
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of hydrogel polyHIPEs from functionalized glycidyl methacrylate†
David
Pahovnik
a,
Janja
Majer
b,
Ema
Žagar
a and
Sebastijan
Kovačič
*a
aNational Institute of Chemistry, Department of Polymer Chemistry and Technology, Hajdrihova 19, 1000 Ljubljana, Slovenia. E-mail: sebastijan.kovacic@ki.si
bUniversity of Maribor, Faculty of Natural Sciences and Mathematics, Koroška 160, 2000 Maribor, Slovenia
First published on 20th July 2016
Abstract
Highly porous hydrogels based on functionalized glycidyl methacrylate (GMA) have been successfully prepared through the high internal phase oil-in-water emulsions. Pre-polymerization functionalization of the GMA monomer, porous structure and water uptake of highly porous hydrogels were investigated. The primary amine groups of tris(2-aminoethyl)amine (TRIS) or 1,2-diaminoethane (EDA) were found to react in three distinct reactions with the GMA, giving a mixture of methacrylate and methacrylamide as the major products with a small amount of the aza-Michael addition product. Elemental analysis revealed nitrogen loadings of 5.3 and 4.6 mmol g−1 for the hydrogel polyHIPEs prepared from the pre-functionalized GMA with TRIS and EDA, respectively. The aminated p(GMA)-based hydrogel polyHIPEs had the densities of around 0.16 g cm−3, void diameters of around 5.5 μm, water uptake up to 15 g g−1 and the specific surface area up to 55 m2 g−1. The water uptake and the specific surface area were found to be between 4 and 5 times higher than the corresponding values for the conventional GMA-based polyHIPEs prepared from the water-in-oil HIPEs. These results demonstrate a highly efficient pre-polymerization functionalization method for the preparation of GMA-based hydrogel polyHIPEs.
1. Introduction
PolyHIPEs are hydrophilic or hydrophobic porous polymers with a highly interconnected porous structure, resulting from templating within oil-in-water (O/W) or water-in-oil (W/O) high internal phase emulsions (HIPEs).1 HIPEs are a special type of emulsions characterized by a droplet (internal) phase volume fraction exceeding 74.05% of the total emulsion volume, whereas the continuous phase consists of monomers that are polymerized around the droplet phase.2 The synthesis of hydrophilic and hydrogel polyHIPEs is not an easy task. A common procedure is to start from the hydrophobic polyHIPEs, thus taking advantage of highly stabile W/O HIPEs, and enhance polyHIPE hydrophilicity through in situ3,4 or post-polymerization functionalization.5–7 However, in both cases a degree of polyHIPE modification with hydrophilic functional groups strongly depends on the efficiency of the functionalization reaction. Alternative methods to prepare the hydrophilic polyHIPEs are: (i) a simultaneous polymerization of hydrophobic and hydrophilic monomers within the W/O HIPEs8,9 and (ii) direct synthesis of hydrophilic polyHIPEs from either CO2-in-water (C/W)10,11 or O/W12,13 HIPEs. In the first case, the so-called bicontinuous polyHIPEs are obtained where the loss of polyHIPE interconnectivity is inevitable due to filling up of the voids with a second polymer phase, which finally affects the polyHIPE permeability. In the second case, the preparation of the C/W HIPEs demands either specialized equipment10 to achieve the required pressure, or application of specific surface-active agents14 to obtain sufficient emulsion stability. Therefore, the O/W HIPEs are still considered to be effective templates for the synthesis of highly porous hydrophilic or hydrogel polyHIPEs.Poly(glycidyl methacrylate) (pGMA) is a versatile polymer since its pendant epoxide groups react readily and irreversibly with several nucleophiles to generate polyglycerol methacrylates.15 Therefore, it draws researchers’ attention in the fields of polymer chemistry, materials science and biochemistry. GMA-based polyHIPEs have been comprehensively investigated. Several studies on morphology,16 permeability17 and polymerization18 have been undertaken, wherein the GMA-based polyHIPEs were tested for protein separation,19 enzyme immobilization20 or as supports for organic synthesis.21
In previous studies, the GMA-based polyHIPEs were synthesized exclusively from the W/O HIPEs where the GMA monomer was dissolved together with a comonomer in the oil (continuous) phase of HIPE. The reason lies in poor water-solubility of GMA as compared to other acrylate monomers (e.g. acrylic acid, acrylamide or 2-hydroxyethyl methacrylate) that are usually used for the synthesis of hydrophilic or hydrogel polyHIPEs. According to GMA partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Pow) of 0.96 and water-solubility of 50 g L−1 (at 20 °C) it can be considered as a more hydrophobic monomer.22 Polymerization of the oil (continuous) phase of the GMA-based HIPE results in an inherently hydrophobic polyHIPE polymer that needs to be appropriately functionalized before it can be applied in water-borne systems. Up to now, the GMA-based polyHIPEs were functionalized by post-polymerization modification using various nucleophiles in order to tune functionality type on the polyHIPE surface. PolyHIPE post-polymerization functionalization is mostly limited to the surface of the voids, whereas the bulk polymer remains mainly non-functionalized, which is preferred when the purpose of functionalization is to introduce functional groups on the voids’ surface, but it is not enough to significantly alter polymer hydrophilicity.
