
Self-healing of glucose-modified polyurethane networks facilitated by damage-induced primary amines†
Ying
Yang
and
Marek W.
Urban
*
Department of Materials Science and Engineering, Center for Optical Materials Science and Engineering Technologies (COMSET), Clemson University, Clemson, SC 29634, USA. E-mail: mareku@clemson.edu
First published on 28th October 2016
Abstract
Living organisms utilize metabolic processes to achieve adaptive, reproductive, and self-healing functions, which require dynamic and precise control of sequential chemical events. Since traditional synthetic materials do not possess these functions, there is an ongoing quest for the development of self-healing attributes. This study reports self-healing of crosslinked polyurethane networks by damage-induced formation of primary amines. Deliberately modified with methyl α-D-glucopyranoside (MGP) and catalyzed by zinc acetate (Zn(OAc)2), polyurethane (PUR) networks are capable of restoring mechanical properties upon mechanical damage. Self-repair is initially driven by entropic shape recovery facilitating interfacial contacts in damaged areas, followed by covalent reformation of cleaved bonds. Amine functionalities resulting from mechanical damage facilitate covalent re-bonding to form urea linkages responsible for recovery of mechanical properties.
Due to prolonged lifetime and the ability to retain original functionalities, synthetic materials capable of self-repairs are highly attractive. Initial studies utilized the inter-diffusion of polymer chains to obtain self-repair, but local high temperatures were required.1,2 The concepts of embedding microcapsules containing healing agents,3 or incorporating stimuli-responsive nano-objects4 into polymer or other materials were developed later on. Another intriguing approach in obtaining self-healing polymers is to directly modify polymer chains with dynamic bonds. By controlling the dissociation and re-association of these entities via external stimuli, such as temperature,5 pH,6 light,7,8 or redox,9 physical and chemical properties within or near the damaged sites can be altered10 to achieve self-healing. With a few exceptions,7,11–13 the majority of synthetic approaches utilized one or two types of re-bonding mechanisms imbedded into a known polymer matrix to facilitate self-healing, including Diels–Alder reaction,5 transesterification,14 disulfide exchange,15 and other reversible reactions.7,16–23 Supramolecular bonds such as metal–ligand interactions and H-bonding imbedded in polymers also offered self-healing attributes.2,6,8,9,13,24–26 In contrast, even for the most primitive biological species, chemical and physical events responsible for repairs and their living functions exhibit significantly greater degree of complexity. In metabolic processes, molecules used for reconstructing cell components are produced from the breakdown of organic matter, which facilitate the maintenance of living functions. If designed properly, synthetic materials may be able to adapt or respond to external stimuli. One of the intriguing properties is the ability to self-repair upon mechanical damage, which is particularly appealing if reactive groups capable of self-healing are generated upon mechanical damage.
Recently, we developed methyl α-D-glucopyranoside (MGP)-based polyurethane networks that are able to self-repair at room temperature by reacting with atmospheric CO2 and H2O catalyzed by dibutyltin dilaurate (DBTDL).27 Since metal catalysts may play an important role in self-healing of polyurethanes, in this study we utilized MGP modified polyurethane networks catalyzed by zinc acetate (Zn(OAc)2) (MGP-PUR-Zn(OAc)2) to achieve self-healing via the formation of reactive groups generated as a result of mechanical damage. As will be seen, for MGP-PUR-Zn(OAc)2 networks, H2O or CO2 will have no contribution to self-healing, but the catalytic activity of Zn(OAc)2 will govern urethane-to-urea conversion resulting in self-healing and more importantly, the change of the catalyst will lead to entirely different self-healing mechanism. Furthermore, non-toxicity and water solubulity of Zn(OAc)2 make this catalyst highly attractive. Considering an increasing evidence that bond reformations can be facilitated by a combination of physical and chemical events,28 we incorporated these network components with the premise that upon mechanical damage reactive groups will be generated and may facilitate covalent re-bonding. The synthesis of the crosslinked network is illustrated in Fig. 1A, where MGP, and polyethylene glycol (PEG, Mn = 300 g mol−1) were reacted with hexamethylene diisocyanate crosslinker (HDI) in the presence of Zn(OAc)2 (1.2% w/w). The following molar ratios of MGP/PEG = 0.59/1 and NCO/OH = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 were used to achieve fully crosslinked networks with the glass transition temperature (Tg) of 56 °C (Fig. S1A of the ESI†), the Young's modulus of 77.8 MPa at 20 °C, and the molecular weight between crosslinks (Mx) of 4.7 kg mol−1. As will be shown, in contrast to the previous studies, H2O or CO2 showed no contribution to self-healing of MGP-PUR-Zn(OAc)2 networks. As anticipated based on the activation energy of the catalyst, thermal stimuli is required.
:1 were used to achieve fully crosslinked networks with the glass transition temperature (Tg) of 56 °C (Fig. S1A of the ESI†), the Young's modulus of 77.8 MPa at 20 °C, and the molecular weight between crosslinks (Mx) of 4.7 kg mol−1. As will be shown, in contrast to the previous studies, H2O or CO2 showed no contribution to self-healing of MGP-PUR-Zn(OAc)2 networks. As anticipated based on the activation energy of the catalyst, thermal stimuli is required.
