
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot RAFT and fast polymersomes assembly: a ‘beeline’ from monomers to drug-loaded nanovectors†
F.
Mastrotto
a,
A. F.
Breen
a,
G.
Sicilia
a,
S.
Murdan
b,
A. D.
Johnstone
c,
G. E.
Marsh
 a,
C.
Grainger-Boultby
a,
N. A.
Russell
c,
C.
Alexander
a and
G.
Mantovani
*a
a,
C.
Grainger-Boultby
a,
N. A.
Russell
c,
C.
Alexander
a and
G.
Mantovani
*a
aSchool of Pharmacy, University of Nottingham, Nottingham NG7 2RD, UK. E-mail: giuseppe.mantovani@nottingham.ac.uk; Fax: +44 (0)115 846 6296; Tel: +44 (0)0115 823 2348
bDepartment of Pharmaceutics, UCL School of Pharmacy, University College London, 29-39 Brunswick Square, London, WC1N 1AX, UK
cFaculty of Engineering, University of Nottingham, Nottingham NG7 2RD, UK
First published on 20th September 2016
Abstract
Rapid and simple routes to functional polymersomes are increasingly needed to expand their clinical or industrial applications. Here we describe a novel strategy where polymersomes are prepared through an in-line process in just a few hours, starting from simple acrylate or acrylamide monomers. Using Perrier's protocol, well-defined amphiphilic diblock copolymers formed from PEG acrylate (mPEGA480), 2-(acryloyloxy)ethyl-3-chloro-4-hydroxybenzoate (ACH) or 2-(3-chloro-4-hydroxybenzamido)ethyl acrylate (CHB), have been synthesised by RAFT polymerisation in one-pot, pushing the monomer conversion for each block close to completion (≥94%). The reaction mixture, consisting of green biocompatible solvents (ethanol/water) have then been directly utilised to generate well-defined polymersomes, by simple cannulation into water or in a more automated process, by using a bespoke microfluidic device. Terbinafine and cyanocobalamine were used to demonstrate the suitability of the process to incorporate model hydrophobic and hydrophilic drugs, respectively. Vesicles size and morphology were characterised by DLS, TEM, and AFM. In this work we show that materials and experimental conditions can be chosen to allow facile and rapid generation drug-loaded polymersomes, through a suitable in-line process, directly from acrylate or acrylamide monomer building blocks.
Introduction
Polymersomes1 are vesicular assemblies formed from amphiphilic block copolymers, which have rapidly emerged as an important class of nanomaterials, with range of potential applications which include drug delivery, diagnostics and bioimaging.2–4 To date, polymersomes have been the subject of over 500 papers, with at least 11 patents filed within the last two years.5 Compared to phospholipid vesicles, polymersomes typically possess improved membrane stability, as well as superior tuneability of surface chemistry and structural parameters.6An increasingly large area of research is focussed on the use of polymersomes as drug nanovectors in drug delivery.7–10 Recently, Battaglia and coworkers showed delivery of an IgG model cargo to the Central Nervous System (CNS) parenchyma and CNS cells mediated by poly(2-diisopropylamino)ethyl methacrylate)-based pH-responsive polymersomes functionalized with LRP-1-targeting Angiopep-2 and Rabies Virus Glycoprotein (RVG) peptide ligands to facilitate transport through the blood–brain barrier.11
Polymersomes have also been utilised to create nanosized reactors, where entrapped (bio)molecules – e.g. enzymes – are part of multicompartmental arrangements mimicking those found in cell organelles. In an early work Meier and coworkers incorporated β-lactamase within PDMS-PMOXA triblock copolymer vesicles.12 The introduction of a channel-forming protein from the outer cell wall of Gram-negative bacteria (OmpF) into the vesicle membrane allowed passive transport of ampicillin, an enzyme substrate, which was converted in the polymersome aqueous lumen into ampicillinoic acid. Lecommandoux and van Hest engineered multicompartimentalised polymersomes-in-polymersomes systems, mimicking artificial cells and cell organelles.13 These studies, in conjunction with the potential for self-propulsion,14 make polymersomes ideal candidates for the development of polymeric protocells.15,16
In terms of their manufacturing, polymersomes are often assembled from amphiphilic block copolymers synthesised through long synthetic procedures – typically at least two sequential polymerisations, requiring purification of both polymer intermediates and final amphiphilic macromolecules from unreacted monomers and traces of other low molecular weight impurities. Importantly, polymerisation reactions often need to be stopped at moderate monomer conversions to ensure high proportion of chain-end fidelity, which is required for subsequent polymer chain-extension.17–19
In addition to being not fully efficient, this approach invariably results in waste of functional monomers utilised for the polymerisation step, which is undesirable especially when these are particularly expensive, not commercially available, or both. Finally, the processes for the assembly of polymersomes are not always trivial.20 Although a range of techniques have been developed,21 most methods still suffer from low reproducibility and poor scalability, which may complicate industrial application of polymersomes.6 Whilst chemists are increasingly focussing on developing more efficient and greener chemical transformations – i.e. as described by Sharpless and coworkers’ click chemistry philosophy – one could envisage that analogous approaches could be applied not only to individual chemical reactions, but also to entire processes. A clear move in this direction is the work on polymerization-induced self-assembly (PISA) by Armes’ group and others,22–30 where block copolymers nano-objects can be assembled with predictable shape and size during the polymerisation process, by judicious choice of monomers and variation of polymer physicochemical characteristics (e.g. packing parameter P).31
In the present work we aimed to develop a route to self-assembled drug nanovectors which would complement the range of applications inherent to PISA. In this case, drug-loaded nanovesicles are engineered by sequential and quantitative polymerisation of appropriate monomers in one pot, followed by self-assembly of the resulting amphiphilic block copolymers into vesicles, by directly adding the final reaction mixture to a suitable aqueous phase (Chart 1). Key advantages of this approach include the avoidance of intrinsically inefficient purifications of polymer intermediates, the potential for incorporating a wide range of drugs – including those not compatible with radical polymerisation conditions, and, similarly to PISA, facile assembly of nanovectors directly from the polymerisation solution, but without having to remove excess unreacted monomers.
