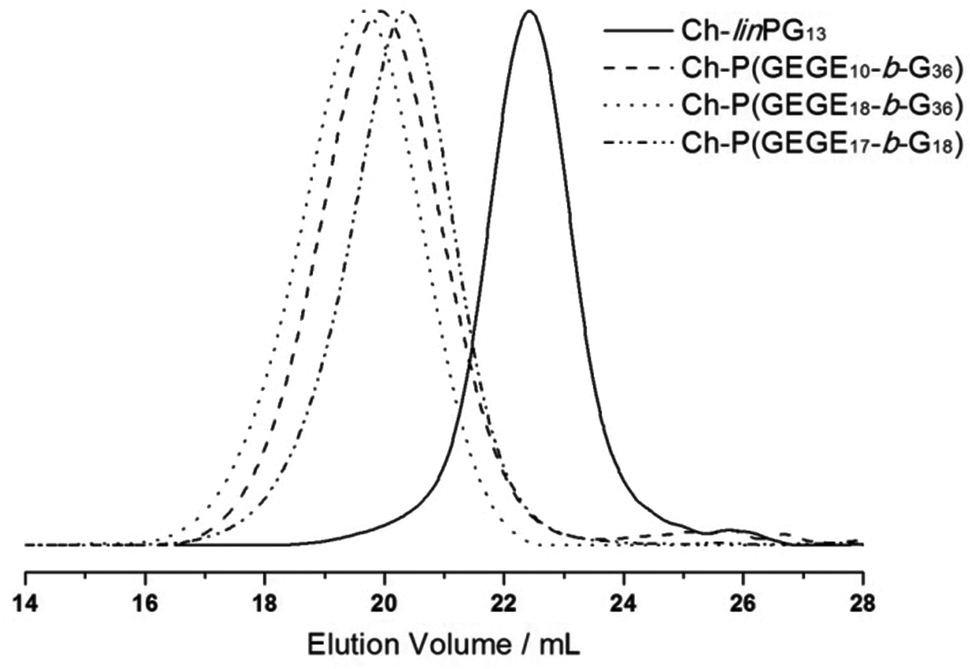 | ||
| Fig. 2 SEC traces (RI detection; DMF) for selected copolymer samples that were synthesized from Ch-linPG13 (black solid line). | ||
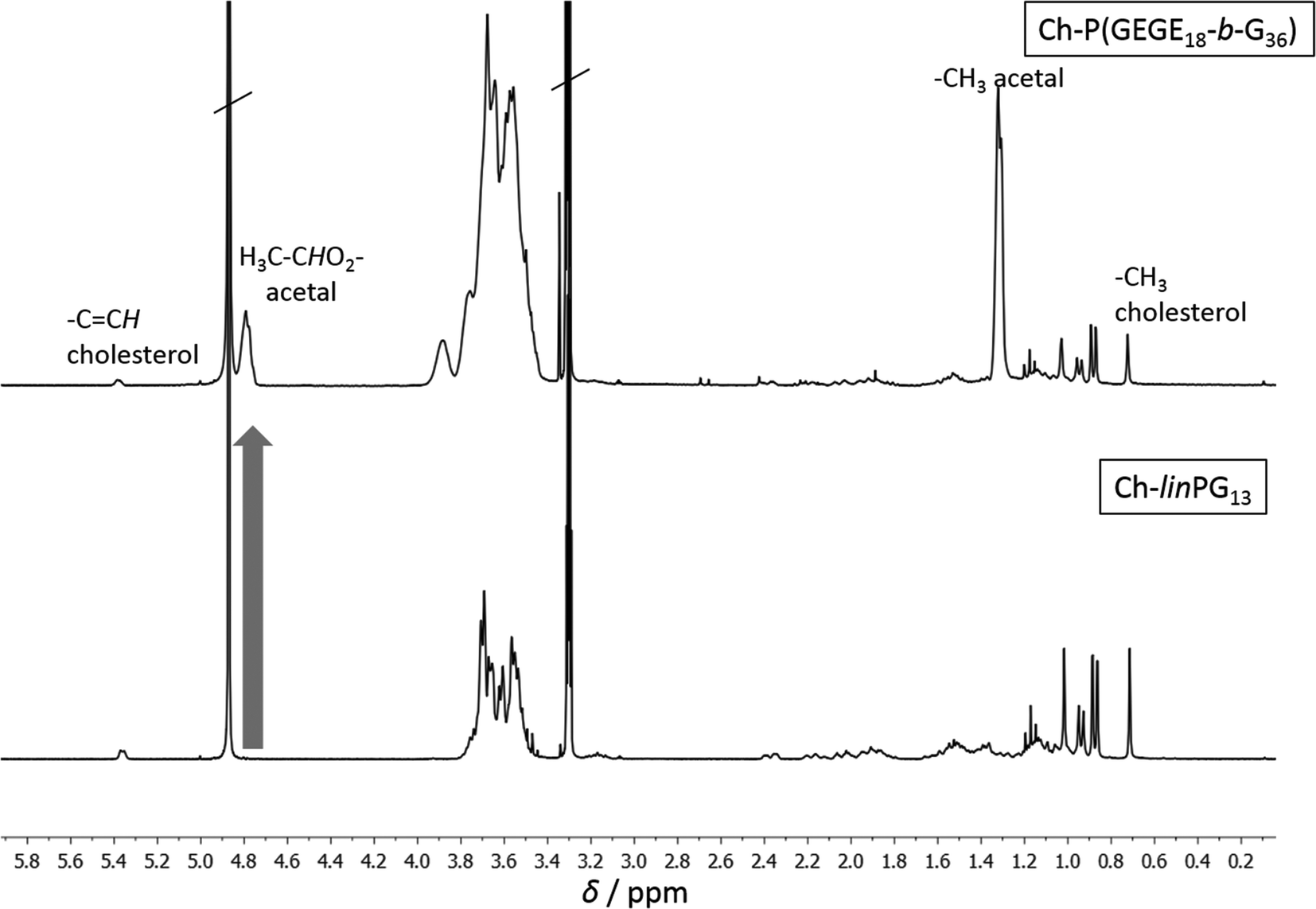 | ||
| Fig. 3 1H NMR spectra of Ch-linPG13 (bottom) and Ch-P(GEGE18-b-G36) (top) in MeOD. | ||
Synthesis of amphiphilic hyperbranched lipids with exactly one acid-cleavable moiety
In a previous publication, we presented the synthesis of glycol-1-(cholesteryloxy)ethylether (Ch-acetal) as a pH-sensitive initiator for the anionic ring-opening polymerization of ethylene oxide.18 This initiator can be accessed via a three-step procedure by, first, synthesizing 2-acetoxyethyl vinyl ether52 which can then be converted with commercially available cholesterol under acid catalysis to introduce the acetal group. After saponification of the acetate protecting group, the hydroxyl-functional initiator glycol-1-(cholesteryloxy)ethylether was obtained, which can be used in the anionic polymerization of various epoxides. It was also shown that the pH-sensitive lipid can be cleaved under acidic conditions.18 Therefore, they are applicable in drug delivery systems, especially for tumor targeting, where the acidic pH of the tumor can be exploited in order to shed the polymer from the vesicle. Here, we expand the idea of exactly one acid-cleavable group in lipids from linear to hyperbranched polyethers. Decomposition products are cholesterol (natural membrane component), biocompatible hyperbranched polyglycerol and acetaldehyde. Since usually the polymer has a molecular weight below 6000 g mol−1 excretion by the kidney would be feasible after detachment from the hydrophobic anchor.The abovementioned EEGE monomer contains an acetal protecting group, which is suitable for the oxyanionic ring-opening polymerization, but has to be cleaved under acidic conditions to obtain the linear polyglycerol as a macroinitiator for the hyperbranched structure. This synthetic route is not suitable in combination with the pH-sensitive initiator glycol-1-(cholesteryloxy)ethylether. Therefore, a different method had to be explored. The first monomer used for experiments with this objective was benzyl glycidyl ether. It is a commercially available glycidyl derivative and can be deprotected under catalytic