DOI:
10.1039/C6PY01083K
(Paper)
Polym. Chem., 2016,
7, 5730-5738
High open circuit voltage polymer solar cells enabled by employing thiazoles in semiconducting polymers†
Received
21st June 2016
, Accepted 24th August 2016
First published on 25th August 2016
Abstract
We investigate the effect of introducing thiazole π-spacers in wide bandgap semiconducting copolymers based on benzothiadiazole with either benzodithiophene or indacenodithiophene. Thiazole-containing polymers show similar optical bandgaps but drastically downshifted frontier orbital levels compared to the corresponding thiophene derivatives as a result of the more electronegative imine nitrogen. Consequently, they provide an enhanced open-circuit voltage (Voc) of about 0.2 V in bulk heterojunction polymer solar cells. Notably, the Voc approaches 0.95 V for polymers with an optical bandgap below 1.75 eV. Despite the favourable energetics, the performance of the thiazole-containing polymers in solar cells is currently restricted by the relatively low molecular weight, which results in a sub-optimal bulk heterojunction morphology.
Introduction
Polymer solar cells (PSCs) are an emerging renewable energy technology that has the prospect of achieving efficient, large-area, flexible photovoltaic modules via roll-to-roll processing or ink-jet printing.1,2 The core component of a PSC is its bulk-heterojunction (BHJ) active layer, which typically consists of a semiconducting polymer as the electron donor and a fullerene derivative as the electron acceptor.3 Tremendous progress has been achieved in the past few years with power conversion efficiencies (PCEs) exceeding 11% in both single- and multi-junction cells.4–6 These achievements strongly benefited from the development of new semiconducting conjugated polymers.7–9 By employing alternating donor–acceptor (D–A) units in the polymer backbone, creating new building blocks, incorporating proper peripheral functionalities, and optimizing alkyl side chain patterns, the electronic structures, energy levels, optical properties, and aggregation behaviour of conjugated polymers can be well controlled and rationally designed.10–14 Recently, wide bandgap conjugated polymers with optical bandgaps (Egs) of ≥1.70 eV have received growing attention, since they are key components for use in multi-junction PSCs and in non-fullerene PSCs where the complementary absorption is of particular importance.15–24 One of the most important advantages of wide bandgap polymers is their potential to offer a high open-circuit voltage (Voc), which is desirable when the polymers are used in the front cell of multi-junction PSCs or under low light intensity, e.g. in indoor applications.25 Taking into account the widely recognized 0.6 eV limit of photon energy loss (Eloss = Eg − eVoc) for efficient charge generation, wide bandgap organic solar cells can provide a maximum attainable Voc of ≥1.1 V, theoretically.26 Presently, the typical Eloss in the wide bandgap subcell is ≥0.9 eV,5,6 and hence there is sufficient room for increasing the Voc of wide bandgap solar cells while maintaining high external quantum efficiency (EQE).
It is well known that the Voc of PSCs is proportional to the energy level of the highest occupied molecular orbital (HOMO) of the semiconducting polymer donor. Generally, polymers possessing deeper-lying HOMO levels can produce a higher Voc.27 Conjugated polymers derived from the widely used electron accepting unit benzo[2,1,3]thiadiazole (BT) and weak electron donating units such as carbazole,28 silafluorene,29 benzo[1,2-b:4,5-b′]dithiophene (BDT),19,30 or indaceno[1,2-b:5,6-b′]dithiophene (IDT)31 linked via two thiophene π-spacers usually produce an Eg of 1.7–1.9 eV, which is an ideal value for solar cells that need complementary optical absorption. However, PSCs made from these polymers usually suffer from a large Eloss of ≥0.9 eV. It is well known that the replacement of an sp2 carbon (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–) by a more electron negative imine nitrogen (N–) within an aromatic unit increases the electron affinity.32–36 For example, Zaborova et al. demonstrated that switching from thiophene to thiazole in conjugated small molecules does not only downshift the frontier orbital levels, but also keeps the Eg almost unchanged.37 Recently, our group showed that incorporating thiazole into conjugated polymers can enable high EQE at very low Eloss.38 In addition, it is expected that the thiazole ring can reduce the steric hindrance between the neighbouring rings, leading to the improved coplanarity of the polymers and closer interchain packing.39 We thus speculate that the replacement of thiophene by a thiazole ring as a π-spacer will be an effective method for creating highly efficient wide bandgap polymers with high Voc and reduced Eloss.