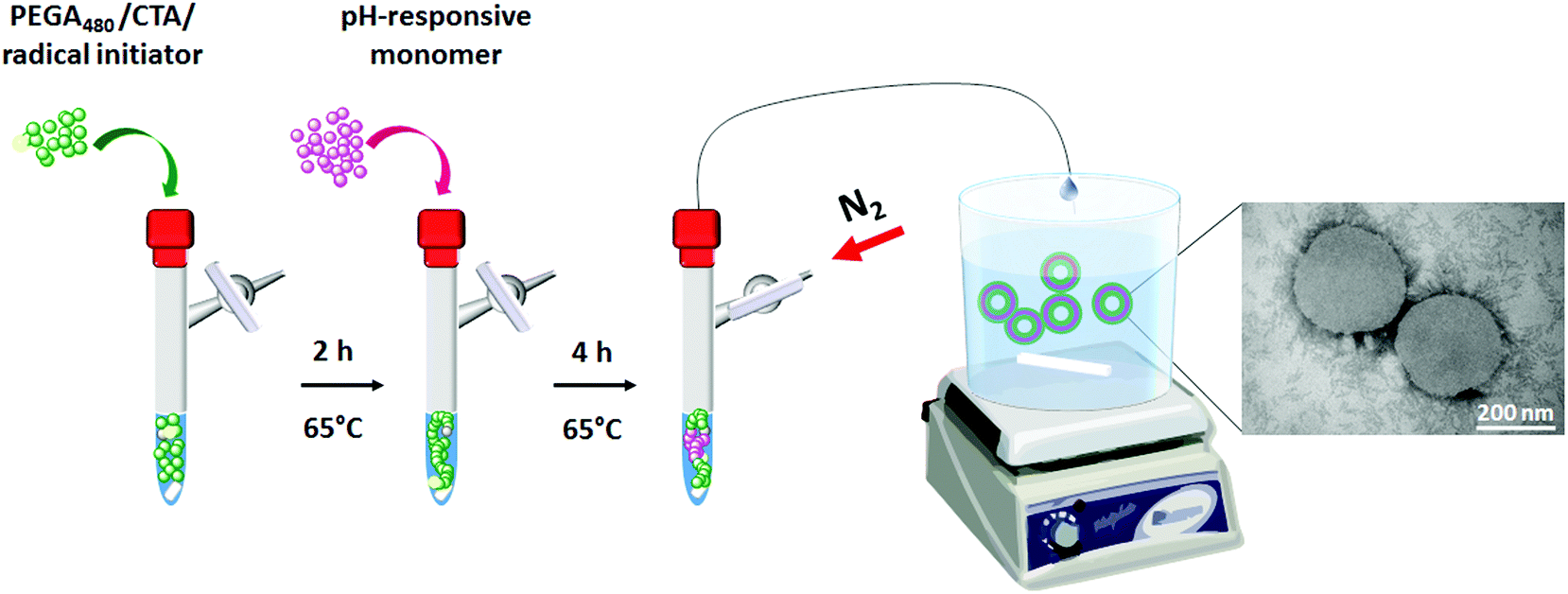 | ||
| Chart 1 In line engineering of mPEGA12a-b-ACH36 or mPEGA12a-b-CHB36 polymeric vesicles via one-pot RAFT and in line post-polymerisation assembly. | ||
For the polymerisation step, recent studies by Perrier and co-workers showed that well-defined multiblock copolymers can be obtained by RAFT polymerisation in one pot by sequential and quantitative polymerisation of suitable monomers in only a few hours18,32,33 or even minutes.34 The monomers were selected based on previous studies, where we showed that block copolymers based on novel pH-responsive monomers bearing substituted aromatic alcohols could be effectively assembled into well-defined nanovesicles, whilst incorporating specific drugs with high efficiency.35,36
Experimental
Materials
Terbinafine free base was extracted from a basic aqueous solution of terbinafine hydrochloride (Sigma-Aldrich ≥98%) with dichloromethane (see ESI†). VA-044 was purchased from Wako Pure Chemical Industries Ltd. All the other solvents and reagents were purchased from Sigma-Aldrich or Fisher Scientific and used as received. Spectra/Por 8 kDa MWCO and SnakeSkin 3.5 kDa MWCO dialysis tubing were obtained from Fisher Scientific and VWR International, respectively. Cellulose membranes (0.45 and 0.22 μm) were purchased from Merck Millipore. 1H–13C HSQC NMR experiments were carried out to facilitate peaks assignment. Yields were not optimized.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 vol/vol). Evaporation of the solvent from the relevant fractions yielded the desired monomer ACH (2) as a white solid. CHB (3) was synthesised using THF instead of CH2Cl2 as the solvent and was purified by flash chromatography (silicagel 60, 35–70 μm, eluents CH2Cl2/EtOAc, gradient 9.5:0.5 to 1:1, vol/vol).
:0.5 vol/vol). Evaporation of the solvent from the relevant fractions yielded the desired monomer ACH (2) as a white solid. CHB (3) was synthesised using THF instead of CH2Cl2 as the solvent and was purified by flash chromatography (silicagel 60, 35–70 μm, eluents CH2Cl2/EtOAc, gradient 9.5:0.5 to 1:1, vol/vol).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHH), 6.14 (dd, J = 17.3, 10.4 Hz, 1H, CHCH2), 6.42 (dd, J = 17.3, 1.4 Hz, 1H, CHCHH), 7.07 (d, J = 8.6 Hz, 1H, CH aromatic), 7.9 (dd, J = 8.5, 2.0 Hz, 1H, CH aromatic), 8.03 (d, J = 2.0 Hz, 1H, CH aromatic). 13C {1H} NMR (100 MHz, CDCl3, δ, ppm): 62.43 (1C, CH2O), 63.00 (1C, CH2O), 116.24 (1C, C aromatic), 120.23 (1C, C aromatic), 123.29 (1C, C aromatic), 128.08 (1C, CHCH2), 130.52 (1C, C aromatic, CH), 131.21 (1C, C aromatic), 131.65 (1C, CH2CH), 155.76 (1C, C aromatic), 165.13 (1C, CO), 166.03 (1C, CO).
CHH), 6.22 (dd, 1H, J = 17.1, 3.7 Hz, CHCHH), 6.27 (dd, 1H, J = 17.1, 8.3 Hz, CHCH2), 6.95 (d, 1H, J = 8.5 Hz, CH aromatic), 7.82 (m, 1H, CH aromatic), 7.98 (m, 1H, CH aromatic). 13C {1H} NMR (100 MHz, MeOD, δ ppm): 39.55 (1C, CH2NH), 64.51 (1C, CH2O), 117.1 (1C, C aromatic, CH), 121.8 (1C, C aromatic, CCl), 123.18 (1C, C aromatic, CCO), 126.96 (1C, CH2C), 130.95 (1C, C aromatic, CH), 131.83, (1C, CHCH2), 132.79 (1C, C aromatic, CH), 159.16 (1C, C aromatic, C–OH), 166.83 (1C, CO), 168.45 (1C, CO).
CHH), 6.14 (dd, J = 17.3, 10.4 Hz, 1H, CHCH2), 6.42 (dd, J = 17.3, 1.4 Hz, 1H, CHCHH), 7.07 (d, J = 8.6 Hz, 1H, CH aromatic), 7.9 (dd, J = 8.5, 2.0 Hz, 1H, CH aromatic), 8.03 (d, J = 2.0 Hz, 1H, CH aromatic). 13C {1H} NMR (100 MHz, CDCl3, δ, ppm): 62.43 (1C, CH2O), 63.00 (1C, CH2O), 116.24 (1C, C aromatic), 120.23 (1C, C aromatic), 123.29 (1C, C aromatic), 128.08 (1C, CHCH2), 130.52 (1C, C aromatic, CH), 131.21 (1C, C aromatic), 131.65 (1C, CH2CH), 155.76 (1C, C aromatic), 165.13 (1C, CO), 166.03 (1C, CO).
CHH), 6.22 (dd, 1H, J = 17.1, 3.7 Hz, CHCHH), 6.27 (dd, 1H, J = 17.1, 8.3 Hz, CHCH2), 6.95 (d, 1H, J = 8.5 Hz, CH aromatic), 7.82 (m, 1H, CH aromatic), 7.98 (m, 1H, CH aromatic). 13C {1H} NMR (100 MHz, MeOD, δ ppm): 39.55 (1C, CH2NH), 64.51 (1C, CH2O), 117.1 (1C, C aromatic, CH), 121.8 (1C, C aromatic, CCl), 123.18 (1C, C aromatic, CCO), 126.96 (1C, CH2C), 130.95 (1C, C aromatic, CH), 131.83, (1C, CHCH2), 132.79 (1C, C aromatic, CH), 159.16 (1C, C aromatic, C–OH), 166.83 (1C, CO), 168.45 (1C, CO).