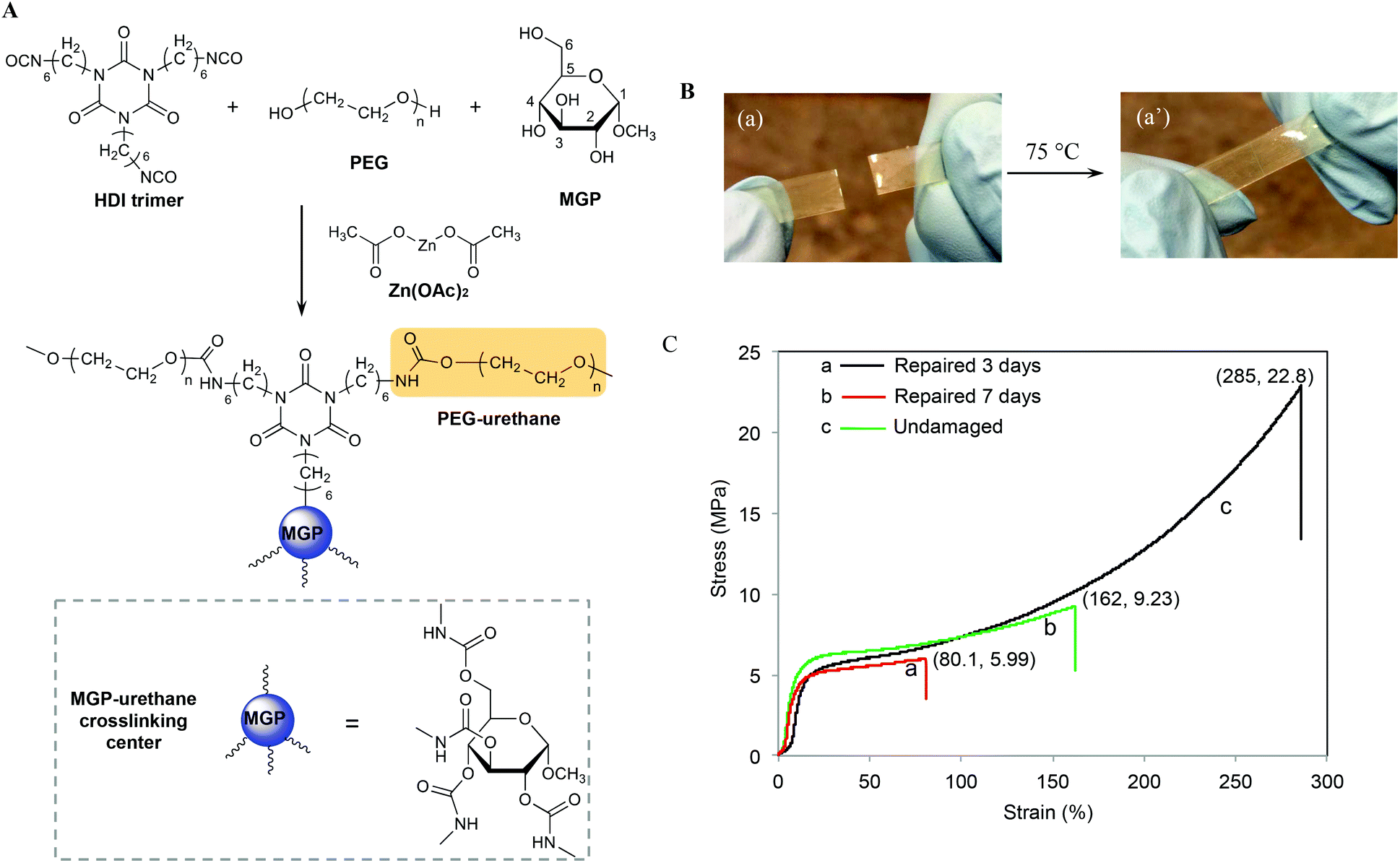 | ||
| Fig. 1 A – Synthesis of MGP-PUR-Zn(OAc)2 networks composed of methyl α-D-glucopyranoside (MGP), polyethylene glycol (PEG), and HDI trimmer crosslinker; B – optical images of self-healing at 75 °C of MGP-PUR-Zn2 networks; C – stress–strain curves of MGP-PUR-Zn(OAc)2 films after healing at 75 °C for 3 (a) and 7 days (b). For reference, curve c was recorded from undamaged film. | ||
When MGP-PUR-Zn(OAc)2 network was cut into two separate pieces and physically reattached, self-healing occurred at 75 °C. This is illustrated in Fig. 1B. The recovery of the mechanical strength was determined by measuring the tensile stress as a function of the time allowed for self-repair. As shown in Fig. 1C, after 72 h (3 days), the tensile stress recovery was 5.99 MPa at 80.1% strain (curve a), and reached 9.23 MPa and 162% strain after 7 days (curve b). Extending the recovery time further does not significantly impact tensile strength and strain. Compared to undamaged MGP-PUR-Zn(OAc)2 networks with a tensile strength of 22.8 MPa, ∼40% of tensile strength was recovered. However, the tensile strength recovery is temperature dependent. At 40 °C, only 1.75 MPa of tensile strength was recovered after 7 days. At 20 °C, no repairs are observed. It should be noted that the ability of MGP-PUR-Zn(OAc)2 to recover 9.23 MPa tensile strength is significantly greater compared to the majority of the reported polymers10 and the recovery to the original shape occurs without melting. For comparison, neat polyurethane crosslinked in the presence of Zn(OAc)2 (PUR-Zn(OAc)2) (without MGP), exhibits no self-healing despite of the lower Tg (3 °C, Fig. S1B; ESI†). The amount of MGP was also varied and the self-healing efficiency as a function of the reactant stoichiometry are provided in the ESI (Table S1†).