hydrogenation. Removal of the benzyl groups is usually accomplished with hydrogen pressure in the presence of a palladium catalyst. However, this approach did not lead to successful hydrogenation without cleavage of the acetal group, although different reaction conditions were tested (variation in temperature, pressure, Pd/C vs. Pd(OH)2/C catalyst, solvent mixtures). An altered synthetic route had to be introduced, which is presented in Scheme 2.
After deprotonation with cesium hydroxide, glycol-1-(cholesteryloxy)ethylether was used as an initiator for the oxyanionic ring-opening polymerization of allyl glycidyl ether (AGE). This reaction was carried out in bulk to suppress isomerization of the allyl groups at elevated temperatures.53,54Table 2 summarizes the obtained characterization data for Ch-acetal-PAGE and supports quantitative polymerization of AGE. The number of repeating units calculated from 1H NMR spectroscopy (20 AGE groups, see discussion below, Fig. 4) is in agreement with the theoretical number of 20 AGE units. The allyl groups were functionalized in a thiol–ene coupling reaction, using excess 2-mercaptoethanol and azobisisobutyronitrile (AIBN) as the radical initiator. The thiol–ene coupling was quantitative according to 1H NMR spectroscopic characterization. This amphiphile already represents a novel multihydroxy-functional polyether with exactly one acetal group. Furthermore, it can be used as a macroinitiator for the hypergrafting process of glycidol in order to increase the amount of hydroxyl groups, as shown in Scheme 2. Molecular weights calculated by 1H NMR spectroscopy of the thiolether-containing hyperbranched polymer were around 6500 g mol−1. Table 2 summarizes all characterization data of the abovementioned copolymers. Column 4 illustrates that the molecular weights obtained from SEC (DMF, PEG standard) are lower than the theoretical molecular weights and the molecular weights calculated from 1H NMR spectra. This trend is more pronounced for the hyperbranched structure due to the influence of the hyperbranched, globular polyglycerol on the hydrodynamic radius of the polymers compared to linear PEG. Values of Đ were below 1.2 (last column, Table 2). Fig. 4 depicts the SEC traces for all three polymer species, revealing narrow and monomodal molecular weight distributions.