CH–) by a more electron negative imine nitrogen (N–) within an aromatic unit increases the electron affinity.32–36 For example, Zaborova et al. demonstrated that switching from thiophene to thiazole in conjugated small molecules does not only downshift the frontier orbital levels, but also keeps the Eg almost unchanged.37 Recently, our group showed that incorporating thiazole into conjugated polymers can enable high EQE at very low Eloss.38 In addition, it is expected that the thiazole ring can reduce the steric hindrance between the neighbouring rings, leading to the improved coplanarity of the polymers and closer interchain packing.39 We thus speculate that the replacement of thiophene by a thiazole ring as a π-spacer will be an effective method for creating highly efficient wide bandgap polymers with high Voc and reduced Eloss.
Herein, we report the synthesis and characterization of two novel wide bandgap polymers (BDT-DTzBT and IDT-DTzBT) with BT as an acceptor unit and BDT or IDT as donor units, linked via thiazole (Tz) π-spacers and compare them to the polymers with thiophene (T) as a π-spacer. The underlying principles of the specific molecular design are outlined in Scheme 1. The experimental results show that the replacement of thiophene by thiazole can indeed effectively downshift the frontier orbital levels of polymers and increase the Voc of the resulting PSCs and suggest that efficient PSCs can be achieved in high molecular weight thiazole-containing polymers.
 |
| | Scheme 1 Design rationales for conjugated polymers from thiophene-substituted BT to thiazole-substituted BT. | |
Results and discussion
Synthesis
The chemical structures and synthetic route of the thiazole-containing polymers, BDT-DTzBT and IDT-DTzBT, are shown in Scheme 2. For direct comparison, the corresponding thiophene-containing analogues, BDT-DTBT and IDT-DTBT, were also synthesized. The polymers were synthesized by standard Stille polymerization from the corresponding bis(trimethyltin) and dibrominated monomers. We found that the thiazole-based polymers consistently have a lower molecular weight than their corresponding thiophene-containing analogues. The number-average molecular weight (Mn) decreases in the series BDT-DTzBT < BDT-DTBT, IDT-DTzBT < IDT-DTBT, as determined with gel permeation chromatography (GPC) performed at 140 °C using ortho-dichlorobenzene (o-DCB) as the eluent (Table 1). The GPC traces are shown in Fig. S1 (ESI†). In particular, the Mn of BDT-DTzBT (18.6 kDa) is quite low. The lower molecular weights of BDT-DTzBT and IDT-DTzBT lead to higher solubility. Consequently, BDT-DTzBT and IDT-DTzBT are readily soluble in both chloroform and chlorinated aromatic solvents at room temperature, while BDT-DTBT and IDT-DTBT are only soluble in chlorinated aromatic solvents upon heating. Despite testing various different conditions for the polymerization of BDT-DTzBT (ESI Table S1†), we were unable to enhance the molecular weights. We speculate that this is caused by a lower intrinsic reactivity of the thiazole-containing dibrominated monomer (M4) or by chelation of the palladium catalyst by the adjacent nitrogen atoms on thiazole-substituted BT moieties (Scheme S1†), which slows down or terminates the polymerization reaction.
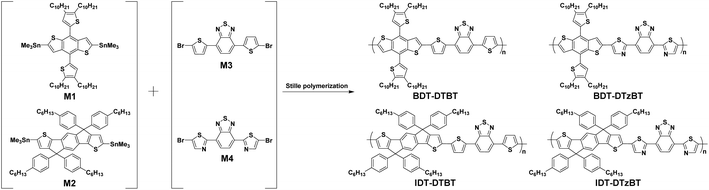 |
| | Scheme 2 Chemical structures and the synthesis route of BDT-DTBT, BDT-DTzBT, IDT-DTBT, and IDT-DTzBT. | |
Table 1 Molecular weight, optical properties, and energy levels of BDT-DTBT, BDT-DTzBT, IDT-DTBT, and IDT-DTzBT
| Polymer |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (kDa) a (kDa) |
PDIa |
λ
max (nm) |
E
optg (eV) |
HOMO (eV) |
LUMO (eV) |
E
CVg (eV) |
ΔELUMOb (eV) |
| Solution |
Film |
|
Determined by GPC at 140 °C using o-DCB as the eluent.