To set the stage for the analysis of molecular events responsible for damage–repair cycle using Raman and ATR FTIR spectroscopy, Fig. 2 illustrates optical images of scratched (A) and repaired (A′) MGP-PUR-Zn(OAc)2 networks. While Raman images of damaged (A) and repaired (A′) areas recorded with 1 μm2 spatial resolution are shown in Fig. 2B and B′, the corresponding spectra collected inside (area 1/1′ of Fig. 2B/B′) and outside (area 2/2′ of Fig. 2B/B′) scratch are shown in Fig. 2C/C′, Traces 1/1′ and 2/2′. The color scale represents the Raman intensity distribution of the 1756 cm−1 band. The Raman spectra were normalized to the 1440 cm−1 band, which is attributed to asymmetric C–H deformation vibration of the aliphatic segments. Table S2 of the ESI† provides detailed tentative band assignments. Upon damage, the C–H and C–C band intensities associated with –N at 1328 and 1067 cm−1 increases with respect to the C–H and C–C vibrations associated with –O at 1285 and 1036 cm−1. These changes are attributed to the cleavage of urethane linkages (NHC(O)O), and there are two possible mechanisms. If cleavages occur at urethane (O)C–N bond, generating –R–O–(O)C˙ and –R–NH˙ radicals, upon releasing of CO2 and H abstraction, –CH3 (or –CH2–) and –R–NH2 are formed at the cleaved chain ends. If cleavages occur at urethane O–C(O) bond, –R–NH–(O)C˙, and –R–O˙ are generated. The former may be converted into –R–NH2 by reaction with H2O at the presence of catalyst, releasing CO2, whereas the latter leads to the formation of –R–OH groups. Based on spectroscopic data, the two mechanisms will lead to the formation of –NH2 groups. The decrease of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration of HDI trimer ring (1756 cm−1), increase of symmetric deformation vibration of aliphatic C–H (1305 cm−1), as well as decrease of aliphatic C–C vibrational bands (1094 cm−1) are attributed to either cleavage or conformational changes of the HDI trimmer segments upon mechanical damage. As shown in Traces 1′ and 2′ in Fig. 2C′, after repair, no spectroscopic changes are observed in the repaired area which is likely attributed to the Raman detection limits. Therefore, further analysis will be conducted using ATR FTIR.
O vibration of HDI trimer ring (1756 cm−1), increase of symmetric deformation vibration of aliphatic C–H (1305 cm−1), as well as decrease of aliphatic C–C vibrational bands (1094 cm−1) are attributed to either cleavage or conformational changes of the HDI trimmer segments upon mechanical damage. As shown in Traces 1′ and 2′ in Fig. 2C′, after repair, no spectroscopic changes are observed in the repaired area which is likely attributed to the Raman detection limits. Therefore, further analysis will be conducted using ATR FTIR.
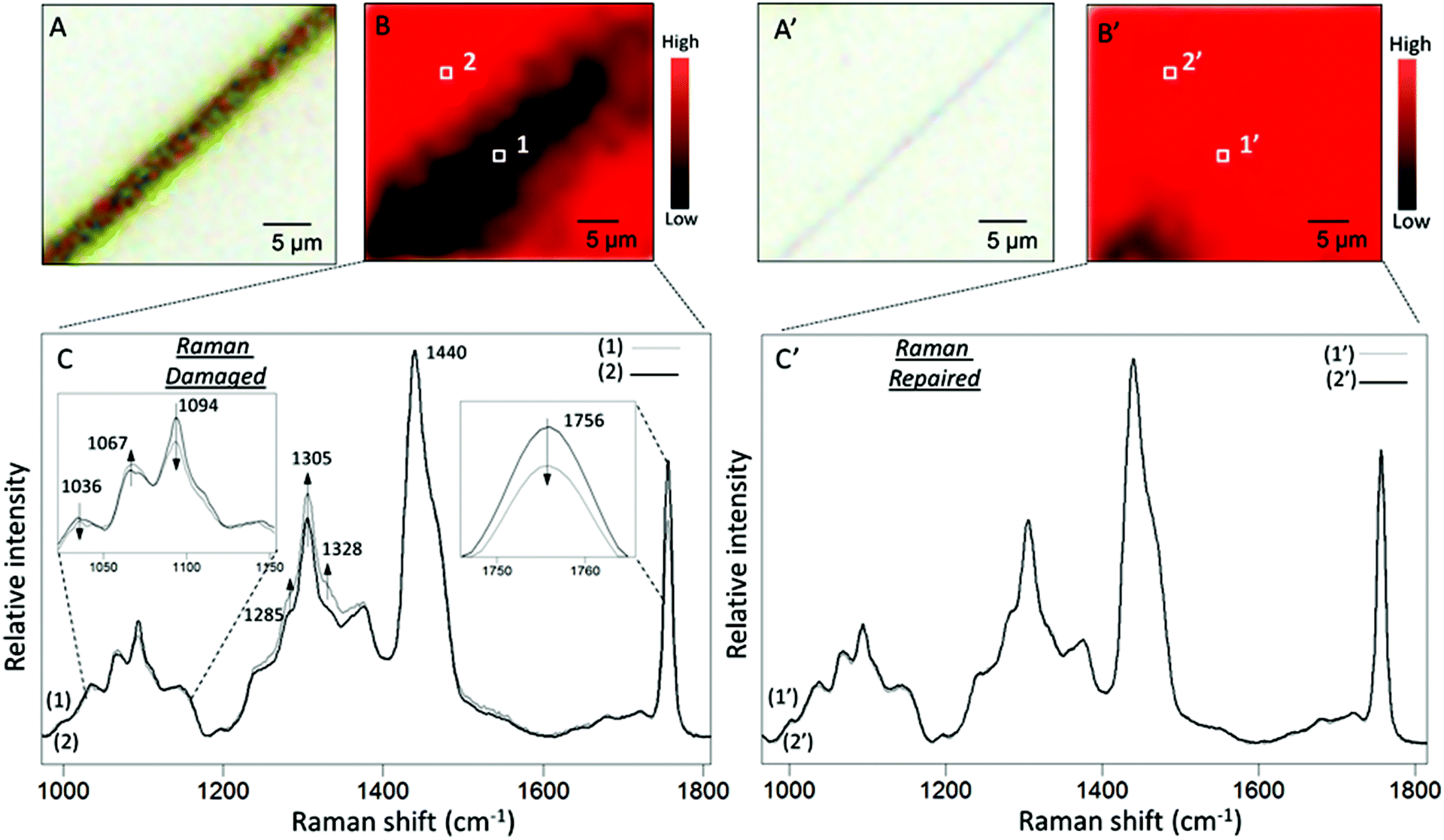 | ||
| Fig. 2 A – Optical image of MGP-PUR-Zn(OAc)2 film immediately after damage; B – Raman imaging of the area shown in A; the color scale represents intensity distribution of the 1756 cm−1 band; C – Raman spectra collected (1) and (2) collected from inside (area 1 marked in B) and outside (area 2 marked in B) damaged area; the band at 1756, 1094 and 1067 cm−1 are enlarged and shown in the inserts for visual effects; A′ – optical image of MGP-PUR-Zn(OAc)2 film after self-healing at 75 °C for 7 days; B′ – Raman imaging of the area shown in A′. The color scale represents intensity distribution of the 1756 cm−1 band; C – Raman spectra (1′) and (2′) collected from inside (area 1′ marked in B′) and outside (area 2′ marked in B′) damaged area. | ||