General one-pot polymerisation procedure: synthesis of mPEGA12a-b-ACH36 (6), block copolymers
The synthesis of the pH-responsive block copolymers was performed via an adaptation of the one-pot RAFT polymerisation strategy described by Perrier and co-workers.18 Monomer conversion after each step are shown in Table 1.| Polymer | Code | [M]/[CTA]/[I] | Conv.%a | M n, NMR (kDa) | M n, SEC (kDa) | Đ |
|---|---|---|---|---|---|---|
| a Obtained from 1H NMR analysis. b Number of repeating unit of ACH or CHB monomers, DPACH and DPCHB respectively, were calculated by 1H NMR by comparing the integral of monomer aromatic proton peak at 6.93 (I6.93) ppm, and the methoxy peak of mPEGA repeating units at 3.23 ppm (I3.23) set as 3. DPACH (or DPCHB) = 12 × (I6.93/(I3.23/3). Mn, NMR was calculated as: (−DPACH (or DPCHB) × MWmonomer) + Mn, macroCTA). c Obtained from SEC analysis in DMF + 0.1% LiBr (PMMA standards). d Obtained from SEC analysis in CHCl3 (polystirene standards). | ||||||
| mPEGA 12a | 4 | 12:1:0.004 |
95 | 5.7 | 6.5c | 1.05c |
| mPEGA 12b | 5 | 12:1:0.004 |
98 | 5.3 | 6.1d | 1.13d |
| mPEGA 12a -b-ACH 36 | 6 | 36:1:0.02 |
94 | 15.5b | 14.9c | 1.18c |
| mPEGA 12b -b-CHB 36 | 7 | 36:1:0.034 |
95 | 15.2b | 15.1d | 1.17d |
:solvent vol:vol ratio was found to be critical to achieve the required degree of control over the chain end-group fidelity, in agreement with what described by Perrier and co-workers.18,32–34 Here a 2.0 M monomer concentration was used in all cases, and when liquid monomers were used, their volume was taken into account when calculating the solvent required to achieve the required monomer concentration). The tube was sealed with a rubber septum, placed in an ice bath, stirred for 5 min to allow temperature equilibration, deoxygenated by gentle Argon bubbling for 15 minutes, and finally placed in an oil bath pre-heated at 70 °C. 0.332 μmol of VA-044 (59 μL of a 1.82 mg mL−1 deoxygenated stock solution in HPLC-grade water) were then added via gas-tight syringe to the reaction tube (t0). After 2 h near-quantitative conversion of mPEGA(480) into mPEGA12a macro-CTA was confirmed by 1H NMR of an aliquot taken from the reaction mixture, which was also analysed by SEC (DMF + 0.1% LiBr as the mobile phase) to estimate Mn and polydispersity index of the mPEGA12a polymer intermediate.
In a separate Schlenk tube ACH monomer (803 mg, 2.96 mmol) was dissolved in 1.5 mL of water:EtOH (1:2 vol/vol); the tube was placed in an ice bath, stirred for 5 min to allow temperature equilibration, and deoxygenated by gentle Argon bubbling for 15 minutes. This solution was then cannulated under Argon into the polymerisation tube containing mPEG12a macro-CTA, and the resulting solution heated at 70 °C under stirring. The polymerisation was started by addition of a previously degassed VA-044 solution in HPLC-grade water (96 μL, 0.54 μmol) via gas-tight syringe. After 2 h at 70 °C ACH monomer conversion was checked by 1H NMR in DMSO-d6. Three sequential additions of degassed fresh initiator solution (96 μL), each followed by 2 h stirring at 70 °C were performed, until conversion finally reached 94%.
In line assembly of polymersomes
In a typical procedure, at the end of the polymerisation run the reaction mixture was diluted with EtOH to a final polymer concentration of 35 mg mL−1 while maintaining the temperature at 40 °C, under stirring. The resulting polymer solution was then directly cannulated into DI water (EtOH:H2O 1:35 vol/vol, final polymer concentration 1.0 mg mL−1).
The polymersomes formation occurred instantaneously, as confirmed by DLS. To remove the residual ethanol, the vesicles suspension was transferred into a dialysis bag (3.5 kDa MWCO) and dialyzed against 5 L of DI water for 4 hours at room temperature with at least 4 water exchanges. Alternatively, the beaker containing the polymersomes suspensions were left to stir overnight to allow the organic solvent to evaporate. Samples were analysed by DLS and TEM.
Polymersomes assembly by nanoprecipitation
For comparative purposes, assembly of polymersomes was also performed by nanoprecipitation on purified block copolymers. Briefly, at the end of the polymerisation reaction mixtures were diluted with acetone (10 mL) and precipitated twice in petroleum ether (200 mL). 5 mg mL−1 polymer solutions were then prepared in DMSO, and diluted to 1 mg mL−1 by addition of either water or PBS. To remove the organic solvent, vesicles suspensions were transferred into a dialysis bag (3.5 kDa MWCO) and dialyzed against 5 L of DI water or PBS for 24 hours at room temperature with at least 4 solvent exchanges. Vesicles size was analysed by DLS and, for samples prepared in PBS, by atomic force microscopy (AFM).Polymersome assembly via a microfluidic device
Polymersomes were also assembled using a bespoke microfluidic device. Two syringes feeding into the device were driven simultaneously by a dual Cole-Palmer 789210C syringe pump. A 10 mL syringe (internal diameter 15.8 mm) delivered at 12 times the volume flow rate of the 1 mL syringe (internal diameter 4.6 mm). The 1 mL syringe was maintained at 50 °C by a heating jacket and contained an ethanolic solution (12 mg mL−1) of mPEGA12b-b-CHB36. This solution was injected into the device at a flow rate of 10 μL min−1 (Flow A, Fig. 4), via a 27G needle (internal diameter 0.21 mm) inserted into a PTFE tube (ID 0.5 mm). The tube ensured a leak-free fit while allowing the needle to be removed as required. Concurrently, water was introduced in the device via the 10 mL syringe, through another ID 0.5 mm PTFE tube, at a flow rate of 120 μL min−1 (Flow B, Fig. 4). The tubes led to a mixing junction placed on a 60 °C hotplate, and the mixed flow (Flow C, 130 μL min−1, Fig. 4) excited the device to a collection vial via an ID 1 mm tube. The experiment was repeated twice and at least 3 different samples were collected and analysed by DLS.Drug loading procedure
Terbinafine free base and cyanocobalamin co-loaded polymersomes were assembled following a procedure analogous to that described in the “in line assembly of polymersomes” section. Typically, terbinafine free base (10% w/w drug/polymer) and cyanocobalamin (10% w/w drug/polymer) were respectively added to the polymer ethanolic/water solution and/or the aqueous phase before the assembly procedure. To remove unentrapped cyanocobalamin and residual organic solvent the polymersomes suspension was dialysed with 8 kDa MWCO dialysis membrane against 5 L of DI water, with 4 water exchanges in 5 hours. Unentrapped terbinafine, precipitated after dialysis, was removed by centrifugation (1500 rpm, 5 minutes) followed by filtration through a 0.45 μm mixed cellulose ester membrane.Drug loading was then quantified via a RP-HPLC method (see ESI†). Loading reported for each polymer is the average of two independent experiments, each prepared and analysed in triplicate.