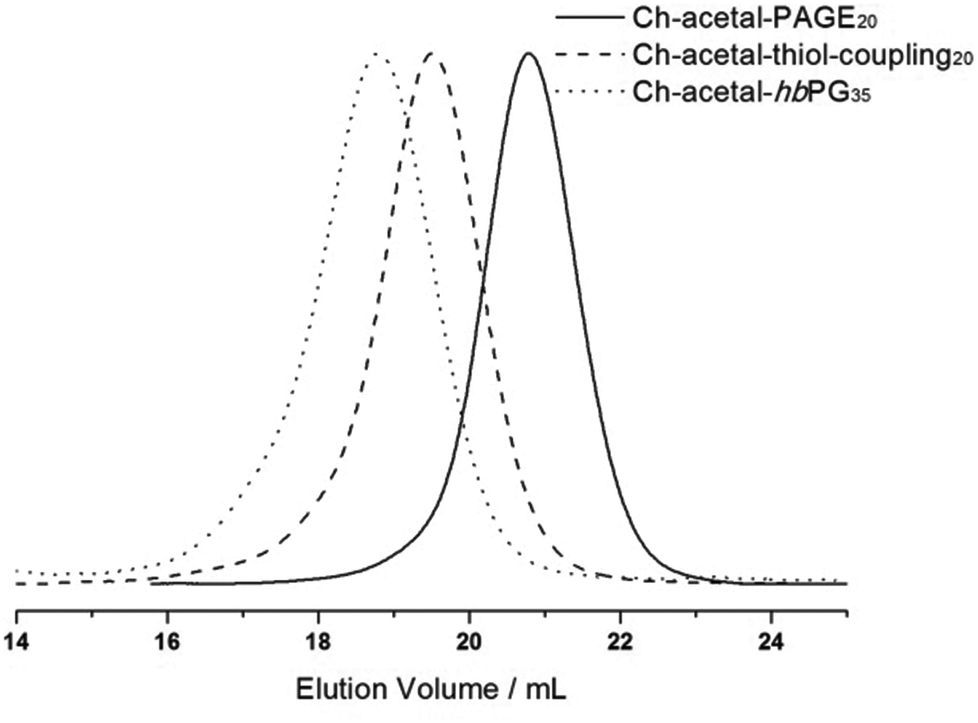 | ||
| Fig. 4 SEC traces (RI detection; DMF) for the copolymers with one single acetal group; allyl containing polyether Ch-acetal-PAGE20 (black solid line), thiolether-containing lipid Ch-acetal–thiol-coupling20 (dashed line), hyperbranched lipid Ch-acetal-hbPG35 (dotted line). | ||
| Composition | M theon | M NMRn | M SECn | Đ SEC |
|---|---|---|---|---|
| g mol−1 | ||||
| M theon: theoretical molecular weights; MNMRn: molecular weights calculated from 1H NMR via end group analysis; MSECn: molecular weights obtained from size exclusion chromatography using PEG standards for calibration; ĐSEC = Mw/Mn: dispersity determined from SEC via PEG calibration. | ||||
| Ch-Acetal-PAGE20 | 2750 | 2750 | 2050 | 1.09 |
| Ch-Acetal–thiol-coupling20 | 4320 | 4320 | 3430 | 1.20 |
| Ch-Acetal-hbPG35 | 6500 | 6900 | 5000 | 1.20 |
Molecular weights, degrees of polymerization, and the degree of functionalization with 2-mercaptoethanol were calculated from 1H NMR spectroscopy. The spectra are shown in Fig. 5, in which the allyl functional polymer Ch-acetal-PAGE20 (red, top) was measured in CD2Cl2, Ch-acetal–thiol-coupling20 (green, middle) was measured in THF-d8, and Ch-acetal-hbPG35 (blue, bottom) was measured in DMSO-d6. The degree of polymerization for the PAGE copolymer was calculated by comparing the integration of the methyl group of cholesterol (0.72 ppm) with the resonance of the allyl proton at 5.93 ppm. Quantitative conversion was observed, and no isomerization of the allyl groups was found. Between 5.17 and 5.39 ppm an overlap of the allyl group and the cholesterol double bond resonances occurs. The acetal group was retained and its signal was detected at 4.82 ppm. The degree of functionalization for Ch-acetal–thiol-coupling20 (green, middle) after thiol–ene coupling was determined by comparing the cholesterol methyl group at 0.72 ppm with the resonances at 1.83 ppm and 2.62 ppm. These two resonances correspond to the methylene groups adjacent to the sulfur (2.62 ppm) and the next but one methylene group. Here, the acetal group at 4.82 ppm and the cholesterol double bond at 5.34 ppm are still intact. After the hypergrafting of glycidol the spectrum for Ch-acetal-hbPG35 was obtained in DMSO-d6 (blue, bottom) which clearly shows that the signals of the methylene groups near the sulfur are still present. The grey frame highlights the acetal resonance, which remains at 4.82 ppm in