Fig. 3A illustrates ATR FTIR spectra collected from damaged (Trace a) and undamaged (Trace b) areas of MGP-PUR-Zn(OAc)2. Trace c is the subtraction of Traces (a–b), which amplifies the detected intensity changes. The results of the same analysis performed after self-repair are shown in Traces a′ (repaired), b′ (undamaged), and c′ (subtraction trace) of Fig. 3A′. The IR spectra were normalized to 1535 cm−1 before subtraction in order to identify the intensity changes in 1600–1500 cm−1 amide vibrational regions. Upon damage, the decrease of the CO at 1680 cm−1 (Fig. 3A, Trace c) and C–N at 1243 cm−1 bands due to urethane linkages are observed, indicating cleavage of NHC(O)O bonds. Furthermore, the increase of the 1081 and 1005 cm−1 bands due to C–OH vibrations are attributed to MGP–OH formation, which again, results from NHC(O)O cleavage. The net result is the generation of –NH2 groups upon mechanical damage manifested by the appearance of the band at 1627 cm−1 due to N–H vibrations. Thus, after mechanical damage, cleavage of urethane linkages and H-bonding occur, which leads to the formation of –NH2 and MGP–OH functional groups.
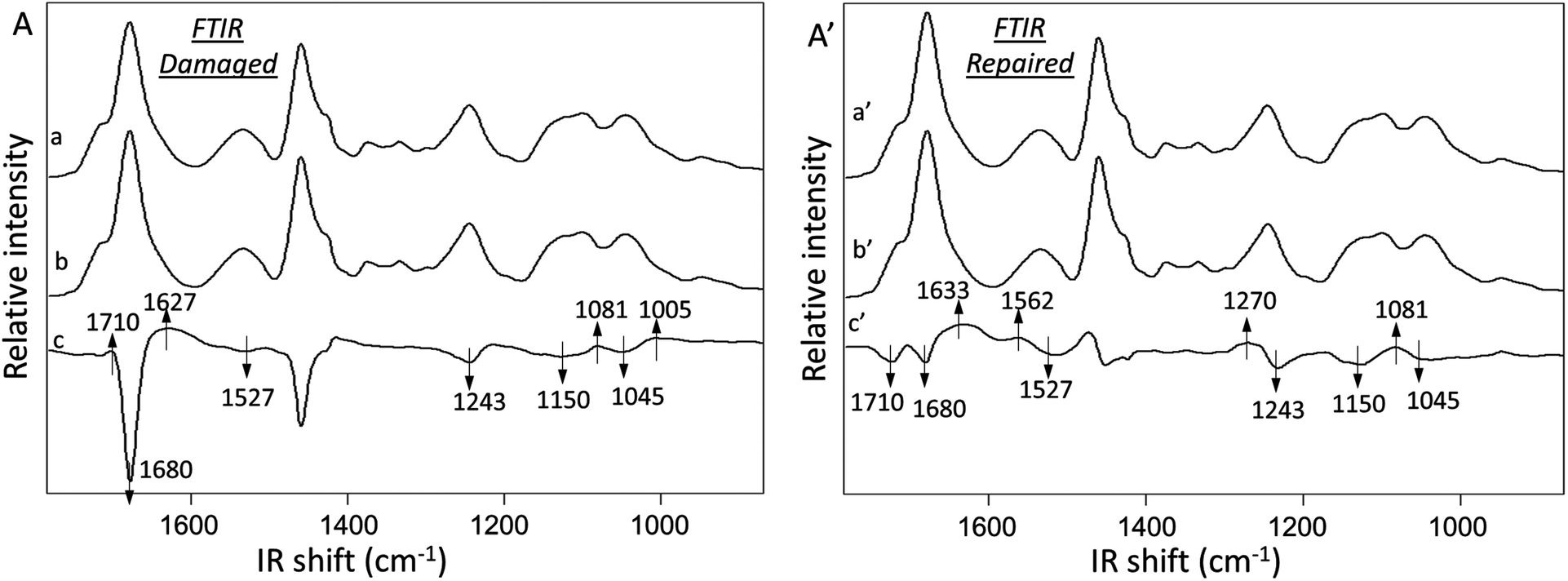 | ||
| Fig. 3 A – ATR-FTIR spectra collected from severely damaged (trace a) and undamaged (trace b) MGP-PUR-Zn(OAc)2 areas, respectively. Trace c is the subtraction spectrum a–b; A′ – ATR-FTIR spectra collected from repaired (a′) and undamaged areas (b′) of MGP-PUR-Zn(OAc)2 film after repairing at 75 °C for 7 days. Trace c′ is the subtraction spectrum a′–b′. The damage was produced by severely scratching the film surface with a razor blade. | ||
The analysis of MGP-PUR-Zn(OAc)2 networks upon self-repair is shown in Fig. 3A′. IR bands responsible for the urethane free CO, H-bonded CO stretching, N–H deformation, C–N stretching, and C–O–C stretching vibrations at 1715, 1680, 1527, 1243, 1150, and 1045 cm−1, respectively, decrease (Trace c′). However, the bands at 1633, 1562, 1270, and 1081 cm−1 due to CO stretching, N–H deformation, C–N stretching, and C–O–C stretching vibrations characteristic of urea bonds, increase. These observations indicate urethane-to-urea conversion during repair, which is further magnified by doubling the amount of urea linkages after 7 days at 75 °C in comparison to a 3-day repair. As anticipated, when the temperature is lowered to 40 °C, significantly less urea is formed (Fig. S2A; ESI†), which is a result of lower reactivity of the reactants, as will be demonstrated later, and limited mobility of the chains. These results explain the lower healing efficiency of MGP-PUR-Zn(OAc)2 at 40 °C. In contrast, urea formation does not occur for the same PUR-Zn(OAc)2 networks without MGP (Fig. S2B; ESI†), which lead to the question of the role of MGP molecules during self-repair.