Loading Capacity (LC) and Encapsulation Efficiency (EE) were calculated as:
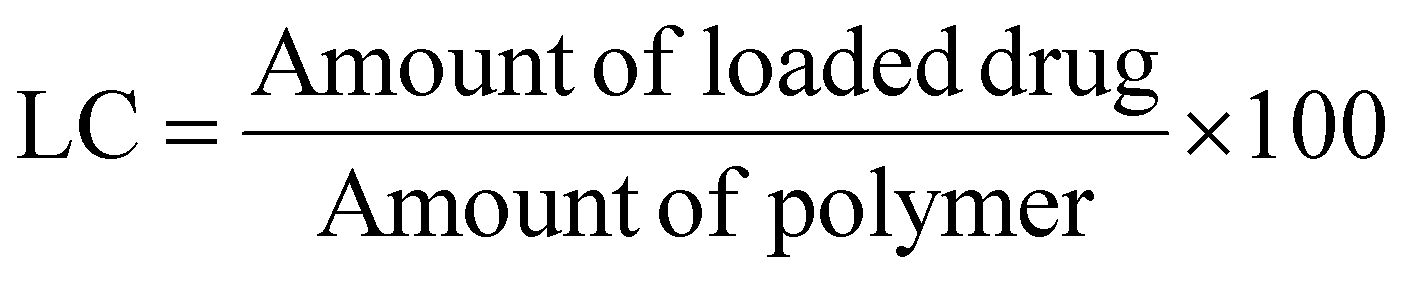

Polymersome size analysis
Particles size analyses were carried out at 25 °C by Dynamic Light Scattering (DLS) using a Zetasizer Nano spectrometer (Malvern Instruments Ltd) equipped with a 633 nm laser at a fixed angle of 173°. Samples were kept at constant temperature (25 °C) during all the experiments. Polymeric vesicles were prepared as described in “in line assembly of polymersomes”, by nanoprecipitation or via microfluidic device. Residual EtOH was removed either by solvent evaporation or by dialysis with a 3.5 kDa MWCO dialysis membrane before analysis. Results are reported as the z-average. All experiments were performed in triplicate.Transmission electron microscopy (TEM)
Transmission Electron Microscopy (TEM) was performed on a FEI TecnaiTM 12 Biotwin transmission electron microscope. 20 μL of 1 mg mL−1 polymersomes suspension in water were placed on a carbon coated copper grid. After 5 minutes the excess of volume was wicked away with filter paper, the samples were negatively stained with 3% uranyl acetate in distilled water and analysed. 191 and 207 individual vesicles were measured from the digitised images for mPEGA12a-b-ACH36 and mPEGA12b-b-CHB36, using imageJ 1.45 analysis software.Atomic force microscopy (AFM)
Atomic Force Microscopy topography images of mPEGA12a-b-ACH36 and mPEGA12a-b-CHB36 polymersomes were obtained in liquid at room temperature using a Bruker Icon FastScan Bio operating in PeakForce nanomechanical tapping mode in fluid using ScanA-syst-Fluid + probes (resonant mechanical frequency: 120–180 kHz, spring constant: 0.7 N m−1). Images were acquired under ScanAsyst auto control using scan rates of 1.60 Hz, with a resolution of 512 × 512 pixels. Small pieces (0.5 cm × 0.5 cm) of freshly cleaved mica were used as substrates. 10 mM MgCl2 solution was incubated with cleaved mica for 30 min. Subsequently, the mica was washed with distilled water and blow-dried under nitrogen stream at room temperature. Polymersomes prepared by the nanoprecipitation method followed by dialysis against PBS were diluted with additional PBS to a final polymer concentration of 500 μg mL−1 and filtered through a 0.45 μm syringe filter. The diluted polymersome solution (100 μL) was pipetted on the mica substrate and then imaged. Image data were analysed using Nanoscope Analysis software (Version 1.5, Bruker).Results and discussion
In line polymersomes: synthesis of ACH acrylate and CHB acrylamide-based copolymers mPEGA12a-b-ACH36 and mPEGA12b-b-CHB36
Accordingly, in this work the required monomers were prepared in one step, by DCC-mediated coupling reaction of 3-chloro-4-hydroxybenzoic acid with (hydroxyethyl)acrylate (HEA) or (hydroxyethyl)acrylamide (HEAM), to give acrylate and acrylamide monomers ACH (2) and CHB (3), respectively (Scheme 1, and ESI, Fig. S5–S8†). Unlike our previous studies where methacrylates were utilised,35,36 in this work the analogous acrylate/acrylamide monomers, characterised by higher propagation rate coefficient (Kp),37,38 required for subsequent one-pot rapid multiple chain extensions at close to full monomer conversion, as described by Perrier and co-workers, were synthesised. To prove the versatility of the strategy developed in this study, both acrylate and acrylamide monomers were utilised.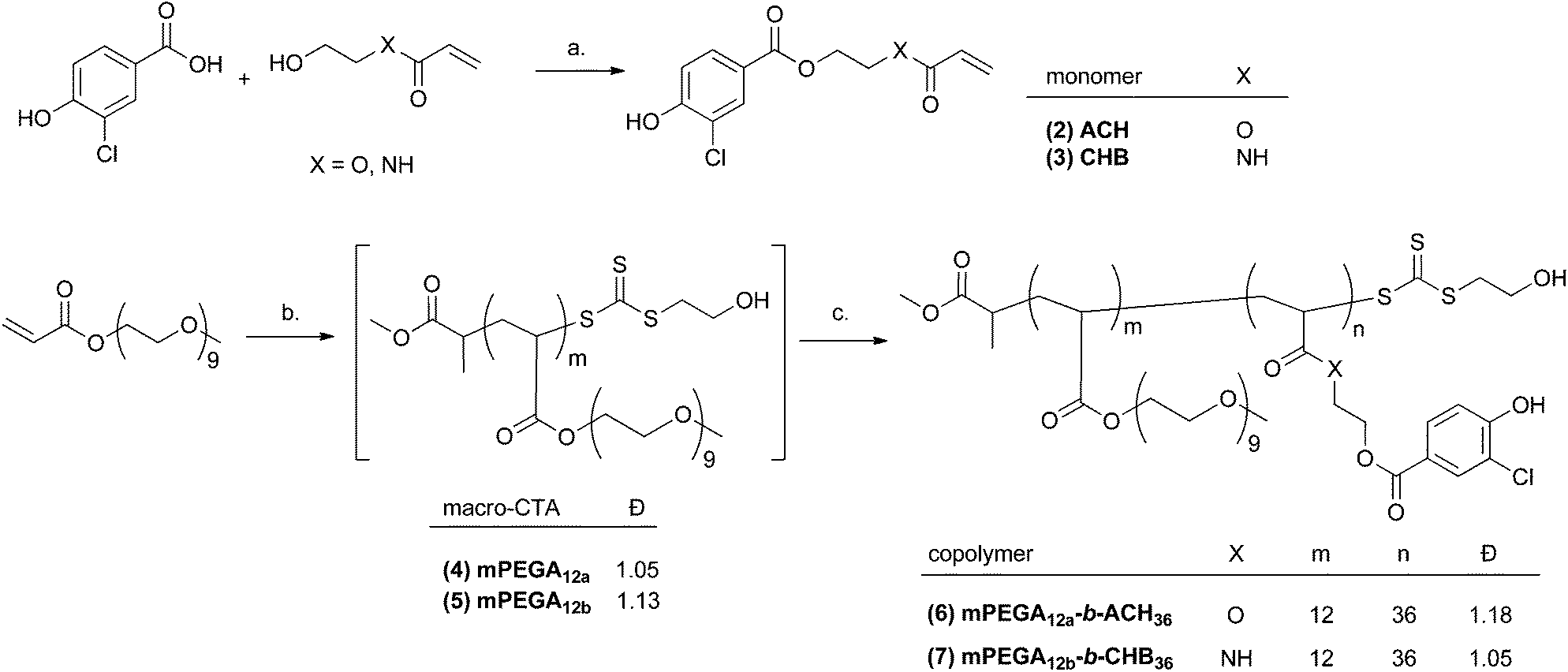 | ||
| Scheme 1 One-pot RAFT synthesis of amphiphilic mPEGA12a-b-ACH36 and mPEGA12b-b-CHB36 block copolymers. Reagents and conditions: a. DCC, DMAP, 16 h, ambient temperature. Solvent: XO: CH2Cl2. XNH: THF; b. MHP (CTA), VA-044, EtOH/H2O, 70 °C. c. XO: mPEGA12a macro-CTA, ACH monomer, VA-044, EtOH/H2O, 70 °C; XNH: mPEGA12b macro-CTA, CHB monomer, VA-044, EtOH/H2O, 70 °C. | ||
To successfully prepare multiblock copolymers via this one-pot RAFT process, a number of parameters – e.g. choice of the solvent, initiator, monomer concentration, temperature and [CTA]/[I] ratio – are key for maintaining near-perfect chain-end fidelity, thus ensuring “livingness” of the polymerisation.18
A mixture of water/EtOH was chosen as a suitable “green” solvent39 for the subsequent polymerisation step. Water is known to increase the propagation rate coefficient (Kp), and thus the polymerisation rate (Rp), of vinyl monomers,40 which is critical for efficient in situ RAFT synthesis of multiblock copolymers,18 whilst EtOH was required to solubilise the monomers ACH (2) and CHB (3), and the (methyl 2-((((2hydroxyethyl)thio)carbonothioyl)thio)propanoate), (MHP) chain-transfer agent. The latter was chosen due to the higher hydrolytic stability of trithiocarbonates compared to other suitable CTAs – e.g. dithioesters – and their ability to minimise initial rate retardation sometimes observed in RAFT polymerisations.41 In the one-pot protocol optimised in this work a macro-CTA was first synthetised using commercially available poly[ethylene glycol(480)]methyl ether acrylate, mPEGA(480) and 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044) as the water soluble initiator [I], at 70 °C (Scheme 1). VA-044 is characterised by a high decomposition rate (t1/2 2 h