every stadium of the reaction route. The resonances of the hydroxyl groups appear between 5.00 ppm and 5.20 ppm, whereas the cholesterol double bond shows a peak at 5.32 ppm. The integral of the polyether backbone signals between 3.20 ppm and 3.80 ppm is used to calculate the number of glycidol groups by subtraction of the polyether signals originating from the precursor (macroinitiator). Residual CH2Cl2 originates from washing the polymer to remove traces of NMP.
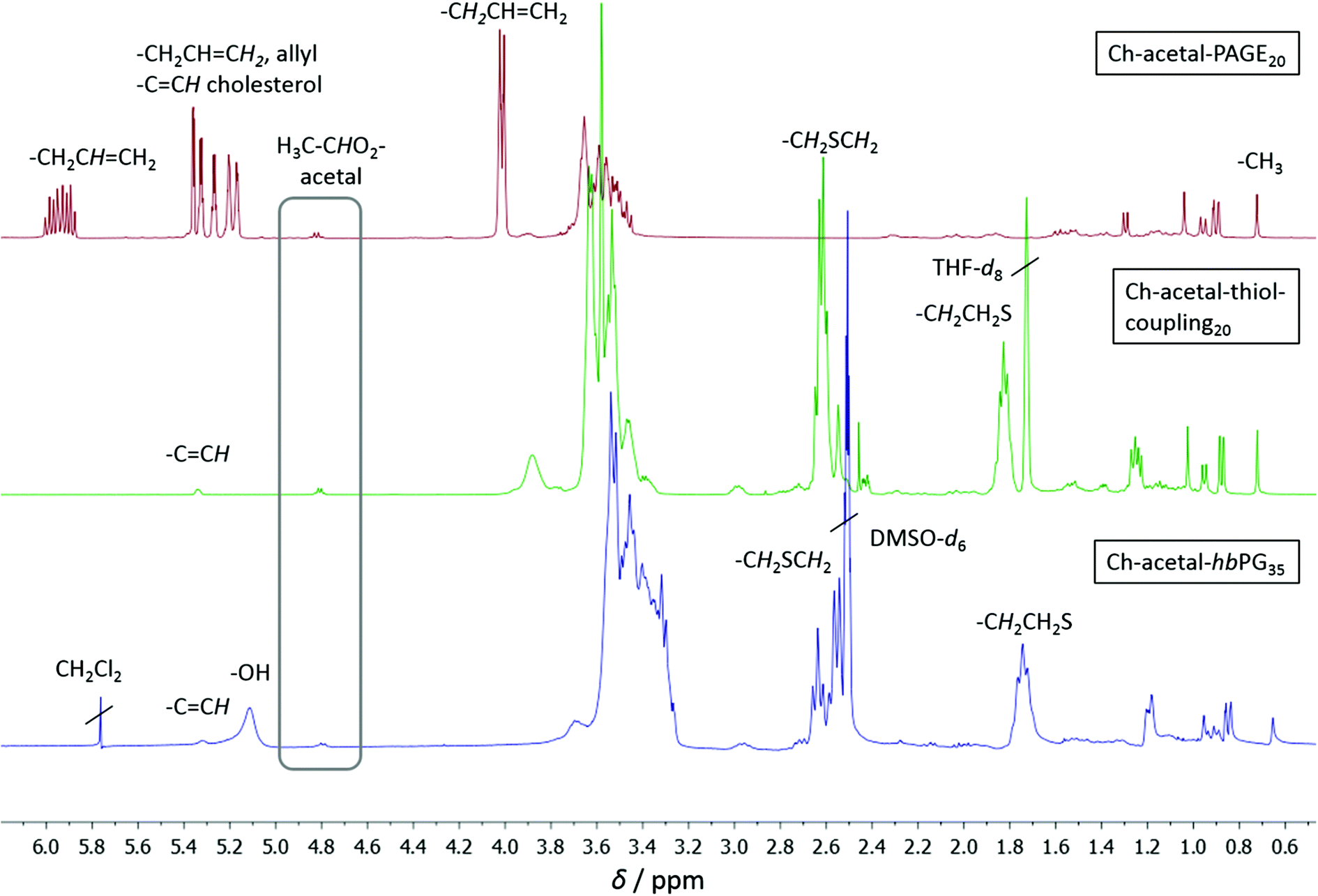 | ||
| Fig. 5 1H NMR spectra for the copolymers with one single acetal group; top to bottom: red: Ch-acetal-PAGE20 in CD2Cl2; green: Ch-acetal–thiol-coupling20 in THF-d8; blue: Ch-acetal-hbPG35 in DMSO-d6. | ||
Incorporation of the acetal containing cholesteryl initiator in every polymer chain is crucial for the behavior of the resulting lipid and was verified via matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-ToF MS) for Ch-acetal-PAGE20 and Ch-acetal–thiol-coupling20, as shown in Fig. 6. The use of the lipophilic initiator permits anchoring of the polymers in the liposomal membranes. Fig. 6a shows the distribution of Ch-acetal-PAGE20, whereas Fig. 6b depicts the distribution of Ch-acetal–thiol-coupling20. In this spectrum, a very small subdistribution was detected, which can be attributed to non-coupled, residual Ch-acetal-PAGE20 (marked with an asterisk). An overlap of both spectra is shown in Fig. 6c. The peak differences translate to exactly 114 g mol−1, which represents the molecular weight of the allyl glycidyl ether repeating unit (Fig. 6a). On the other hand, the molecular weight of 192 g mol−1, which corresponds to the hydroxyl functional repeating units (C8O3SH16), is detectable in the red spectrum in Fig. 6b (Ch-acetal–thiol-coupling20). In summary, the MALDI-ToF characterization confirms the structures of the novel lipids.
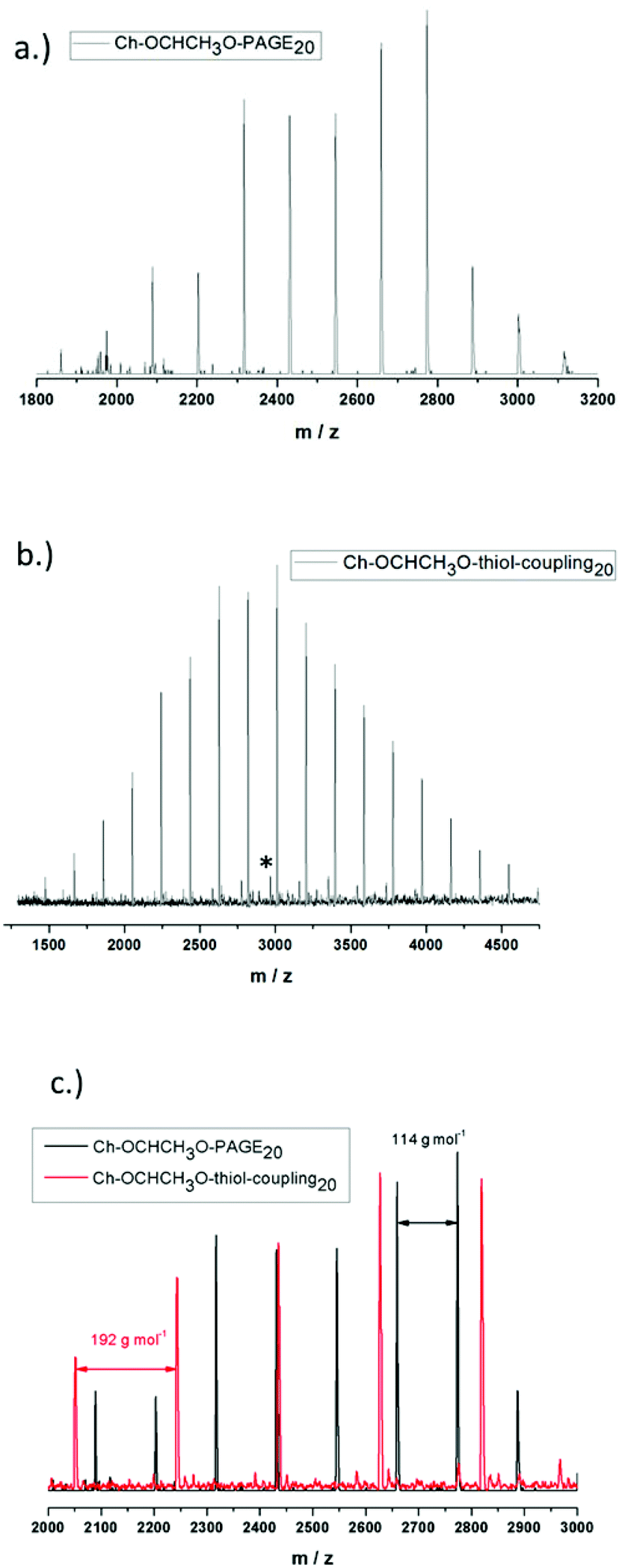 | ||
| Fig. 6 MALDI-ToF spectra for (a) Ch-acetal-PAGE20 (b) Ch-acetal–thiol-coupling20 (c) overlay of both spectra. | ||
Degradation of hyperbranched lipid with multiple pH-responsive moieties