Pow) of 0.96 and water-solubility of 50 g L−1 (at 20 °C) it can be considered as a more hydrophobic monomer.22 Polymerization of the oil (continuous) phase of the GMA-based HIPE results in an inherently hydrophobic polyHIPE polymer that needs to be appropriately functionalized before it can be applied in water-borne systems. Up to now, the GMA-based polyHIPEs were functionalized by post-polymerization modification using various nucleophiles in order to tune functionality type on the polyHIPE surface. PolyHIPE post-polymerization functionalization is mostly limited to the surface of the voids, whereas the bulk polymer remains mainly non-functionalized, which is preferred when the purpose of functionalization is to introduce functional groups on the voids’ surface, but it is not enough to significantly alter polymer hydrophilicity.
In this paper we investigated a synthetic approach to the GMA-based hydrogel polyHIPEs through the O/W HIP emulsions. A pre-polymerization modification of the GMA monomer for the preparation of hydrogel polyHIPEs via O/W HIPEs was compared with the post-polymerization modification of p(GMA) polyHIPE prepared from the W/O HIPE as well as with the unmodified p(GMA) polyHIPE. In the pre-polymerization functionalization approach, the GMA monomer was functionalized beforehand by two different amines, that is 1,2-diaminoethane (EDA) and tris(2-aminoethyl)amine (TRIS), which enabled us to prepare the O/W HIPEs that were subsequently polymerized. In the post-polymerization functionalization approach, p(GMA) polyHIPE was synthesized from the W/O HIPE. Afterwards, it was functionalized either with EDA or TRIS to prepare the post-polymerization functionalized polyHIPEs. Non-functionalized p(GMA) polyHIPE represents a reference sample. The effect of pre- vs. post-polymerization functionalization on the porous structure, specific surface area, chemical properties and water absorption capability of the GMA-based polyHIPEs is discussed herein.
2. Experimental
2.1 Materials
Glycidyl methacrylate (GMA, Sigma-Aldrich), ethylene glycol dimethacrylate (EGDMA, Sigma-Aldrich), N,N′-methylenebisacrylamide (MBAA, Sigma-Aldrich), poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (Pluronic® L121, Sigma-Aldrich), Triton™ X-705 solution (Sigma-Aldrich), α,α′-azoisobutyronitrile (AIBN, Fluka), ammonium persulfate (APS, Sigma-Aldrich), N,N,N′,N′-tetramethylethylenediamine (TMEDA), 2,6-di-tert-butyl-4-methyl-phenol (BHT, Sigma-Aldrich), calcium chloride dihydrate (CaCl2·2H2O, Sigma-Aldrich), toluene (Merck), N,N-dimethylformamide (DMF, Merck), chloroform (J. T. Baker), diethyl ether (Merck), 1,2-diaminoethane (EDA, Fluka), and tris(2-aminoethyl)amine (TRIS; Sigma-Aldrich) were all used as received.2.2 Pre- and post-polymerization functionalization
:1) and finally with ethanol. Afterwards the obtained polyHIPEs were dried under vacuum.
2.3 PoyHIPE synthesis
:1) for 24 hours and then with ethanol for further 24 h. Afterwards, the obtained monolith was dried under vacuum at room temperature.