at 70 °C), which again is key to speeding up the polymerisation without losing the chain livingness.32,33 mPEGA(480) was chosen for the synthesis of the first hydrophilic block because of its aqueous solubility, biocompatibility and ability to generate polymers with prolonged plasma half-life and low immunogenicity.42–44 After an initial series of experiments required to identify suitable reaction conditions (i.e. monomer concentration and [CTA]/[I] molar ratio) which would allow to reach near-quantitative monomer conversion whilst retaining chain-end fidelity, a [CTA]/[I] ratio of 250:1 and a monomer concentration of 2.0 M were chosen. The degree of polymerisation (DP) for the p(mPEGA) macro-CTA was targeted to 12. After 2 h the reaction reached a monomer conversion >95%, as assessed by 1H NMR, by comparing the decrease of the signal for the vinyl proton at ∼5.96 ppm to that of the mPEG terminal methoxy group at 3.24 ppm (see ESI, Fig. S9 and S10†). SEC analysis of the macro-CTA (mPEG12a, mPEG12b, Table 1 and Fig. 1), using DMF + 0.1% LiBr as the mobile phase showed unimodal, symmetric, and narrow molecular weight distributions (Đ < 1.13), with Mn, SEC in good agreement with the theoretical one (Table 1, polymers 4 and 5). Two different macro-CTAs, mPEGA12a and mPEG12b, both with DP = 12 were synthesised and directly utilised without isolation or purification for the synthesis of mPEGA12a-b-ACH36(6) and mPEGA12b-b-CHB36(7), respectively. Importantly, both reactions led to the desired mPEGA12 macro-CTAs with near-quantitative monomer conversion, thus validating the reproducibility of this process. Both acrylate ACH (2) and acrylamide CHB (3) monomers have limited solubility in water, hence for the second polymerisation step they were first dissolved in EtOH, then the resulting solutions were diluted with water to a final EtOH/water volume ratio of 2:1. To ensure full monomer conversion in the one-pot RAFT with higher EtOH content, a larger amount of radical intitiator was required;33 in this work for the second block a [macro-CTA]/[I] ratio of 150:1 was initially chosen in both cases. A DP of 34 for both ACH and CHB was targeted, to provide final copolymers with a hydrophilic block weight fraction (f = 35% ± 10%)45 suitable for self-assembling into vesicles.2 Indeed, when considering the Mn provided by 1H NMR analysis the calculated f values were 0.358 and 0.357 for mPEGA12a-b-ACH36 and mPEGA12b-b-CHB36, respectively.
 | ||
| Fig. 1 SEC traces and 1H NMR spectra (DMSO-d6) of non-purified mPEGA12 macro-CTA and final block copolymers for A. mPEGA12a-b-ACH36(6), and B. mPEGA12b-b-CHB36(7). | ||
Following the addition of ACH (2) or CHB (3) monomers and VA-044 radical initiator, a monomer conversion of 80–90% was observed after 2 hours. Additional radical initiator was necessary to achieve >95% of monomer conversion (Table 1, polymer 6 and 7, see ESI Fig. S11 and S12†), resulting in a final [macro-CTA]/[I] ratio of 1:50 or 1:30 for mPEGA12a-b-ACH36 and mPEGA12b-b-CHB36, respectively. A possible explanation for this is that the second block grown here is longer than most of those prepared by Perrier and coworkers for which near-full conversion could be achieved under the conditions reported,18,27,32–34 although other side-processes – e.g. very minimal H transfer from ACH (2) or CHB (3) monomers – cannot in theory be ruled out at this stage. Nevertheless, pleasingly, SEC analysis (Table 1 and Fig. 1) showed that very well defined block-copolymers with a final Đ of 1.18 (mPEGA12a-b-ACH36) and 1.17 (mPEGA12b-b-CHB36), were synthesised indicating good control over the polymerisation process. The DP of the second block was determined by 1H NMR, by comparing the integral of the OCH3 signal of mPEGA repeating units in the hydrophilic block at 3.24 ppm, with that of the aromatic proton of ACH and CHB repeating units at 6.93 ppm (ESI, Fig. S11 and S12†). A DPNMR of 36 was found for the second block of both mPEGA12a-b-ACH36(6), and mPEGA12b-b-CHB36(7), slightly higher than the expected 34. This can be explained considering that a minimal amount of mPEGA12 macro-CTA was withdrawn at the end of the first polymerisation step for 1H NMR and SEC analysis, thus very marginally increasing the monomer:macro-CTA molar ratio.
Polymersomes in-line assembly, drug loading and characterization
At the end of the polymerisation runs the reaction mixtures were diluted with additional EtOH to a polymer concentration of 35 mg mL−1 (total volume 37 mL) to facilitate the subsequent manipulations and assembly of mPEGA12a-b-ACH36(6) and mPEGA12b-b-CHB36(7) into their corresponding polymersomes. The resulting solutions were kept at 40 °C to ensure full polymer solubilisation and reduced viscosity of the polymer solutions. To assemble non drug-loaded vesicles these solutions were directly cannulated under stirring into DI water (final polymer concentration of 1 mg mL−1).Polymersomes assembly was confirmed by DLS analysis of the water/ethanol mixture immediately after the cannulation process. DLS traces showed the presence of nanoassemblies with an average diameter of 136 ± 2 nm and 89 ± 11 nm for mPEGA12a-b-ACH36, and mPEGA12b-b-CHB36, respectively.