The acetal-containing polymer Ch-P(GEGE17-b-G18) was investigated using SEC measurements with respect to its acidic degradation at room temperature. For this purpose, the sample was dissolved in buffer solution (pH 2) and the sample was stirred for 24 h. Of course, pH 2 is not relevant for the degradation in tumor tissue, but for proof-of-concept studies, acidic hydrolysis can be demonstrated. Fig. 7 depicts the SEC traces of the macroinitiator Ch-linPG13 (dotted line), the acetal-containing polymer Ch-P(GEGE17-b-G18) (black line), and the degraded product of Ch-P(GEGE17-b-G18) (red line). As expected, the SEC trace for the degraded product shifts to higher elution volume, i.e. lower molecular weights. Furthermore, signals in the lower molecular weight range are observed which can be attributed to “polyether-arms” (fragments) that were cleaved under acidic conditions. The SEC trace for the cleaved polymer does not overlap with the macroinitiator (dotted line). This can be explained by the fact that during the addition of GEGE two different hydroxyl groups are formed. One forms at the end of the acid-labile acetal moiety, and a second one is covalently attached to the macroinitiator. As the likelihood for the addition of the following monomers (GEGE or glycidol) is the same for both hydroxyl groups, only half of the growing arms will be degradable afterwards. Hence, the formation of small fragments and a higher molecular weight mode compared to the macroinitiator, is detected.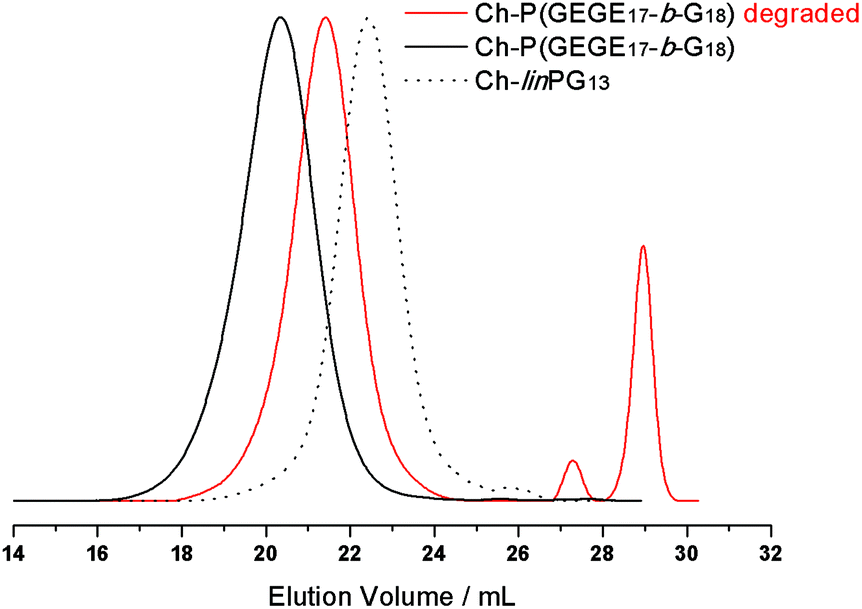 | ||
| Fig. 7 SEC traces (RI detection; DMF) for the macroinitiator Ch-linPG13 (dotted line), the acetal-containing polymer Ch-P(GEGE17-b-G18) (black line), and the degraded product of Ch-P(GEGE17-b-G18) (red line). | ||
pH-Cleavage of Ch-acetal-PEG-CH2-C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) CH and shedding from the liposome
CH and shedding from the liposome
As mentioned above, ideally, a polymer coating should be stable under physiological conditions (pH 7.4), but is cleaved at lower pH, thus enabling membrane–membrane fusion. As a proof-of-principle, we investigated alkyne-functionalized α-(1-(cholesteryloxy)ethoxy)-ω-hydro-PEG46 (Ch-acetal-PEG46-CH2-CCH) in liposome formulations with respect to its shedding properties close to in vivo conditions. To this end, a fluorescent dye (Atto 488 azide) was attached to the polymer after liposome preparation via click-chemistry and acetal cleavage was studied via fluorescent spectroscopy (schematic representation Fig. 8). The crucial point is the protonation of the acetal groups that are located near the phospholipid membrane. As discussed below, it could be demonstrated that acetal-cleavage takes place, although the PEG layer may impede cleavage, slowing down acetal degradation.