The presence of MGP in MGP-PUR-Zn(OAc)2 networks generate urethane linkages in C2, C3, and C4 positions of MGP labeled in Fig. 1A. These MGP-urethanes linkages exhibit reduced stability due to their secondary carbon–oxygen bond and the presence of bulky pyranoside groups.29 Thus, the urethane carbonyl groups of these moieties are more reactive toward nucleophiles. Therefore, the –NH2 generated at damaged sites, as identified by spectroscopic analysis, are responsible for the reactions with MGP-urethanes in the presence of Zn(OAc)2 catalyst, and lead to the formation of urea linkages. To examine this hypothesis, a model molecule was synthesized by reacting MGP with four equivalent hexyl isocyanates (HI). In turn, this molecule was reacted with a diamine, 2,2′-(ethylenedioxy)bis(ethylamine) (EDBEA) catalyzed by Zn(OAc)2 under various temperatures. The results of these experiments showed that the urethanes linked to MGP react with –NH2 groups to form urea linkages in the presence of Zn(OAc)2 at 75 °C (Schemes S1, S2 and Fig. S3, S4; ESI†). Notably, these reactions do not occur without Zn(OAc)2, or by replacing –NH2 with –OH. It should be noted that the PEG-urethane compounds synthesized by reacting PEG with two equivalent amounts of hexyl isocyanates are unreactive toward –NH2 under the same conditions, indicating that PEG-urethane linkages exhibit enhanced stability compared to the MGP-urethanes.
In view of these experimental data, the following self-healing mechanism for MGP-PUR-Zn(OAc)2 networks is elucidated. Upon mechanical damage, urethane bond cleavage and formation of –NH2 groups are the primary events. Upon physical reattachment of two surfaces, there is a possibility of H-bonding during early stages of self-healing, but Raman/IR measurements could not confirm its formation. However, indirect evince suggests that when two physically reattached surfaces are exposed to 110 °C, they become detached, suggesting that the H-bonding (5–10 kcal mol−1; dissociation temperature ∼110–120 °C) could be responsible for reattachment at the early stages of self-healing at 75 °C. Nevertheless, covalent re-bonding manifested by the urethane-to-urea conversion, as shown in Fig. 4A, is responsible for the recovery of mechanical strength at the repaired interfaces. When temperature is increased to 75 °C, the –NH2 groups generated during damage react with MGP-urethane bonds through nucleophilic attack, whereby Zn(OAc)2 catalyzes the reaction by activating carbonyl groups via metal–ligand coordination. As a result, cleaved chains are re-connected by urea linkages (Fig. 4A). This process is also schematically depicted in Fig. 4B. It should be noted that since primary amine groups are generated by mechanical damage, and are not present in the undamaged networks, thermal stability of undamaged material is unaffected. Thermogravimetric analysis (TGA) (Fig. S5; ESI†) show 2% weight loss at 243 °C. Notably, further increased Zn(OAc)2 concentration levels compromise the repair efficiency due to strong coordination between Zn–polar sites, thus limiting chain mobility (Table S3 and Fig. S6; ESI†). For lower Zn(OAc)2 concentrations, the healing efficiency was lower, which is attributed to slower reaction rates (Fig. S6†). The formation of thermodynamically stable urea linkages due to urethane-to-urea conversion can only occur once, thus self-healing is limited to one cycle.