Ethanol was removed by either dialysis or slow evaporation at room temperature, and the resulting assemblies were characterised by dynamic light scattering (DLS) and transmission electron microscopy (TEM). The in line polymersomes assembly process led to homogeneous dispersions of polymeric vesicles in water, (Fig. 2A), with hydrodynamic diameters estimated by DLS of 122 ± 20 nm or 41 ± 0.18 nm, for mPEGA12a-b-ACH36(6) and mPEGA12b-b-CHB36(7), respectively, with narrow PDI (<0.23). Similar results in terms of vesicle size and polydispersity were obtained regardless of the method used for removal of traces of ethanol in the polymersomes suspension (data not shown).
 | ||
| Fig. 2 Characterisation of in-line assembled mPEGA12a-b-ACH36 and mPEGA12b-b-CHB36 polymersomes (A) DLS analysis of one representative sample. Size shown refers to distribution of hydrodynamic diameter measured by DLS at 173° in water, at 25 °C. Size (nm) is reported as z-average mean. N = 2. (B) TEM images of polymersomes stained with 3% uranyl acetate solution. | ||
To test the efficiency of the in-line process in assembling well-defined vesicles, conventional nanoprecipitation of purified mPEGA12a-b-ACH36(6) and mPEGA12b-b-CHB36(7) was also performed. Accordingly, polymers were dissolved in DMSO (5.0 mg mL−1) and water was added dropwise to the resulting solution until a 1.0 mg mL−1 concentration was reached.
This procedure led to the formation of very similar size vesicles in the case of mPEGA12a-b-ACH36 (122 ± 9 nm), but larger assemblies in the case of mPEGA12b-b-CHB36 (172 ± 2 nm ESI, Fig. S13†), a difference that could be ascribed to the different procedures and conditions used for their assembly.46
Analysis of the TEM images performed (ESI Fig. S14 and S15†) on polymersomes prepared via the in-line procedure and stained with 3% uranyl acetate solution gave results consistent with the size estimated by DLS for both mPEGA12a-b-ACH36, with an average core diameter of 126 ± 75 nm, and mPEGA12b-b-CHB36 (44 ± 5 nm).
To gain further insight into the morphology of these vesicles in solution, mPEGA12a-b-ACH36 and mPEGA12a-b-CHB36 polymersomes were analysed by AFM in PeakForce tapping mode (Fig. 3). A major advantage of this method is that the sample can be imaged in solution, thus obtaining a more realistic representation of the vesicles morphology in comparison to microscopy techniques performed on dry samples (i.e. TEM). Furthermore, the use of tapping instead of contact mode avoid the flattening caused by the pressure exterted, allowing a more accurate estimation of vesicle size and three-dimensional shape.
 | ||
| Fig. 3 Representative examples of vesicle morphology as seen by AFM analysis of polymersomes prepared from 1. mPEGA12a-b-ACH36 and 2. mPEGA12a-b-CHB36. A. AFM height image of polymersomes adsorbed on a mica surface in PBS solution (0.50 mg mL−1); B. The corresponding topographic 3D rendered image showing the distribution of polymersome heights; C. List of the mean height, mean area and mean diameter of adsorbed polymersomes at positions a–e or 1–4. D. Arrows highlight some of the collapsed vesicle structures observed during these measurements. | ||
Immobilisation of the polymersomes on the mica surface, a pre-requisite to perform the analysis, was achieved by electrostatic interactions.47 As described by Li et al.,48 negatively charged polymers can be easily immobilized on a mica surface, also negatively charged, via electrostatic interaction mediated by Mg2+.
Our previous studies35 showed that at physiological conditions (pH 7.4) polymers containing 3-chloro-4-hydroxybenzoate repeating units are negatively charged due to the deprotonation of part of their aromatic alcohol pendant groups. Accordingly, 1.0 mg mL−1 polymersome suspensions were prepared by nanoprecipitation in PBS buffer. The average sizes detected were around 80 nm for mPEGA12a-b-ACH36 and 45 nm for mPEGA12a-b-CHB36 (Fig. 3). The height of both polymeric aggregates was found to be around 14–17 nm, thus smaller than their diameter, which can be ascribed to a partial collapse of their hollow structure, thus confirming their vesicular morphology.49 Partially collapsed structures with typical ‘deflated’ balloon’ morphology could also be visualized (Fig. 3, insert 1D), thus further confirming the vesicular nature of these supramolecular assemblies.
Larger sizes were estimated by DLS analysis of the polymersomes in PBS (170 and 70 nm for mPEGA12a-b-ACH36 and mPEGA12a-b-CHB36, respectively, Fig. S16 ESI†) in comparison to those observed by AFM analysis. It has already been shown that the vesicles hydrodynamic diameter estimated by DLS can be larger than the geometrical diameter because of the hydration layer of the polymer shell.50 Liang et al. have discussed a similar discrepancy observed when analysing EggPC/cholesterol vesicles by both DLS and AFM and ascribed it to a potential partial fragmentation of larger vesicle upon adsorption on mica, and/or the intrinsic higher sensitivity of DLS for larger vesicle, which results in an overestimation of the mean hydrodynamic diameter.51 The latter phenomenon has also been described when AgNPs53 and polystyrene nanoparticles52 were utilised to compare these two analytical techniques.
To establish an automated and robust process to generate polymersomes reproducibly and with very low polydispersity, a microfluidic system was then engineered (Fig. 4). Indeed, microfluidic strategies have been successfully employed to fabricate soft micro- and nano-assemblies54,55 To this aim, polytetrafluoroethylene (PTFE) tubing were secured with gaskets inside the screw-tight caps of a 3-way connector. An ethanolic solution (12 mg mL−1) of mPEGA12b-b-CHB36 maintained at 50 °C was injected via pump syringe at 10 μL min−1 (Flow A, Fig. 4) into a junction where DI water entered via another ID 0.5 mm tube at a flow rate of 120 μL min−1 (Flow B, Fig. 4). The mixed liquids (Flow C, Fig. 4) exited the junction via an ID 1 mm tube and were collected for size analysis. A range of A and B flow rates were tested in preliminary experiments to optimize the solvent mixing inside the connector. Polymersomes were obtained with this methodology at a rate of 0.12 mg min−1, and were analysed by DLS before and after ethanol evaporation.
 | ||
| Fig. 4 Automated assembly of mPEGA12b-b-CHB36 polymersomes: schematic representation of the microfluidic device utilised in this study. | ||
The average size in the two cases was found to be very similar, with a diameter for the vesicles in pure water of 37 ± 7 nm and a PDI of 0.15 ± 0.04. One of the major advantages offered by this technique is the possibility of finely tuning the ratio of the two solvents in the device by independently modifying the solvent flow rates via syringe pumps, and to minimise the use of organic solvent required to induce the formation of homogeneous vesicles. The accurate control of the flow rate also resulted in very consistent batch to batch reproducibility (Fig. S17 ESI†), in a process that could easily be scaled up for large scale production.