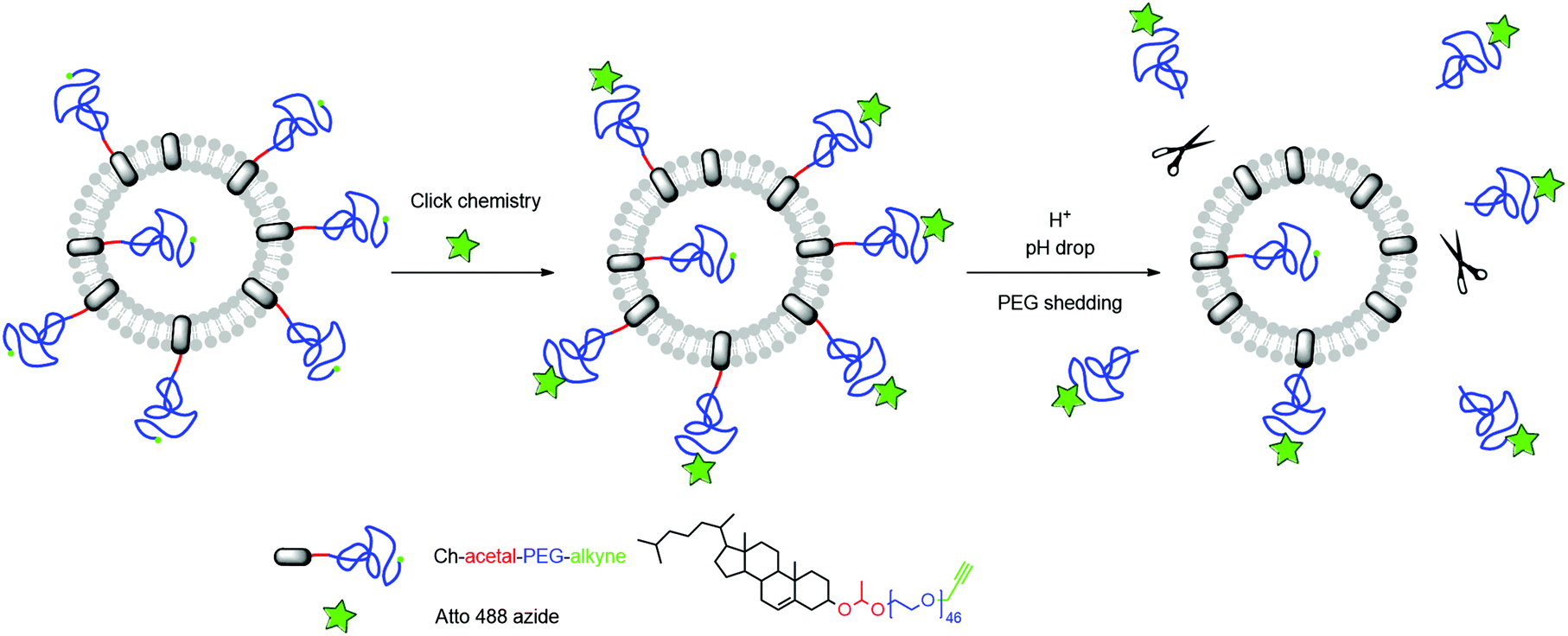 | ||
| Fig. 8 Schematic representation of the liposome functionalization and subsequent acidic cleavage of the PEG-coating. | ||
Alkyne-containing amphiphiles were incorporated into liposomes in a molar fraction of 5 mol%. Liposomes were prepared via dual centrifugation, resulting in z-average hydrodynamic radii (±standard deviation (SD) of 3 measurements) of 170 ± 2.8 nm (μ2/Γ2 = 0.22) and 154 ± 2.2 nm (μ2/Γ2 = 0.10) for amphiphiles with and without acetal groups, respectively. Low values for μ2/Γ2 indicated narrow size distributions as obtained from cumulant analysis of DLS data.
After functionalization of the terminal alkyne group with Atto 488 azide, fluorescent liposomes enabled the observation of the acidic shedding. It turned out that after dilution in PBS and during ongoing equilibration the fluorescence emission decreased to a certain value. This observation is believed to be due to changes in the microenvironment of the fluorophores, e.g., during the partial transition from vesicles to micelles. The cleavage process was then observed after acidification, leading to a strong increase of fluorescence emission intensity, as shown in Fig. 9.
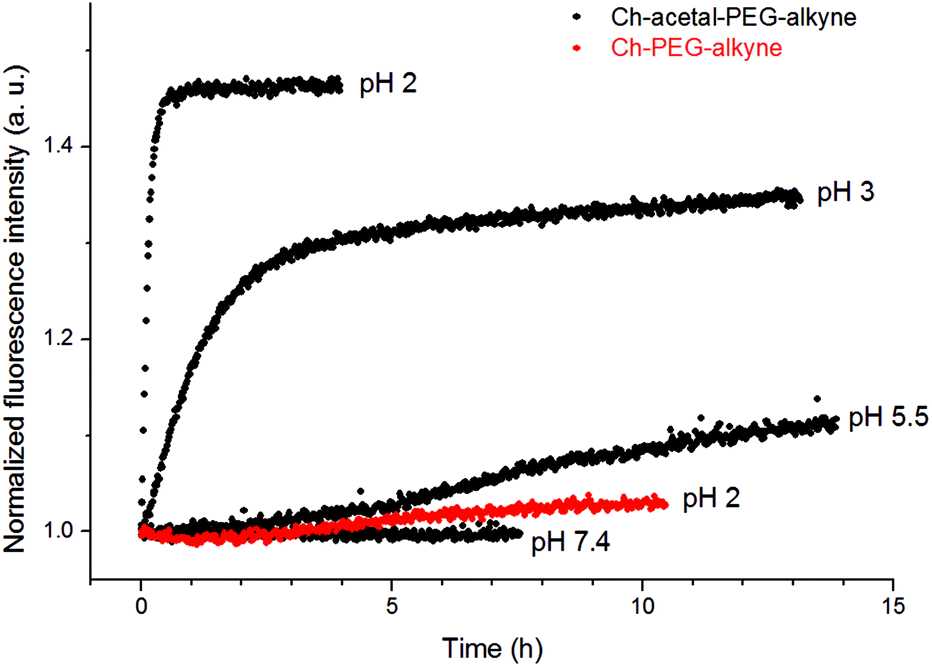 | ||
| Fig. 9 Fluorescence emission increases during acidic cleavage of acetal-containing amphiphiles (Ch-acetal-PEG46-alkyne, black). Amphiphiles without acetal groups (Ch-PEG41-alkyne, red) are stable at low pH. | ||
As expected, a low pH value of 2 led to a fast cleavage within several minutes and therefore high fluorescence emission, while the liposome system without cleavable amphiphiles showed no substantial increase (red data points). However, higher pH values provided degradation within hours (pH 3) or even days (pH 5.5). The increase in fluorescence intensity can be explained by both less thermal relaxation of fluorophores due to surface-related interactions and a potentially lower inner filter effect. While the complex microenvironment within liposomes led to rather qualitative results, the kinetics of the cleavage were in good agreement with earlier measurements.18 Although the cleavage at pH 5.5 was slow, it demonstrates a potential mechanism to shed the liposomal coating in lysosomal compartments. Therefore, the incorporation of acid-labile groups might enable both the elimination of polymeric moieties and the interaction between liposomal and cellular membranes.