2.4 Characterization
The morphology investigations were performed with a scanning electron microscope (SEM) (Carl Zeiss, SUPRA 35 VP microscope, Germany). A piece of each sample was cryogenically fractured and mounted on a carbon tab for better conductivity and a thin layer of gold was sputtered on the sample's surface prior the scanning analysis (see details in the ESI†). The FT-IR spectra were recorded on a Perkin-Elmer Spectrum One instrument (Perkin-Elmer, Inc., Waltham, MA, USA) upgraded with a Universal ATR Accessory with a diamond Top-plate-ZnSe. Spectra were recorded in a range of 650–4000 cm−1 at a resolution of 4 cm−1. Data acquisition and processing were performed using a PE Software Spectrum. 1H NMR spectra were recorded in DMSO-d6 with five drops of trifluoroacetic acid added, while 13C NMR, and 1H–13C gradient Heteronuclear Single Quantum Coherence adiabatic version (gHSQCad) spectra were recorded in DMSO-d6 on a Varian Unity Inova 300 instrument (Oxford, UK) in the pulse Fourier-transform mode. Tetramethylsilane (TMS, δ = 0) was used as an internal chemical-shift standard. Nitrogen sorption measurements were performed on an IMI-100 manometric gas sorption analyzer (Hiden Isochema, Inc., Warrington, UK) at 77 K in a range of relative pressure values from 10−6 to 1. The as-prepared samples were degassed at 150 °C for 16 h prior to the measurements. The specific surface areas were determined by the BET (Brunauer Emmett Teller) method based on the obtained sorption isotherms. The densities of functionalized GMA monomers were determined by a Gay-Lussac pycnometer at 23 °C.3. Results and discussion
3.1 Pre-polymerization functionalization of GMA monomer
The primary amine groups of TRIS and EDA react in three distinct reactions with GMA (cf.Scheme 1). The first reaction is addition of the amine to the epoxide group, leading to the secondary amine functionalized methacrylate derivatives. This reaction is typical of post-polymerization modification of the GMA-based polyHIPEs. The second reaction involves aminolysis of the GMA ester group that leads to the methacrylamide derivatives, which can be copolymerized with the amine functionalized methacrylate derivatives formed in the first reaction. The amine functionalized methacrylate derivatives exhibit improved water-solubility as compared to the methacrylamide derivatives and thus help in the preparation of the O/W type of HIPE through improvement of emulsion stability. The third reaction is the undesired aza-Michael reaction since by addition of the amino monomer to the activated double bond, a β-amino acid is formed which is not able to polymerize in the next step. Since the multifunctional amine modifiers were used for GMA modification and since the secondary amine formed in the epoxide reaction or the aza-Michael reaction can further react with any of the three functional groups of either unreacted or reacted GMA, a mixture of multifunctional, branched, water-soluble monomers was obtained. A ratio between the extents of individual reactions depends on the reaction conditions used, and was evaluated from the 1H NMR spectra of the reaction products. Due to the complex mixtures of reaction products formed and significant overlapping of the signals for methylene groups of TRIS or EDA with the signals of glycidyl derivatives we initially focused on the identification of the products by 1H–13C 2D NMR (gHSQCad) (cf.Fig. 1). First, a model reaction was performed where TRIS reacted with GMA in an equimolar ratio of the amino and the epoxide groups at 60 °C for 60 h. The signals of each characteristic functional group were identified, i.e. the double bond in methacrylate