ΔELUMO = ELUMO (polymer) − ELUMO ([70]PCBM).
|
| BDT-DTBT |
32.6 |
2.8 |
628 |
652 |
1.68 |
−5.60 |
−3.57 |
2.03 |
0.67 |
| BDT-DTzBT |
18.6 |
3.4 |
592 |
600 |
1.73 |
−5.69 |
−3.80 |
1.89 |
0.44 |
| IDT-DTBT |
93.7 |
6.5 |
600 |
608 |
1.75 |
−5.55 |
−3.72 |
1.83 |
0.52 |
| IDT-DTzBT |
46.7 |
2.7 |
618 |
608 |
1.73 |
−5.71 |
−3.92 |
1.79 |
0.32 |
Optical properties
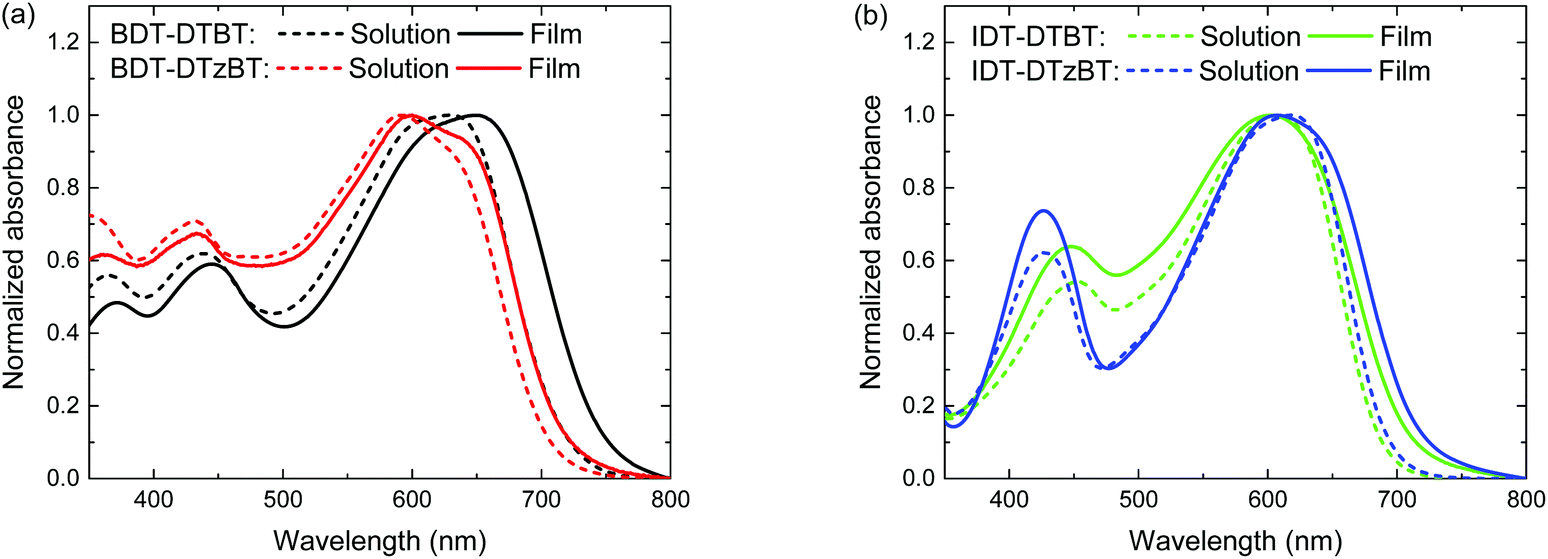
UV-vis absorption spectra of the polymers both in solutions and in thin films are shown in Fig. 1 and the relevant data are listed in Table 1. All polymers show a bathochromically shifted absorption in the solid-state compared to the solution as a result of aggregation. As expected, the thiazole-containing polymers show a similar Eg (∼1.70 eV) to their thiophene-containing analogues in thin films (Table 2). BDT-DTzBT shows some vibronic fine structures, which suggests coplanarity along the conjugated main. A similar vibronic fine structure is not observable in IDT-DTzBT, which is attributed to the bulky hexylphenyl substituents on the IDT unit that impede the aggregation and close packing of the polymer chains in the solid state. IDT-DTzBT, which possesses a reasonably high molecular weight, shows a higher absorption coefficient than IDT-DTBT (Fig. S2 and S3, ESI†). In contrast, the absorption coefficient of BDT-DTzBT is significantly lower than that of BDT-DTBT (Fig. S2 and S3, ESI†), which may be caused by the too low molecular weight of the former.