Conclusion
In the present study, we showed the incorporation of multiple cleavable groups by combining the epoxide inimer GEGE and glycidol in the oxyanionic ring-opening polymerization with cholesterol as the initiator. Random or block copolymers (Ch-P(GEGEx-co-Gy) or Ch-P(GEGEx-b-Gy)) were synthesized with narrow molecular weight distributions, and the degradation of a block copolymer was investigated by SEC at pH = 2. Furthermore, hyperbranched polyglycerol with a single acetal moiety was introduced using glycol-1-(cholesteryloxy)ethylether as an initiator for the polymerization of AGE, subsequent thiol–ene coupling of 2-mercaptoethanol and polymerization of glycidol. To the best of our knowledge, this represents the first synthesis of acetal-containing hyperbranched lipids for liposome preparations. The polymers obtained are promising with respect to the combination of the following properties: (i) steric stabilization of liposomes, (ii) multifunctionality, (iii) acidic degradability, and (iv) biocompatibility. Degradability in liposome formulations was proven by using the linear analogue (1-(cholesteryloxy)ethoxy)-ω-hydro-PEG46-CH2-CCH functionalized with a fluorescent label (Atto 488 azide) at pH between 7.4 and 2.0. A strong pH-dependence for the shedding process was observed. We believe that acid-labile lipids with a high number of functionalities and multiple cleavable groups mark a promising development step for polyether-based lipids for future biomedical applications.
Acknowledgements
S. S. M. is a recipient of a fellowship through the Excellence Initiative (DFG/GSC 266). T. F. is grateful to the Max Planck Graduate Center with the Johannes Gutenberg-Universität Mainz (MPGC) for a fellowship and financial support. M. W. thanks the MPGC for a fellowship and financial support. This work was supported by the collaborative research center SFB 1066 (Project A7) by the German Research Foundation (DFG). We like to thank Ulrike Kemmer-Jonas for technical assistance and Dr Elena Berger-Nicoletti for MALDI-ToF MS measurements.References
- S. Amselem, A. Gabizon and Y. Barenholz, J. Pharm. Sci., 1990, 79, 1045–1052 CrossRef CAS PubMed
.
- R. Duncan and R. Gaspar, Mol. Pharmaceutics, 2011, 8, 2101–2141 CrossRef CAS PubMed
- B. S. Pattni, V. V. Chupin and V. P. Torchilin, Chem. Rev., 2015, 115, 10938–10966 CrossRef CAS PubMed
- A. Sharma and U. S. Sharma, Int. J. Pharm., 1997, 154, 123–140 CrossRef CAS
- M. C. Woodle, K. K. Matthay, M. S. Newman, J. E. Hidayat, L. R. Collins, C. Redemann, F. J. Martin and D. Papahadjopoulos, Biochim. Biophys. Acta, 1992, 1105, 193–200 CrossRef CAS
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS PubMed
- G. Blume and G. Cevc, Biochim. Biophys. Acta, 1990, 1029, 91–97 CrossRef CAS
- M. L. Immordino, F. Dosio and L. Cattel, Int. J. Nanomed., 2006, 1, 297–315 CrossRef CAS PubMed
- R. L. Hong, C. J. Huang, Y. L. Tseng, V. F. Pang, S. T. Chen, J. J. Liu and F. H. Chang, Clin. Cancer Res., 1999, 5, 3645–3652 CAS
- Z. Amoozgar and Y. Yeo, WIREs Nanomed. Nanobiotechnol., 2012, 4, 219–233 CrossRef CAS PubMed
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed
- I. Mellman, R. Fuchs and A. Helenius, Annu. Rev. Biochem., 1986, 55, 663–700 CrossRef CAS PubMed
- T. L. Andresen, S. S. Jensen and K. Jørgensen, Prog. Lipid Res., 2005, 44, 68–97 CrossRef CAS PubMed
- A. de Milito and S. Fais, Future Oncol., 2005, 1, 779–786 CrossRef CAS PubMed
- A. Asokan and M. J. Cho, J. Pharm. Sci., 2002, 91, 903–913 CrossRef CAS PubMed
- E. Yuba, C. Kojima, A. Harada, T. S. Watarai and K. Kono, Biomaterials, 2010, 31, 943–951 CrossRef CAS PubMed
- K. Remaut, B. Lucas, K. Braeckmans, J. Demeester and S. C. de Smedt, J. Controlled Release, 2007, 117, 256–266 CrossRef CAS PubMed
- C. Dingels, S. S. Müller, T. Steinbach, C. Tonhauser and H. Frey, Biomacromolecules, 2013, 14, 448–459 CrossRef CAS PubMed
- E. R. Gillies, A. P. Goodwin and J. M. J. Fréchet, Bioconjugate Chem., 2004, 15, 1254–1263 CrossRef CAS PubMed
- J. Shin, P. Shum and D. H. Thompson, J. Controlled Release, 2003, 91, 187–200 CrossRef CAS PubMed