and methacrylamide moieties, which are important for further (co)polymerization reactions, together with their corresponding methyl groups, and the methyl group of the products formed by the aza-Michael reaction. By changing the reaction conditions we were able to change the ratio between the different types of reactions as indicated by quantification of the extent of individual reaction from the integrals of the signals belonging to the double bonds and the methyl groups of particular reaction products in the 1H NMR spectra (cf. Fig. S1†). Optimal reaction conditions under which a repeat composition of the final mixtures was obtained, containing the least amount of the products formed by the aza-Michael addition reaction, included three equivalents of glycidyl methacrylate per EDA or TRIS amino groups, and conducting the reaction at 60 °C for 18 h (cf.Table 1).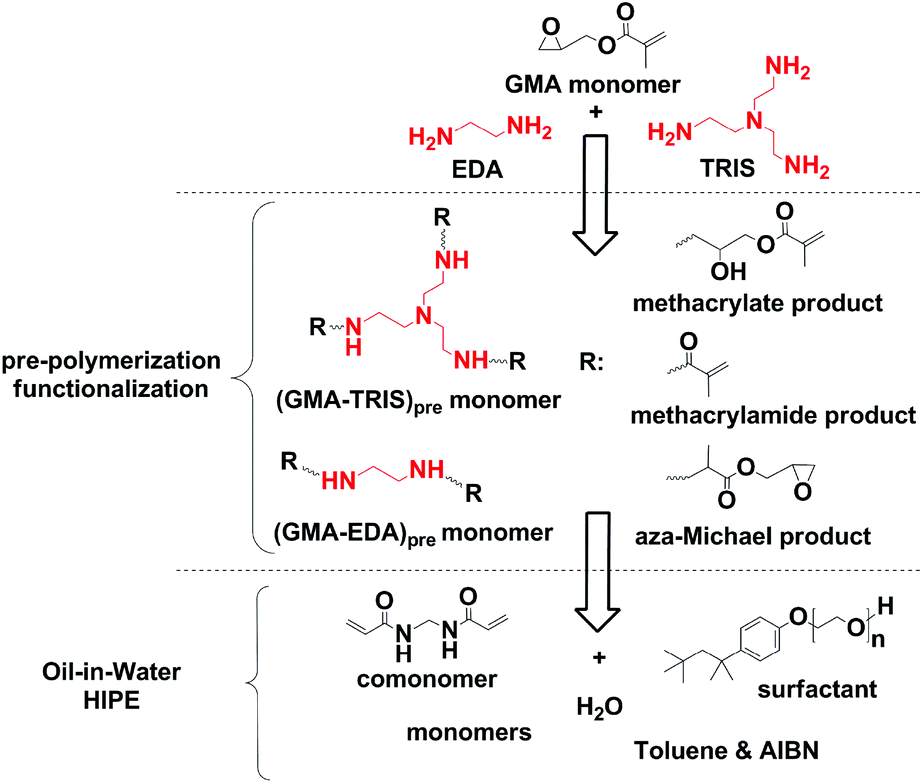 | ||
| Scheme 1 Reaction scheme and products formed by GMA functionalization together with corresponding HIPE formulation. | ||
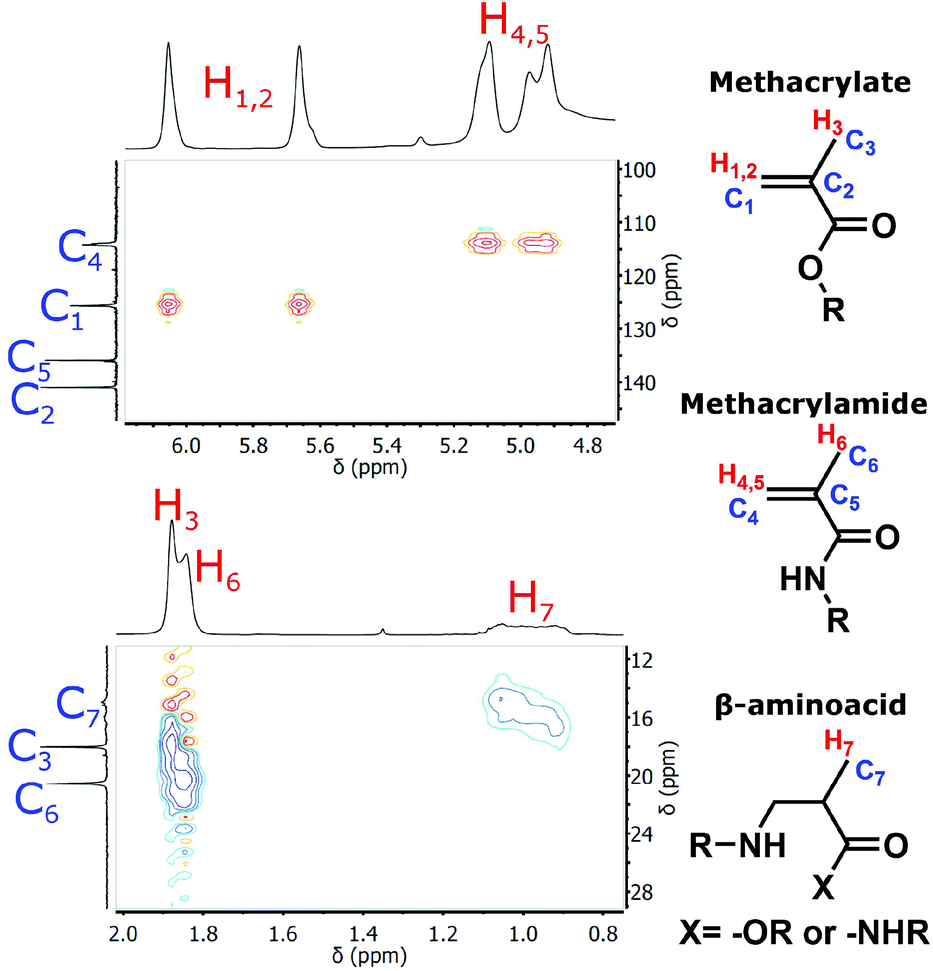 | ||
| Fig. 1 Enlarged 1H–13C 2D NMR (gHSQCad) spectrum of TRIS modified GMA. | ||
3.2 Preparation of HIPEs and characterization of polyHIPEs prepared from functionalized GMA monomers
Pre-polymerization functionalization of GMA as disclosed herein is a method to enhance its hydrophilicity and expand its suitability for the preparation of O/W HIPEs. A critical issue in the preparation of hydrophilic or hydrogel polyHIPE polymers is to find a sufficiently kinetically stable O/W HIPE until the continuous phase is polymerized. Unfortunately, this is not an easy task since the O/W HIPEs demand a high surfactant concentration (between 20 and 30 wt%), and sometimes even the use of a mixture of surfactants in order to strengthen the oil/water interface, which ultimately make the system unsustainable and, therefore, neglected as compared to the W/O HIPEs. In this work, we used the continuous (aqueous) phase, consisting of functionalized (GMA-TRIS)pre or (GMA-EDA)pre monomer, MBAA crosslinker, and Triton™ X-705 non-ionic surfactant (19 vol% according to the continuous phase). First, the APS was used as an initiator, however, the polymerization in the (aqueous) continuous phase started immediately after it had been dissolved at room temperature. This is most probably a consequence of the tertiary amine functional groups in the monomer structure (cf.Scheme 1) that might act similarly as the TMEDA (reducing agent).23 Instead of APS, we therefore selected AIBN as an initiator and dissolved it in the internal (toluene) phase (cf. Table S1†). The (GMA-TRIS)pre and (GMA-EDA)pre O/W HIPEs turned out to be very stable even without the addition of any electrolyte (visually no phase separation after 48 h at 50° C was noticed). This was ascribed to a low partitioning of functionalized monomers between the phases (opposed to neat GMA in W/O HIPE),24 since (GMA-TRIS)pre and (GMA-EDA)pre monomers contain several functional groups such as hydroxyl, amino and amide groups (cf.Scheme 1). For comparison, we also synthesized a conventional, non-functionalized p(GMA) polyHIPE from the W/O HIPE according to the procedure reported elsewhere.21 All synthesized polyHIPEs were white monoliths which were Soxhlet washed and vacuum dried.The polymerization yields (YPH) were 46, 48 and 79% for the polyHIPEs synthesized from the (GMA-TRIS)pre, (GMA-EDA)pre and original GMA, respectively. Low polymerization yields of the polyHIPEs prepared from the functionalized monomers, i.e. (GMA-TRIS)pre and (GMA-EDA)pre, are most probably a consequence of a combination of the interface initiation on the one hand (as schematically illustrated in Fig. S2†), and the retarded polymerization on the other hand. Previous studies have demonstrated that locus of initiation greatly impacts the properties of polyHIPEs and provides among others lower polymerization yields when the polymerization is interfacially initiated.25 On the other hand, amines are known to be effective chain-transfer agents in radical polymerization.23,26 Due to an abundance of the amine groups present in the structure of (GMA-TRIS)pre and (GMA-EDA)pre monomers, polymerization of the continuous (monomer) phase is most likely retarded as a result of the chain transfer reaction, which further affects the low polymerization yields of polyHIPEs prepared from the (GMA-TRIS)pre and (GMA-EDA)pre monomers.
The determined polyHIPEs densities (ρPH) were 0.16, 0.18 and 0.21 g cm−3 for the polyHIPEs prepared from the (GMA-TRIS)pre, (GMA-EDA)pre and original GMA, respectively, and were expected from a ratio of the overall porogen to the monomer content. Densities of the corresponding reference bulk hydrogels (ρbHG) and polymers (ρP) are listed in Table 2. Representative porous microstructures for the polyHIPEs prepared from the (GMA-TRIS)pre, (GMA-EDA)pre and GMA were determined by SEM and are shown in Fig. 2 and S3–S5.† PolyHIPEs’ structure in general consists of “voids”, the volume vacated by the droplets of the dispersed phase, and “windows”, the openings through the walls surrounding the voids. The structures in Fig. 2 resemble the polyHIPE structure since they consist of voids with a diameter from 2 to 6.5 μm (Table 1). The walls of these voids further contain the openings, which look like porous nodular structures rather than spherical windows, but they still form interconnections in-between the voids. On the other hand, the hydrogel walls in themselves have a nanoscale porous structure with smaller macropores between 70 and 150 nm (cf.Fig. 2). A similar structural hierarchy was disclosed in our previous study on polyacetylene based polyHIPE foam.27
 | ||
| Fig. 2 Scanning electron micrographs of (A) (GMA-TRIS)pre polyHIPE morphology, (B) (GMA-TRIS)pre polyHIPE porous walls and (C) (GMA-EDA)pre polyHIPE morphology. | ||
| p(GMA-TRIS)pre | p(GMA-EDA)pre | p(GMA) | |
|---|---|---|---|
| a Yield of HIPE polymerization. b Dry polyHIPE density. c Dry (hydro)gel density. d Bulk polymer density determined by a pycnometer. e Total polyHIPE porosity. f Total bulk hydrogel porosity. g Porosity from polyHIPE void structure. h Porosity from porous polyHIPE hydrogel walls. i Void diameter determined from SEM pictures of broken samples. j Specific surface area. | |||
| YPHa, % | 46 | 48 | 79 |
| ρ PH , g cm−3 | 0.16 | 0.18 | 0.21 |
| ρ HG , g cm−3 | 0.63 | 0.59 | 1.08 |
| ρ P , g cm−3 | 1.18 | 1.13 | 1.20 |
| P PH-T , % | 86 | 84 | 82 |
| P bHG-T , % | 44 | 47 | 10 |
| P PH-V , % | 75 | 70 | 80 |
| P PH-HG , % | 11 | 14 | 2 |
| d v ± σi, μm | 6.5 ± 2 | 4.5 ± 1 | 2 ± 1 |
| SSAj, m2 g−1 | 55 | 37 | 9 |
The total porosities of polyHIPEs (PPH-T) and the reference bulk hydrogels (PbHGT) were calculated from the polyHIPEs’ or bulk hydrogel densities and densities of the fully dense polymers (cf. ESI†). The total polyHIPEs’ porosities (PPH-T) were found to be similar for the pre-polymerization functionalized polyHIPEs and the non-functionalized p(GMA) polyHIPE, i.e. 86, 84 and 82% for the (GMA-TRIS)pre, (GMA-EDA)pre and GMA polyHIPEs, respectively. On the other hand, significant differences were observed for their total bulk hydrogel porosities (PbHG-T), which were found to be 44, 47 and only 10% for the polyHIPEs prepared from (GMA-TRIS)pre, (GMA-EDA)pre and GMA, respectively (cf.Table 2).