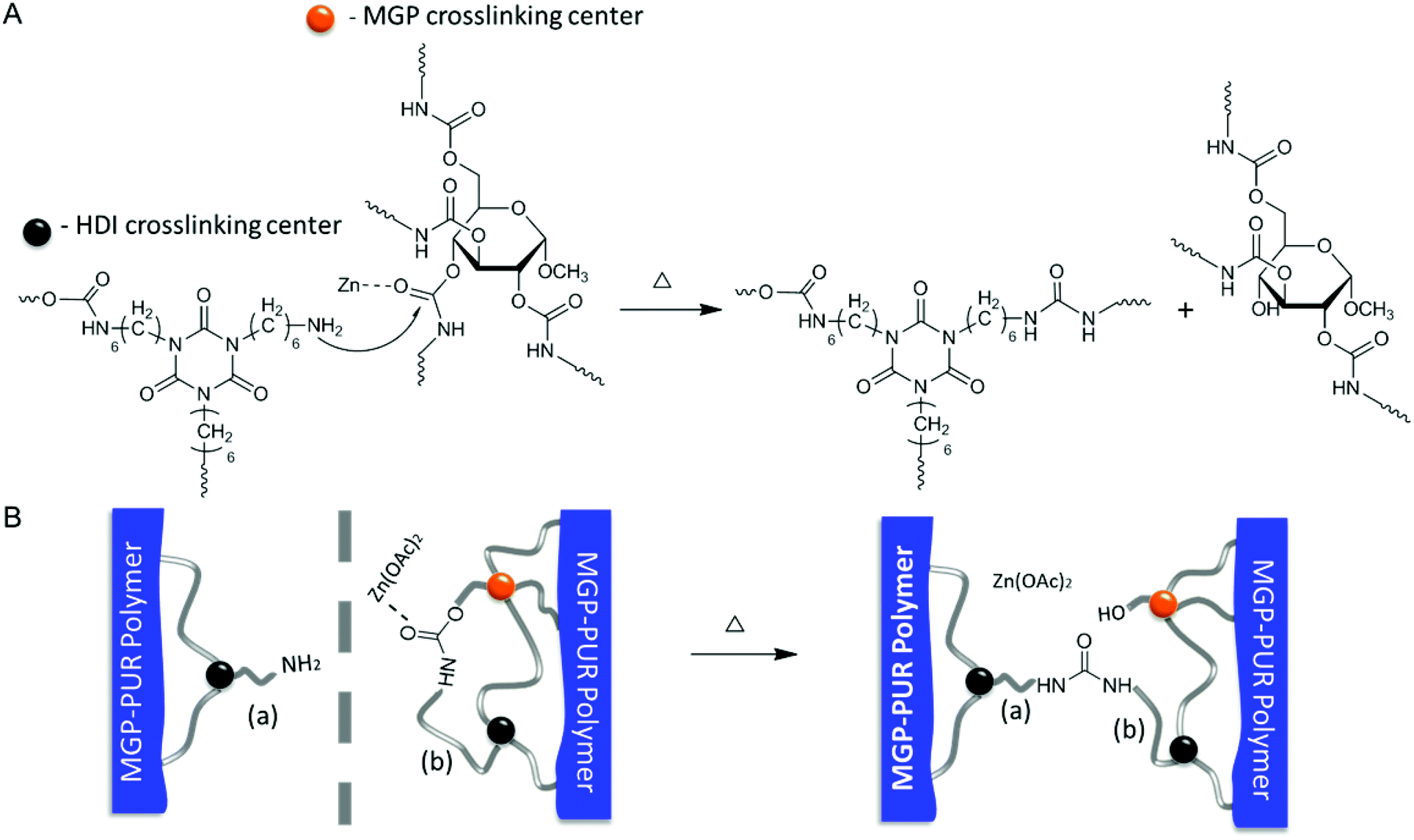 | ||
| Fig. 4 A – Proposed mechanism responsible for thermal healing of Zn(OAc)2-catalyzed MGP-PUR; B – schematic illustration of re-connection of cleaved chains at the area of damage via PUR-to-PUA conversion. | ||
Regardless of the mechanism, self-healing is driven by physical and/or chemical events. For chemical reactions leading to re-bonding, there are two pre-requisites: (i) reactive groups on two mechanically separated surfaces must be physically brought into contact with each other and (ii) reactivity of these groups must lead to re-bonding. The spectroscopic and physical evidence for the same PUR networks modified with oxetane/oxolane-substituted chitosan demonstrated that scratch recovery is driven by a flow of fragmented network components to form a scar.30,31 In contrast, crosslinked MGP-PUR-Zn(OAc)2 networks, due to the built-in elasticity, exhibit different behavior. Fig. 5A illustrates the closure of a 75 μm (width) × 600 (depth) μm scratch in a MGP-PUR-Zn(OAc)2 network as a function of time. A time-lapse video of this process is provided in the ESI (Movie_Shape Recovery†). The recovery takes ∼10 min at 20 °C, but when the temperature is raised >40 °C, the recovery takes <10 s. This shape recovery effect is governed by the rubber elasticity of the network at or above the glass transition temperature (Tg), and is driven by the recovery of the entropic network energy stored during mechanical deformation.32Fig. 5B illustrates the elasticity of MGP-PUR-Zn(OAc)2 networks upon 180% elongation, which within ∼20 s recovers 100% of the original length at 40 °C, but the same process takes ∼5 h at 20 °C. To determine how elastic shape recovery along with covalent bond reformations contribute to self-healing, two MGP-PUR-Zn(OAc)2 specimens were partially cut and allowed to self-repair for 7 days at 20 and 75 °C without physical reattachment. After that time, tensile measurements were conducted and the results are summarized in Fig. 5C. As seen, the rupture of the specimen repaired at 75 °C requires a load of 63.6 N (9.3 MPa). This value matches the rupture force/stress of a specimen cut into two separate pieces (Fig. 1B), and repaired by temporarily pressing damaged sites against each other, followed by exposure to 75 °C (Fig. 1C, Trace b). For the same specimen kept at 20 °C for 7 days, 50.4 N load is required to rupture. For reference, a specimen cut without repair will rupture at 38 N loads. These results indicate that the elastically driven shape recovery facilitating interfacial contact may enable covalent re-bonding if reactive groups such as primary amines are generated. The sequence of the events responsible for this behavior is depicted in Fig. 5D.