Finally, the efficiency of the in-line assembly process to incorporate hydrophilic and hydrophobic drugs within the vesicle structures was then investigated. In general, therapeutics are entrapped within polymersomes either during their preparation, or by introduction of drug molecules into pre-formed vesicles.21 For the latter, injection56 or electroporation57 techniques are typically employed. In the former case, hydrophobic molecules can be incorporated into polymeric films utilised to prepare polymersomes, whilst hydrophilic drugs can be introduced into the aqueous phase used to rehydrate these films.58 Other commonly used techniques include solvent exchange mediated encapsulation,59 and the use of microfluidics to load small molecules or proteins while forming vesicles.60
In this present study two model molecules, terbinafine, a hydrophobic antifungal drug which is very poorly soluble in water in its free base form (<2 μg mL−1, as assessed by RP-HPLC, data not shown), and hydrophilic cyanocobalamine (Vit B12, solubility in water ca. 10–11 mg mL−1)61 were chosen as model drugs for incorporation within vesicle lipophilic membrane and aqueous lumen, respectively. In addition to its hydrophobic nature, terbinafine was chosen due to the presence of a tertiary alkyl amine in its structure, which by forming ionic pairs with the weakly acidic 3-chloro-4-hydroxybenzoic ester repeating units (pKa 7–7.5)36 in the hydrophobic blocks of mPEGA12a-b-ACH36 and mPEGA12b-b-CHB36 could lead to more efficient drug entrapment. This phenomenon was observed in our previous studies using analogous block copolymers, where we utilised tamoxifen, a hydrophobic drug with a basic tertiary amine group, which gave drug loading capacity as high as 18%.36 In typical experiments, at the end of the polymerisation reaction an ethanolic solution of terbinafine free base (35 mg mL−1) was added via syringe to the polymerisation mixture to give a 10% w/w drug/polymer solution, at 40 °C. Cyanocobalamine was dissolved in the aqueous medium used for the subsequent assembly of mPEGA12a-b-ACH36(6) and mPEGA12b-b-CHB36(7)-based polymersomes, again in a 10% w/w drug/polymer ratio. The polymer–terbinafine solutions were then cannulated into water–cyanocobalamine solutions, to generate the desired drug-loaded mPEGA12a-b-ACH36 and mPEGA12b-b-CHB36-based polymersomes. As observed in previous experiments, rapid self-assembly into vesicles with no visible precipitation was observed, suggesting that the method was robust, and that rapid formation of polymersomes under these conditions was not negatively affected by the presence of loaded molecules. The excess of cyanocobalamine was removed by dialysis against water using a 6–8 kDa MWCO membrane, while traces of unentrapped terbinafine were subsequently removed after the dialysis step by centrifugation, followed by filtration through a 0.45 μm filter. Drug loading was quantified by RP-HPLC (see ESI†) on the resulting purified vesicles.
Both mPEGA12a-b-ACH36 and mPEGA12b-b-CHB36, block copolymers displayed a higher cyanocobalamine loading capacity (LC) (Table 2). Interestingly, the acrylamide based polymer mPEGA12b-b-CHB36 showed better drug encapsulation for both the hydrophilic cyanocobalamine and lipophilic terbinafine. Vesicles were analysed by DLS (Fig. 5) to investigate how drug loading affected their size and size distribution. As we observed earlier with similar polymers,36 loaded polymersomes showed a smaller hydrodynamic radius than the empty assemblies, decreasing to ∼72 nm for mPEGA12a-b-ACH36 while an increase in size to ∼80 nm was observed for mPEGA12b-b-CHB36, as assessed by DLS analysis.
 | ||
| Fig. 5 (A) Loaded polymersomes (1.0 mg mL−1) size distributions of hydrodynamic diameter measured by DLS at 173° in water at 25 °C. Size (nm) is reported as z-average; (B) terbinafine/cyanocobalamine-loaded mPEGA12a-b-ACH36 and mPEGA12b-b-CHB36 polymersomes. | ||
One possible explanation for this phenomenon is a different membrane packing caused by the presence of terbinafine and potential ion pairing between the drug and the weakly acidic 3-chloro-4-hydroxybenzoate of ACH and 3-chloro-4-hydroxybenzamide of CHB polymer repeating units. The observed high entrapment efficiency of cyanocobalamine may be due, at least in part, to additional electrostatic interactions between the partially deprotonated 3-chloro-4-hydroxybenzoate/benzamide repeating units and positively charged CoIII centres.
Conclusions
In this study we devised an in line synthetic/assembly strategy which enables facile engineering of drug-loaded polymersomes in only a few hours, starting from acrylate/acrylamide monomers. Using Perrier's one-pot multiple chain extension RAFT polymerisation strategy, amphiphilic mPEGA12a-b-ACH36 and mPEGA12b-b-CHB36 copolymers could be prepared with narrow molecular weight distributions at very high monomer conversion. Reaction conditions – concentration of monomers, CTA:radical initiator molar ratio, nature of the reaction solvent – were optimised to attain good control over the polymerisation process and to avoid intrinsically inefficient purification of reaction intermediates. Importantly, the process was developed using ethanol:water as the reaction medium, in view of a potential application of this process in biological/biomedical settings. The final reaction mixtures could then be directly utilised to assemble well defined polymeric vesicles incorporating both model hydrophilic and hydrophobic drugs in the nanocarriers aqueous lumen and hydrophobic membrane, respectively.
Similarly to click techniques where sub-families of chemical reactions are selected based on a range of favourable features, and PISA, where monomers and macromolecular parameters are chosen to direct supramolecular assembly, our results suggest that when appropriate reagents and conditions are utilised, in line processes could become a viable general route for efficient, scaleable and cost-effective assembly of polymersome nanovectors.
Data access statement
All raw data created during this research are openly available from the corresponding author (giuseppe.mantovani@nottingham.ac.uk) and at the University of Nottingham Research Data Management Repository (https://rdmc.nottingham.ac.uk/) and all analysed data supporting this study are provided as ESI† accompanying this paper.Acknowledgements
The authors thank the UK Engineering and Physical Sciences Research Council (EPSRC: Grants EP/H006915/1 and EP/H005625/1 Leadership Fellowship (C. A.); EP/L01646X (A. F. B.); EP/I01375X/1 (G. E. M.)). The authors also thanks the Advanced Microscopy Unit (School of Life Science, University of Nottingham) and Ms Denise McLean for assistance with TEM images acquisition.Notes and references
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CAS.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- D. E. Discher and F. Ahmed, Annu. Rev. Biomed. Eng., 2006, 8, 323–341 CrossRef CAS PubMed.
- G. Battaglia, in Nanotechnologies for the Life Sciences, Wiley-VCH Verlag GmbH & Co. KGaA, 2007, DOI:10.1002/9783527610419.ntls0250.
- K. Schillén, K. Bryskhe and Y. S. Mel'nikova, Macromolecules, 1999, 32, 6885–6888 CrossRef.
- R. Bleul, R. Thiermann and M. Maskos, Macromolecules, 2015, 48, 7396–7409 CrossRef CAS.
- J. S. Lee and J. Feijen, J. Controlled Release, 2012, 161, 473–483 CrossRef CAS PubMed.
- F. Meng, Z. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS PubMed.
- L. Guan, L. Rizzello and G. Battaglia, Nanomedicine, 2015, 10, 2757–2780 CrossRef CAS PubMed.
- R. P. Brinkhuis, F. P. J. T. Rutjes and J. C. M. van Hest, Polym. Chem., 2011, 2, 1449–1462 RSC.
- X. Tian, S. Nyberg, P. S. Sharp, J. Madsen, N. Daneshpour, S. P. Armes, J. Berwick, M. Azzouz, P. Shaw, N. J. Abbott and G. Battaglia, Sci. Rep., 2015, 5, 11990 CrossRef CAS PubMed.