Recently, Silverstein et al. disclosed that the total porosity of hydrogel polyHIPEs consists of two types of porosities, i.e. the porosity from the polyHIPE voids (PPH-V) and the porosity within the polyHIPE hydrogel walls (PPH-HG).28 The PPH-T, PPH-V and PPH-HG for the p(GMA-TRIS)pre, p(GMA-EDA)pre and p(GMA) polyHIPEs were calculated using eqn (S1)–(S3) published elsewhere (cf. ESI†).28 As expected, for the p(GMA-TRIS)pre polyHIPE the PPH-T and the PPH-V values were determined to be 86 and 75%, respectively, indicating predominant contribution of the voids to the overall porosity. Similar was observed for the p(GMA-EDA)pre polyHIPE with the PPH-T and PPH-V values of 84 and 70%, respectively. The remainder of porosity, PPH-HG, originated from the porous hydrogel walls and was calculated to be 11 and 14% for p(GMA-TRIS)pre and p(GMA-EDA)pre polyHIPEs, respectively (cf.Table 2). In the case of p(GMA) polyHIPE, the PPH-T, PPH-V and PPH-HG values were 82, 80 and 2%, respectively. While there is no significant deviation in terms of PPH-T between the polyHIPEs prepared from the (GMA-TRIS)pre, (GMA-EDA)pre and GMA, there are differences in the PPH-V and PPH-HG. In the p(GMA) polyHIPE, the PPH-V dominates overall porosity whereas the PPH-HG is only 2%, which is reflected in the relatively low specific surface area of 9 m2 g−1. On the other hand, the PPH-HG increases to 11% for the p(GMA-EDA)pre polyHIPE and up to 14% for the p(GMA-TRIS)pre polyHIPE, reflecting an increase in the specific surface area up to 37 and 55 m2 g−1, respectively (cf.Table 2 and Fig. S6†).
To corroborate enhanced hydrophilicity of polyHIPEs prepared from the (GMA-EDA)pre and (GMA-TRIS)pre monomers an equilibrium water absorption capacity (WUPH-T) was further evaluated and the values were compared with those obtained for the post-polymerization functionalized polyHIPEs by EDA or TRIS (samples designated by (GMA-EDA)post and (GMA-TRIS)post polyHIPEs), and the non-functionalized GMA polyHIPE. The WUPH-T values were found to be 14.8 and 11.1 g g−1 for the p(GMA-TRIS)pre and p(GMA-EDA)pre polyHIPEs, respectively, and 4.2, 5.3 and 5.8 g g−1 for the p(GMA), p(GMA-TRIS)post and p(GMA-EDA)post polyHIPEs, respectively (cf.Table 3). These values represent two to three times higher water uptake for the p(GMA-EDA)pre and p(GMA-TRIS)pre polyHIPEs, which unequivocally show the improvement of the polyHIPE hydrophilicity prepared by the pre-polymerization functionalization approach. Since water absorption capabilities of hydrogel polyHIPEs combine the water absorption through swelling of the hydrophilic polymer and the capillary action in the nanometre and micrometre-scale porous architecture, we further evaluated other contributions to the WUPH-T of polyHIPEs prepared from the (GMA-TRIS)pre, (GMA-EDA)pre and pure GMA. These include the water uptake within the original void volume (WUPH-V, eqn (S5)), the water uptake by the porous hydrogel wall structure (WUbHG-T, measured), and the water uptake from the “void expansion” which is driven by swelling of the porous hydrogel walls (WUPH-VE, eqn (S6)).28 The WUPH-V, WUbHG-T and WUPH-VE values were 5.0, 7.3 and 2.8 g g−1 for the p(GMA-TRIS)pre polyHIPE, and 3.9, 3.0 and 4.2 g g−1 for the p(GMA-EDA)pre polyHIPE (cf. Table S3†). Due to the hydrophobic nature of the polymethacrylate backbone in the p(GMA) polyHIPE, the WUbHG-T was found to be less than 0.1 g g−1. Obviously in the case of p(GMA) polyHIPE only the WUPH-V contributes to the WUPH-T, meaning that water absorption through the polymer swelling is negligible (cf. Table S3†). Since the post-polymerization functionalization of polyHIPEs is limited mostly to the void surface, the p(GMA-TRIS)post and p(GMA-EDA)post polyHIPEs exhibit the same trend regarding the WUPH-T as it was found for the p(GMA) polyHIPE. The contribution to WUPH-T in the case of p(GMA-TRIS)post and p(GMA-EDA)post polyHIPEs is mainly due to water absorption in the highly interconnected, micrometer-scale voids since the polymethacrylate backbone poorly swells in water (WUbHG-T of p(GMA) < 0.1 g g−1). Therefore, a high water-uptake of the polyHIPEs prepared from the functionalized (GMA-TRIS)pre or (GMA-EDA)pre monomers indicates successful conversion of the primarily hydrophobic polymethacrylate structure into the hydrophilic water-absorbing polyHIPEs.