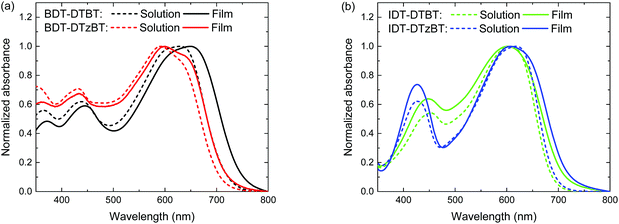 |
| | Fig. 1 Absorption spectra of the polymers in solutions and in thin films: (a) BDT-DTBT and BDT-DTzBT; (b) IDT-DTBT and IDT-DTzBT. | |
Table 2 Performance parameters of polymer:[70]PCBM solar cellsa
| Polymer |
J
sc(EQE)b (mA cm−2) |
V
oc (V) |
FF (−) |
PCEc (%) |
EQEmax (−) |
E
lossd (eV) |
μ
he (cm2 V−1 s−1) |
|
Measured with 100 mW cm−2 white light illumination in a ITO/MoOx(10 nm)/polymer:[70]PCBM/PFN/Al(100 nm) device configuration.
Determined by integrating the EQE with the AM1.5G solar spectrum.
Calculated using Jsc (EQE).
E
loss = Eg − eVoc, where Eg is the optical bandgap of the active layer, using Eg = 1.70 eV for [70]PCBM.49
At an electric field of E = 2 × 105 V cm−1.
|
| BDT-DTBT |
10.9 |
0.73 |
0.60 |
4.8 |
0.60 |
0.95 |
1.3 × 10−4 |
| BDT-DTzBT |
7.4 |
0.92 |
0.63 |
4.3 |
0.42 |
0.78 |
1.3 × 10−5 |
| IDT-DTBT |
9.8 |
0.75 |
0.49 |
3.6 |
0.54 |
0.95 |
4.6 × 10−4 |
| IDT-DTzBT |
10.5 |
0.94 |
0.46 |
4.5 |
0.56 |
0.76 |
3.6 × 10−5 |
Electrochemical properties and energy levels
The effects of replacing thiophene by thiazole as a π-spacer on the electrochemical properties and frontier orbital levels of the polymers were studied by cyclic voltammetry (CV) experiments. As shown in Fig. 2a, the thiazole-containing polymers BDT-DTzBT and IDT-DTzBT show a significant shift of the oxidation and reduction waves by about 0.1–0.2 V towards more positive potentials. The HOMO and lowest unoccupied molecular orbital (LUMO) levels of the polymers were determined from the onsets of the oxidation and reduction waves and using the energy of ferrocene (Fc/Fc+) of −5.23 eV vs. vacuum as the internal standard. The energy level diagram is depicted in Fig. 2b and the data are summarized in Table 1. Notably, the offsets, ΔELUMO, between the LUMO levels of the polymers and that of [6,6]-phenyl-C71-butyric acid methyl ester ([70]PCBM), approach the empirical limit of 0.30 eV for efficient exciton dissociation in PSCs for the thiazole-containing polymers BDT-DTzBT and IDT-DTzBT. Clearly, the replacement of thiophene by thiazole effectively downshifts both HOMO and LUMO levels of the resulting polymers. Such a downshift in frontier orbital levels not only increases the Voc of the resulting PSCs, but also improves the stability of the polymers against oxygen and moisture. These results prove that the substitution of thiazole into the conjugated backbone is an effective method for adjusting energy levels in organic photovoltaic materials.