- C. Nardin, J. Widmer, M. Winterhalter and W. Meier, Eur. Phys. J. E: Soft Matter Biol. Phys., 2001, 4, 403–410 CrossRef CAS.
- R. J. R. W. Peters, M. Marguet, S. Marais, M. W. Fraaije, J. C. M. van Hest and S. Lecommandoux, Angew. Chem., Int. Ed., 2014, 53, 146–150 CrossRef CAS PubMed.
- D. A. Wilson, R. J. M. Nolte and J. C. M. van Hest, Nat. Chem., 2012, 4, 268–274 CrossRef CAS PubMed.
- D. A. Hammer and N. P. Kamat, FEBS Lett., 2012, 586, 2882–2890 CrossRef CAS PubMed.
- N. P. Kamat, J. S. Katz and D. A. Hammer, J. Phys. Chem. Lett., 2011, 2, 1612–1623 CrossRef CAS PubMed.
- J. Vandenbergh, T. de Moraes Ogawa and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2366–2374 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Macromolecules, 2014, 47, 3451–3460 CrossRef CAS.
- N. A. Hadjiantoniou, T. Krasia-Christoforou, E. Loizou, L. Porcar and C. S. Patrickios, Macromolecules, 2010, 43, 2713–2720 CrossRef CAS.
- H. R. Marsden, L. Gabrielli and A. Kros, Polym. Chem., 2010, 1, 1512–1518 RSC.
- L. Messager, J. Gaitzsch, L. Chierico and G. Battaglia, Curr. Opin. Pharmacol., 2014, 18, 104–111 CrossRef CAS PubMed.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707–15713 CrossRef CAS PubMed.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Macromolecules, 2012, 45, 5081–5090 CrossRef CAS.
- C. Gonzato, M. Semsarilar, E. R. Jones, F. Li, G. J. P. Krooshof, P. Wyman, O. O. Mykhaylyk, R. Tuinier and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 11100–11106 CrossRef CAS PubMed.
- V. Ladmiral, M. Semsarilar, I. Canton and S. P. Armes, J. Am. Chem. Soc., 2013, 135, 13574–13581 CrossRef CAS PubMed.
- W. Zhao, G. Gody, S. Dong, P. B. Zetterlund and S. Perrier, Polym. Chem., 2014, 5, 6990–7003 RSC.
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350–355 RSC.
- J. Yeow, O. R. Sugita and C. Boyer, ACS Macro Lett., 2016, 5, 558–564 CrossRef CAS.
- J. Yeow, J. Xu and C. Boyer, ACS Macro Lett., 2015, 4, 984–990 CrossRef CAS.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505–2513 Search PubMed.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Macromolecules, 2014, 47, 639–649 CrossRef CAS.
- G. Gody, R. Barbey, M. Danial and S. Perrier, Polym. Chem., 2015, 6, 1502–1511 RSC.
- F. Mastrotto, S. Salmaso, Y. L. Lee, C. Alexander, P. Caliceti and G. Mantovani, Polym. Chem., 2013, 4, 4375–4385 RSC.
- F. Mastrotto, S. Salmaso, C. Alexander, G. Mantovani and P. Caliceti, J. Mater. Chem. B, 2013, 1, 5335–5346 RSC.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- G. Qi, C. W. Jones and F. J. Schork, Macromol. Rapid Commun., 2007, 28, 1010–1016 CrossRef CAS.
- C. Capello, U. Fischer and K. Hungerbuhler, Green Chem., 2007, 9, 927–934 RSC.
- F. Huo, X. Wang, Y. Zhang, X. Zhang, J. Xu and W. Zhang, Macromol. Chem. Phys., 2013, 214, 902–911 CrossRef CAS.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- P. Caliceti and F. M. Veronese, Adv. Drug Delivery Rev., 2003, 55, 1261–1277 CrossRef CAS PubMed.
- A. Sato, S. W. Choi, M. Hirai, A. Yamayoshi, R. Moriyama, T. Yamano, M. Takagi, A. Kano, A. Shimamoto and A. Maruyama, J. Controlled Release, 2007, 122, 209–216 CrossRef CAS PubMed.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- Although volume fraction is arguably a more appropriate parameter to look at for these types of considerations, weight fraction, easier to calculate, is generally regarded as an acceptable alternative.
- P. Lim Soo and A. Eisenberg, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 923–938 CrossRef.
- G. Battaglia, C. LoPresti, M. Massignani, N. J. Warren, J. Madsen, S. Forster, C. Vasilev, J. K. Hobbs, S. P. Armes, S. Chirasatitsin and A. J. Engler, Small, 2011, 7, 2010–2015 CrossRef CAS PubMed.
- F. Li, T. Ketelaar, M. A. Cohen Stuart, E. J. Sudholter, F. A. Leermakers and A. T. Marcelis, Langmuir, 2008, 24, 76–82 CrossRef CAS PubMed.
- G.-Y. Liu, L.-P. Lv, C.-J. Chen, X.-S. Liu, X.-F. Hu and J. Ji, Soft Matter, 2011, 7, 6629–6636 RSC.
- H. Fissan, S. Ristig, H. Kaminski, C. Asbach and M. Epple, Anal. Methods, 2014, 6, 7324–7334 RSC.
- X. Liang, G. Mao and K. Y. Ng, J. Colloid Interface Sci., 2004, 278, 53–62 CrossRef CAS PubMed.
- C. M. Hoo, N. Starostin, P. West and M. L. Mecartney, J. Nanopart. Res., 2008, 10, 89–96 CrossRef CAS.
- E. Tomaszewska, K. Soliwoda, K. Kadziola, B. Tkacz-Szczesna, G. Celichowski, M. Cichomski, W. Szmaja and J. Grobelny, J. Nanomater., 2013, 2013, 10 Search PubMed.
- W. J. Duncanson, T. Lin, A. R. Abate, S. Seiffert, R. K. Shah and D. A. Weitz, Lab Chip, 2012, 12, 2135–2145 RSC.
- S.-H. Kim, J. W. Kim, D.-H. Kim, S.-H. Han and D. A. Weitz, Microfluid. Nanofluid., 2013, 14, 509–514 CrossRef CAS.
- F. Ahmed, R. I. Pakunlu, A. Brannan, F. Bates, T. Minko and D. E. Discher, J. Controlled Release, 2006, 116, 150–158 CrossRef CAS PubMed.
- L. Wang, L. Chierico, D. Little, N. Patikarnmonthon, Z. Yang, M. Azzouz, J. Madsen, S. P. Armes and G. Battaglia, Angew. Chem., Int. Ed., 2012, 51, 11122–11125 CrossRef CAS PubMed.
- G. Liu, S. Ma, S. Li, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomaterials, 2010, 31, 7575–7585 CrossRef CAS PubMed.
- T. Anajafi, M. D. Scott, S. You, X. Yang, Y. Choi, S. Y. Qian and S. Mallik, Bioconjugate Chem., 2016, 27(3), 762–771 CrossRef CAS PubMed.
- J. Thiele, A. R. Abate, H. C. Shum, S. Bachtler, S. Förster and D. A. Weitz, Small, 2010, 6, 1723–1727 CrossRef CAS PubMed.
- X. Wang, L. Wei and L. P. Kotra, Bioorg. Med. Chem., 2007, 15, 1780–1787 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py01292b |
| This journal is © The Royal Society of Chemistry 2016 |