The molecular structure of the p(GMA-TRIS)pre and p(GMA-EDA)pre polyHIPEs was further characterized by means of elemental analysis (EA) and FT-IR spectroscopy. The results were compared with those obtained for the post-polymerization functionalized p(GMA-EDA)post and p(GMA-TRIS)post polyHIPEs and the non-functionalized p(GMA) polyHIPE. FT-IR spectra of all polyHIPE samples show a typical acrylate ester band at 1725 cm−1 (cf.Fig. 3 or S7†). In the case of p(GMA-TRIS)pre and p(GMA-EDA)pre polyHIPEs, the distinctive epoxy bands at 908 and 845 cm−1, typical of the epoxy ring, completely disappeared (cf.Fig. 3). Instead, a new intensive band at 1612 cm−1 appeared in FTIR spectra, indicating the presence of the methacrylamide product. In the case of post-polymerization functionalized p(GMA-EDA)post and p(GMA-TRIS)post polyHIPEs a weak band at 1575 cm−1 typical of the N–H vibration appeared in their FT-IR spectra, indicating almost complete absence of the aminolysis reaction (cf. Fig. S7†). Examination of the elemental composition of the p(GMA-TRIS)pre and the p(GMA-EDA)pre polyHIPEs revealed 7.41 and 6.43% of nitrogen, respectively, corresponding to the nitrogen loadings of 5.3 and 4.6 mmol g−1, respectively (cf. Table S4†). The nitrogen loading of polyHIPEs prepared by post-polymerization functionalization of p(GMA) polyHIPE with TRIS was found to be 4 or 2.5 mmol g−1, depending on the reaction conditions used for their preparation.21,29
 | ||
| Fig. 3 FT-IR spectra of (A) p(GMA) polyHIPE, (B) p(GMA-TRIS)pre polyHIPE and (C) p(GMA-EDA)pre polyHIPE. | ||
4. Conclusions
We have demonstrated a new synthetic approach for the preparation of p(GMA)-based hydrogel polyHIPEs through the O/W HIPE templating. For this purpose we applied the pre-polymerization functionalization approach by which we increased the monomer water-solubility. GMA was thus functionalized by TRIS or EDA, whereby the mixtures of different reaction products were obtained. Such prepared pre-polymerization functionalized p(GMA)-based hydrogel polyHIPEs exhibit improved chemical and morphological properties as compared to those of conventional p(GMA)-based polyHIPEs synthesized from W/O HIPEs. The water uptake of the pre-polymerization functionalized p(GMA)-based hydrogel polyHIPEs is high, i.e. up to 15 g g−1 water, which represents a great improvement as compared to that of the conventional p(GMA)-based polyHIPEs with less than 4 g g−1. Moreover, the specific surface area of up to 55 m2 g−1 is more than five-times larger than that of conventional p(GMA)-based polyHIPEs.5. Conflict of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully acknowledge the financial support of the Ministry of Higher Education, Science and Technology of the Republic of Slovenia, and the Slovenian Research Agency (Program no. P2-0145). The authors wish to thank Mrs Sarah Jurjevec for skillful synthetic work.Notes and references
- H. Zhang and A. I. Cooper, Soft Matter, 2005, 1, 107–113 RSC; N. R. Cameron, Polymer, 2005, 46, 1439–1449 CrossRef CAS.
- M. Silverstein, Prog. Polym. Sci., 2014, 39, 199–234 CrossRef CAS.
- P. Viswanathan, D. W. Johnson, C. Hurley, N. R. Cameron and G. Battaglia, Macromolecules, 2014, 47, 7091–7098 CrossRef CAS.
- S. Kovačič, F. Preishuber-Pflugl, D. Pahovnik, E. Žagar and C. Slugovc, Chem. Commun., 2015, 51, 7725–7728 RSC.
- S. Livshin and M. S. Silverstein, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4840–4845 CrossRef CAS.
- A.-C. Knall, S. Kovačič, M. Hollauf, D. P. Reishofer, R. Saf and C. Slugovc, Chem. Commun., 2013, 49, 7325–7327 RSC.