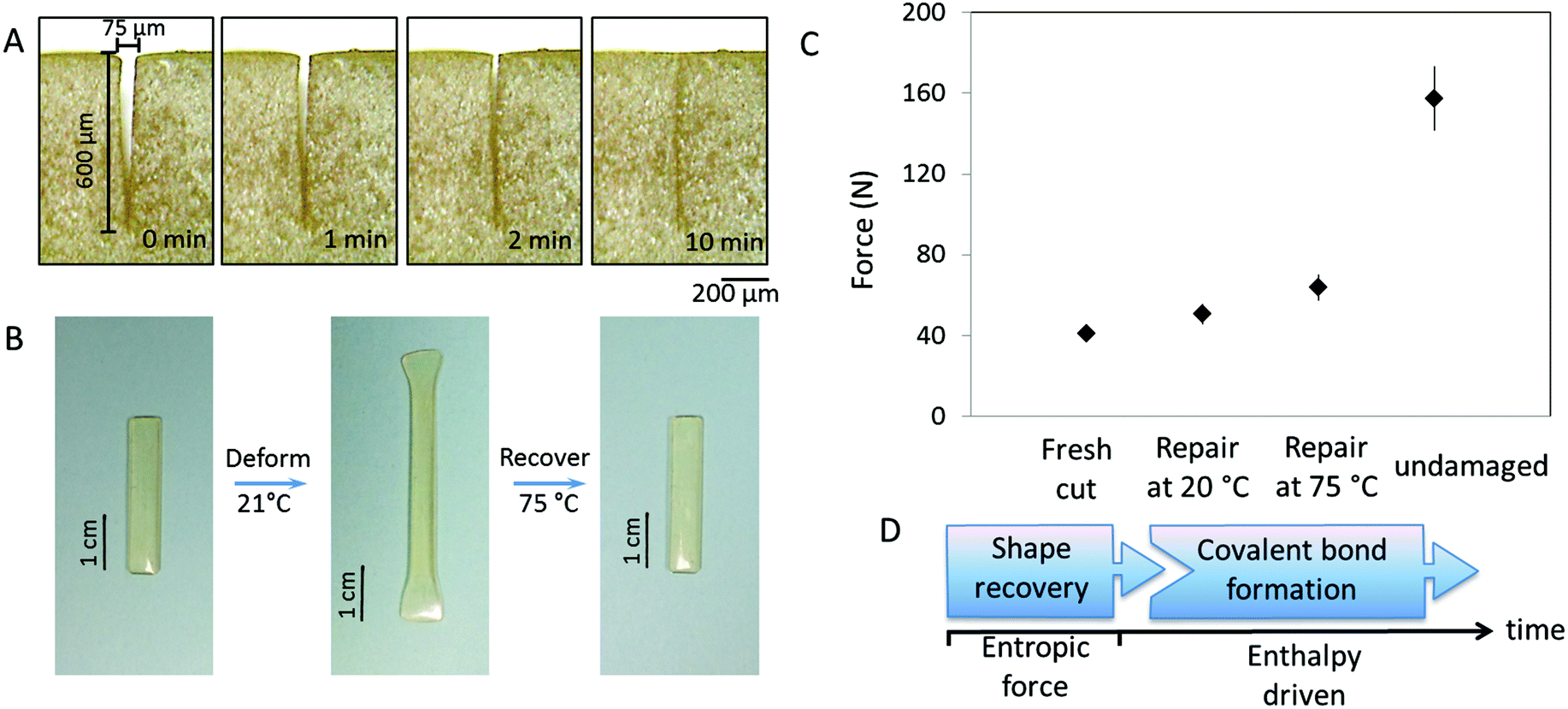 | ||
| Fig. 5 A – Recovery of a 75 × 600 μm (width × depth) cut as a function of time at 20 °C (film thickness is 1 mm). The optical images are taken from the side section of the cut; B – recovery of MGP-PUR-Zn(OAc)2 film after being elongated by 80% at 20 °C; C – maximum tensile force of MGP-PUR-Zn(OAc)2 after being cut by 75 × 600 μm at the center of the film, healing the cut at 20 °C and 75 °C for 7 days, and in the undamaged state; D – schematic illustration of the sequential physical and chemical events responsible for self-healing of MGP-PUR-Zn(OAc)2 network. | ||
In summary, during damage–repair cycle of MGP-PUR-Zn(OAc)2 networks, spatial and temporal synchronization of physical remodeling and chemical reactions are essential to recover mechanical properties. The critical steps to achieve self-repair in elastic networks is the network ability of shape recovery and in situ generation of reactive chain ends that are able to reform covalent bonds. Taking advantages of shape recovery attributed to stored entropic energy, interfacial contacts can be generated at or above the Tg to facilitate self-repair. The use of Zn(OAc)2 catalyst results in entirely different self-healing mechanism in which the final step is covalent rebonding facilitated by amine functionalities resulting from mechanical damages of the network. The presence of nucleophilic amines make the use of this system particularly attractive because their ability to reform covalent linkages, thus making them attractive in other systems.
Experimental
Methyl α-D-glucopyranoside (MGP), polyethylene glycol (PEG, avg. Mn = 300 g mol−1), hexamethylene isocyanate (HI), anhydrous N,N-dimethylformamide (DMF), tetrahydrofuran (THF), zinc acetate (Zn(OAc)2), 2,2′-(ethylenedioxy)bis(ethylamine) (EDBEA), butanediol, were purchased from Sigma Aldrich Co., tri-functional hexamethylene diisocyanate polymer (HDI trimer) N3300a was provided by Covestro.MGP-PUR-Zn(OAc)2 films were prepared by reacting HDI trimer with MGP in DMF in the presence of Zn(OAc)2 catalyst using overhead agitation at 500 rpm with a small four-blade polytetrafluoroethylene (PTFE) impeller in a 50 ml three-neck reaction flask at 25 °C for 10 min. Then PEG was added and continued to react for another 5 min. The molar ratio of HDI trimer/PEG/MGP = 1/0.69/0.41 were utilized while maintaining 38% (w/w) solids. Such mixtures were applied to obtain an approximate film thickness of 1 mm (±0.1 mm) on a PTFE substrate and dried at 75 °C for 5 days.
Control compounds MGP–4HI and MGP-HI were prepared by reacting MGP and HI using 1:4 and 1:1 molar ratios, respectively. The reactions were carried out in DMF at 60 °C for 48 h catalyzed by 1% (w/w) of Zn(OAc)2. The resulting compound was precipitated and washed with water and ethanol, then was dried in vacuum oven at 60 °C for overnight. Control compound PEG–2HI was prepared in a similar manner with a molar ratio of PEG:HI = 1:2 using a 4:1 mixture of THF and DMF as the solvent. The final product was directly dried at 60 °C in vacuum without washing.
Scratches of specimen were created with precisely controlled dimensions using custom built computerized Micro-Cut Instrument that facilitates control of the depth, speed and load during damage. In a typical experiment, a speed of 5 mm s−1 was utilized. For spectroscopic analysis, the depth of the scratches is controlled to be 100 μm, and for shape recovery analysis, the depth of the cuts is 600 μm. Optical images were recorded using Leica DM2500 M microscope.