- J. A. Boomer, M. M. Qualls, H. D. Inerowicz, R. H. Haynes, V. S. Patri, J.-M. Kim and D. H. Thompson, Bioconjugate Chem., 2009, 20, 47–59 CrossRef CAS PubMed
- J. Shin, P. Shum, J. Grey, S.-I. Fujiwara, G. S. Malhotra, A. González-Bonet, S.-H. Hyun, E. Moase, T. M. Allen and D. H. Thompson, Mol. Pharmaceutics, 2012, 9, 3266–3276 CrossRef CAS PubMed
- X. Guo and F. C. Szoka, Bioconjugate Chem., 2001, 12, 291–300 CrossRef CAS PubMed
- C. Masson, M. Garinot, N. Mignet, B. Wetzer, P. Mailhe, D. Scherman and M. Bessodes, J. Controlled Release, 2004, 99, 423–434 CrossRef CAS PubMed
- X. Guo, J. Andrew MacKay and F. C. Szoka Jr., Biophys. J., 2003, 84, 1784–1795 CrossRef CAS PubMed
- R. M. Sawant, J. P. Hurley, S. Salmaso, A. Kale, E. Tolcheva, T. S. Levchenko and V. P. Torchilin, Bioconjugate Chem., 2006, 17, 943–949 CrossRef CAS PubMed
- A. A. Kale and V. P. Torchilin, Bioconjugate Chem., 2007, 18, 363–370 CrossRef CAS PubMed
- D. Chen, X. Jiang, Y. Huang, C. Zhang and Q. Ping, J. Bioact. Compat. Polym., 2010, 25, 527–542 CrossRef CAS
- H. Xu, Y. Deng, D. Chen, W. Hong, Y. Lu and X. Dong, J. Controlled Release, 2008, 130, 238–245 CrossRef CAS PubMed
- C. Clawson, L. Ton, S. Aryal, V. Fu, S. Esener and L. Zhang, Langmuir, 2011, 27, 10556–10561 CrossRef CAS PubMed
- T. M. Allen and E. H. Moase, Adv. Drug Delivery Rev., 1996, 21, 117–133 CrossRef CAS
- A. M. Hofmann, F. Wurm, E. Hühn, T. Nawroth, P. Langguth and H. Frey, Biomacromolecules, 2010, 11, 568–574 CrossRef CAS PubMed
- A. M. Hofmann, F. Wurm and H. Frey, Macromolecules, 2011, 44, 4648–4657 CrossRef CAS
- S. S. Müller, C. Dingels, A. M. Hofmann and H. Frey, Tailored Polymer Architectures for Pharmaceutical and Biomedical Applications, Am. Chem. Soc., 2013, 1135, 11–25 Search PubMed
- D. Wilms, S.-E. Stiriba and H. Frey, Acc. Chem. Res., 2010, 43, 129–141 CrossRef CAS PubMed
- R. K. Kainthan, J. Janzen, E. Levin, D. V. Devine and D. E. Brooks, Biomacromolecules, 2006, 7, 703–709 CrossRef CAS PubMed
- M. Imran ul-haq, B. F. Lai, R. Chapanian and J. N. Kizhakkedathu, Biomaterials, 2012, 33, 9135–9147 CrossRef CAS PubMed
- R. K. Kainthan, S. R. Hester, E. Levin, D. V. Devine and D. E. Brooks, Biomaterials, 2007, 28, 4581–4590 CrossRef CAS PubMed
- C. Siegers, M. Biesalski and R. Haag, Chem. – Eur. J., 2004, 10, 2831–2838 CrossRef CAS PubMed
- R. A. Shenoi, B. F. L. Lai and J. N. Kizhakkedathu, Biomacromolecules, 2012, 13, 3018–3030 CrossRef CAS PubMed
- R. A. Shenoi, J. K. Narayanannair, J. L. Hamilton, B. F. L. Lai, S. Horte, R. K. Kainthan, J. P. Varghese, K. G. Rajeev, M. Manoharan and J. N. Kizhakkedathu, J. Am. Chem. Soc., 2012, 134, 14945–14957 CrossRef CAS PubMed
- R. A. Shenoi, B. F. Lai, M. Imran ul-haq, D. E. Brooks and J. N. Kizhakkedathu, Biomaterials, 2013, 34, 6068–6081 CrossRef CAS PubMed
- C. Tonhauser, C. Schüll, C. Dingels and H. Frey, ACS Macro Lett., 2013, 1, 1094–1097 CrossRef
- U. Massing, S. Cicko and V. Ziroli, J. Controlled Release, 2008, 125, 16–24 CrossRef CAS PubMed
- T. Fritz, M. Hirsch, F. C. Richter, S. S. Müller, A. M. Hofmann, K. A. Rusitzka, J. Markl, U. Massing, H. Frey and M. Helm, Biomacromolecules, 2014, 15, 2440–2448 CrossRef CAS PubMed
- A. O. Fitton, J. Hill, D. E. Jane and R. Millar, Synthesis, 1987, 1140–1142 CrossRef CAS
- P. Geissler and A. Johnson, J. Am. Oil Chem. Soc., 1990, 67, 541–546 CrossRef CAS
- A. Johnson and P. Geissler, J. Am. Oil Chem. Soc., 1990, 67, 123–131 CrossRef CAS
- J. Pollerberg, Fette, Seifen, Anstrichm., 1966, 68, 561–562 CrossRef CAS
- E. Santacesaria, M. Di Serio, R. Garaffa and G. Addino, Ind. Eng. Chem. Res., 1992, 31, 2413–2418 CrossRef CAS
- A. Sunder, R. Hanselmann, H. Frey and R. Mülhaupt, Macromolecules, 1999, 32, 4240–4246 CrossRef CAS
- B. W. Greenland, S. Liu, G. Cavalli, E. Alpay and J. H. Steinke, Polymer, 2010, 51, 2984–2992 CrossRef CAS
- B. F. Lee, M. J. Kade, J. A. Chute, N. Gupta, L. M. Campos, G. H. Fredrickson, E. J. Kramer, N. A. Lynd and C. J. Hawker, Polym. Chem., 2011, 49, 4498–4504 CrossRef CAS PubMed
- A. Alkan, A. Natalello, M. Wagner, H. Frey and F. R. Wurm, Macromolecules, 2014, 47, 2242–2249 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2016 |