- N. Barlik, B. Keskinler, M. M. Kocakerim and G. Akay, J. Appl. Polym. Sci., 2015, 132, 42286–42294 CrossRef.
- S. Kovačič, K. Jerabek and P. Krajnc, Macromol. Chem. Phys., 2011, 212, 2151–2158 CrossRef.
- N. Cohen and M. S. Silverstein, Macromolecules, 2012, 45, 1612–1621 CrossRef CAS.
- R. Butler, C. M. Davies and A. I. Cooper, Adv. Mater., 2001, 13, 1459–1463 CrossRef CAS.
- R. Butler, I. Hopkinson and A. I. Cooper, J. Am. Chem. Soc., 2003, 125, 14473–14481 CrossRef CAS PubMed.
- O. Kulygin and M. S. Silverstein, Soft Matter, 2007, 3, 1525–1529 RSC.
- P. Krajnc, D. Štefanec and I. Pulko, Macromol. Rapid Commun., 2005, 16, 1289–1293 CrossRef CAS; S. Kovačič, D. Štefanec and P. Krajnc, Macromolecules, 2007, 40, 8056–8060 CrossRef; C. Youssef, R. Backov, M. Treguer, M. Birot and H. Deleuze, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2942–2947 CrossRef.
- C. Boyere, A. Favrelle, A. F. Leonard, F. Boury, C. Jerome and A. Debuigne, J. Mater. Chem. A, 2013, 1, 8479–8487 CAS.
- Q. L. Li, W. X. Gu, H. Gao and Y. W. Yang, Chem. Commun., 2014, 50, 13201–13215 RSC.
- S. Yang, L. Zeng, Z. Li, X. Zhang, H. Liu, C. Nie and H. Liu, Eur. Polym. J., 2014, 57, 127–136 CrossRef CAS; A. Barbetta, M. Dentini, L. Leandri, G. Ferraris, A. Coletta and M. Bernabei, React. Funct. Polym., 2009, 69, 724–736 CrossRef.
- I. Junkar, T. Koloini, P. Krajnc, D. Nemec, A. Podgornik and A. Štrancar, J. Chromatogr., A, 2007, 1144, 48–54 CrossRef CAS PubMed.
- S. D. Kimmins, P. Wyman and N. R. Cameron, React. Funct. Polym., 2012, 72, 947–954 CrossRef CAS; S. Yang, L. Zeng, Y. Wang, X. Sun, P. Sun, H. Liu, C. Nie and H. Liu, Colloid Polym. Sci., 2014, 292, 2563–2570 Search PubMed.
- P. Krajnc, N. Leber, D. Stefanec, S. Kontrec and A. Podgornik, J. Chromatogr., A, 2005, 1065, 69–73 CrossRef CAS PubMed; C. H. Yao, L. Qi, H. Y. Jia, P. Y. Xin, G. L. Yang and Y. Chen, J. Mater. Chem., 2009, 19, 767–772 RSC; S. Jerenec, M. Šimic, A. Savnik, A. Podgornik, M. Kolar, M. Turnšek and P. Krajnc, React. Funct. Polym., 2014, 78, 32–37 CrossRef.
- S. D. Kimmins, P. Wyman and N. R. Cameron, Polymer, 2014, 55, 416–425 CrossRef CAS.
- J. Majer and P. Krajnc, Macromol. Symp., 2010, 296, 5–10 CrossRef CAS; S. Huš, M. Kolar and P. Krajnc, J. Chromatogr., A, 2016, 1437, 168–175 CrossRef PubMed.
- http://www.cdc.gov/niosh/ipcsneng/neng1679.html .
- X. Feng, Chin. J. Polym. Sci., 1986, 4, 109–118 Search PubMed.
- I. Pulko, V. Smrekar, A. Podgornik and P. Krajnc, J. Chromatogr., A, 2011, 1218, 2396–2401 CrossRef CAS PubMed.
- I. Gurevitch and M. S. Silverstein, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1516–1525 CrossRef CAS; T. Gitli and M. S. Silverstein, Soft Matter, 2008, 4, 2475–2485 RSC.
- C. H. Bamford and E. F. T. White, Trans. Faraday Soc., 1956, 52, 716–727 RSC.
- E. Slováková, M. Ješelnik, E. Žagar, J. Zedník, J. Sedláček and S. Kovačič, Macromolecules, 2014, 47, 4864–4869 CrossRef.
- M. Ovadia and M. S. Silverstein, Polym. Int., 2016, 65, 280–289 CrossRef CAS.
- S. D. Kimmins, P. Wyman and N. R. Cameron, Polymer, 2014, 55, 416–425 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Recipes, FT-IR and 1H NMR spectra, N2 sorption spectra, water uptake and porosity calculations and SEM pictures. See DOI: 10.1039/c6py01122e |
| This journal is © The Royal Society of Chemistry 2016 |