Raman imaging were recorded using a Renishaw inVia Raman microscope equipped with a computer controlled three-axis encoded (X, Y, Z) motorized stage, a RenCam CCD detector, and a Leica microscope (DM2500 M). The 785 nm diode laser at 30 mW power provided an excitation source. Raman imaging were collected from 27 × 25 μm2 area with spatial resolution of 1 μm2. An acquisition time of 60 s was used.
Micro-attenuated total reflectance Fourier transform infrared spectra (μATR FT-IR) were obtained using an Agilent Cary 680 FT-IR single-beam spectrometer setting at 4 cm−1 resolution. A diamond crystal, and constant contact pressure between crystal and the specimens was used. All spectra were corrected for spectral distortions and optical effects using the Urban–Huang algorithm.14 It should be noted that during a single internal reflection experiment using a diamond crystal, all data were collected from a 1 mm radius within which ∼50 scratches were made. Assuming that the top 20 nm of damaged surface contain 100% of cleaved bonds upon damage, the overall detected signal will consist of ∼99.9% of uncleaved and ∼0.1% of cleaved bonds. While this estimation demonstrates that the anticipated intensity changes are small, the intensity changes are within ∼1% magnitude of the overall band intensities. The subtraction traces shown in Fig. 3 were enlarged to visualize the intensity changes from each experiment which was repeated several times to confirm that the changes are consistent and are due to damage–repair cycle.
Dynamic mechanical analysis (DMA) were performed on a TA Instrument Q800 DMA. Rectangular specimens having a size of ∼15.5 mm × 6 mm × 1 mm were tested at a frequency of 30 Hz and 5 μm amplitude. Dynamic mechanical properties were measured from −70 °C up to 150 °C while heating at 2 °C min−1. Dynamic moduli and mechanical damping (tanδ) were analyzed using TA Universal Analysis 2000. The measured glass transition temperature (Tg) for MGP-PUR-Zn(OAc)2 at maximum tanδ is 56 ± 1 °C. Tensile stretch test was carried out on Instron 4502 at room temperature (20 °C) at a rate of 3 mm min−1. Thermogravimetric analysis (TGA) was performed on TA Instrument Q500. Temperature was ramped from 20 to 550 °C at a heating rate of 10 °C min−1.
Acknowledgements
This work was partially supported by the National Science Foundation under award CMMI 1332964 and J. E. Sirrine Foundation Endowment at Clemson University.References
- R. Wool and K. O'connor, J. Appl. Phys., 1981, 52, 5953–5963 CrossRef CAS.
- S. J. Kalista Jr., T. C. Ward and Z. Oyetunji, Mech. Adv. Mater. Struct., 2007, 14, 391–397 CrossRef.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- C. C. Corten and M. W. Urban, Adv. Mater., 2009, 21, 5011–5015 CrossRef CAS PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef CAS PubMed.
- B. Ghosh and M. W. Urban, Science, 2009, 323, 1458–1460 CrossRef CAS PubMed.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- Y. Yang, X. Ding and M. W. Urban, Prog. Polym. Sci., 2015, 49–50, 34–59 CrossRef CAS.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467–472 CrossRef CAS PubMed.
- N. Roy, E. Buhler and J. M. Lehn, Polym. Int., 2014, 63, 1400–1405 CrossRef CAS.
- Z. Wang, Y. Yang, R. Burtovyy, I. Luzinov and M. W. Urban, J. Mater. Chem. A, 2014, 2, 15527–15534 CAS.
- M. Capelot, D. Montarnal, F. O. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- C.-M. Chung, Y.-S. Roh, S.-Y. Cho and J.-G. Kim, Chem. Mater., 2004, 16, 3982–3984 CrossRef CAS.
- G. Deng, C. Tang, F. Li, H. Jiang and Y. Chen, Macromolecules, 2010, 43, 1191–1194 CrossRef CAS.
- P. Froimowicz, H. Frey and K. Landfester, Macromol. Rapid Commun., 2011, 32, 468–473 CrossRef CAS PubMed.
- C. E. Yuan, M. Z. Rong, M. Q. Zhang, Z. P. Zhang and Y. C. Yuan, Chem. Mater., 2011, 23, 5076–5081 CrossRef CAS.
- A. P. Bapat, D. Roy, J. G. Ray, D. A. Savin and B. S. Sumerlin, J. Am. Chem. Soc., 2011, 133, 19832–19838 CrossRef CAS PubMed.
- K. Imato, M. Nishihara, T. Kanehara, Y. Amamoto, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2012, 51, 1138–1142 CrossRef CAS PubMed.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 Search PubMed.
- S. Ji, W. Cao, Y. Yu and H. Xu, Adv. Mater., 2015, 27, 7740–7745 CrossRef CAS PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. Folmer, J. K. Hirschberg, R. F. Lange, J. K. Lowe and E. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- J. A. Neal, D. Mozhdehi and Z. Guan, J. Am. Chem. Soc., 2015, 137, 4846–4850 CrossRef CAS PubMed.
- Y. Yang and M. W. Urban, Angew. Chem., Int. Ed., 2014, 53, 12142–12147 CrossRef CAS PubMed.
- M. W. Urban, Nat. Chem., 2012, 4, 80–82 CrossRef CAS PubMed.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. O. Ganachaud, Chem. Rev., 2012, 113, 80–118 CrossRef PubMed.
- B. Ghosh, K. V. Chellappan and M. W. Urban, J. Mater. Chem., 2011, 21, 14473–14486 RSC.
- B. Ghosh, K. V. Chellappan and M. W. Urban, J. Mater. Chem., 2012, 22, 16104–16113 RSC.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, New York, 1953, ch. XI, p. 432 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py01221c |
| This journal is © The Royal Society of Chemistry 2017 |