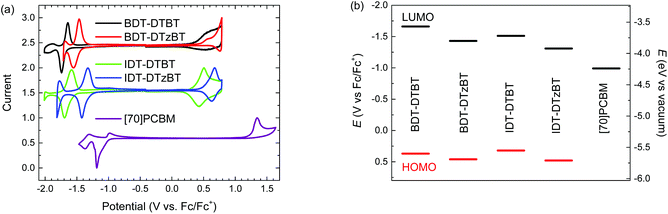 |
| | Fig. 2 Cyclic voltammograms (a) and energy levels (b) of BDT-DTBT, BDT-DTzBT, IDT-DTBT, IDT-DTzBT, and [70]PCBM. A value of −5.23 eV for Fc/Fc+ is used to determine the energy levels vs. vacuum. | |
Photovoltaic properties
The photovoltaic properties of the polymers were evaluated in single-junction PSCs under simulated AM1.5G illumination (100 mW cm−2). EQE calibrated current densities were used to accurately calculate the PCEs of the solar cells. Prior to the optimization of the polymer:fullerene active layer, the effects of the contacts for hole and electron collection on the photovoltaic performance of the PSCs were investigated for BDT-DTzBT and IDT-DTzBT. Employing the commonly used acidic poly(3,4-ethylenedioxythiophene):poly(styrenesulfonic acid) (PEDOT:PSS) can lead to a large barrier for hole extraction and diminished performance via protonation of the basic nitrogen.40 As shown in the ESI (Fig. S4 and S5; Tables S2 and S4†), both BDT-DTzBT and IDT-DTzBT afford the best solar cells when molybdenum oxide (MoOx) and poly[(9,9)-bis(3′-(N,N-dimethylamino)propyl)-2,7-fluorene]-alt-2,7-(9,9-dioctylfluorene)](PFN)41 are used as a hole-collecting layer and an electron-collecting layer, respectively. To enable direct comparison all polymers were therefore tested using an indium tin oxide (ITO)/MoOx/polymer:[70]PCBM/PFN/Al cell architecture. We then extensively optimized the processing conditions (solvent, co-solvent, and polymer-fullerene weight ratio). A summary of the device parameters of the PSCs based on BDT-DTzBT and IDT-DTzBT under various conditions is collected in the ESI (Tables S3 and S5†). The current density–voltage (J–V) characteristics and EQE curves of the optimized PSCs are shown in Fig. 3, and the corresponding device parameters are summarized in Table 2. Device statistics can be found in the ESI (Table S6†).
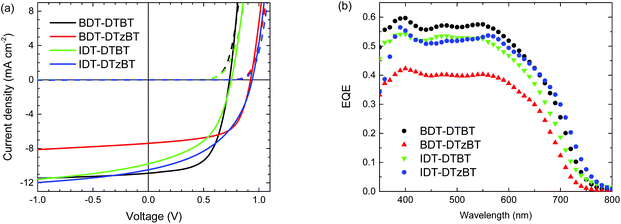 |
| | Fig. 3 (a) Current density–voltage (J–V) characteristics of ITO/MoOx(10 nm)/polymer:[70]PCBM/PFN/Al(100 nm) solar cells in the dark and under AM1.5G illumination (100 mW cm−2). (b) Corresponding EQE spectra, measured with 1 sun equivalent light bias. | |
All the four polymers offer PCEs around 4%–5% in PSCs. The device performance of the BDT-DTBT:[70]PCBM cell in this study is comparable to our previous work where ITO/PEDOT:PSS and LiF/Al were used as hole-collecting and electron-collecting contacts, respectively.19 Compared to the thiophene-containing polymers, the Voc of solar cells based on the thiazole-containing polymers is enhanced by about 0.2 V. Consequently, a high Voc approaching 0.95 V was achieved by BDT-DTzBT and IDT-DTzBT. We note that such a high value is rarely observed for the polymers with an Eg below 1.75 eV.42–45 Correspondingly, the Eloss of the PSCs reduces from ∼0.95 eV for BDT-DTBT and IDT-DTBT to ∼0.75 eV for BDT-DTzBT and IDT-DTzBT (Table 2). These results are consistent with the energy level alignment of the polymers (Fig. 2) and confirm the validity of our design of replacing thiophene by thiazole to increase the Voc. However, Fig. 3 also shows that BDT-DTzBT produces a lower PCE than BDT-DTBT due to a substantially lower short-circuit current density (Jsc) and EQE. We attribute this to the combined effect of the smaller optical absorption coefficient (Fig. S2 and S3, ESI†) and the lower molecular weight of BDT-DTzBT. The beneficial effect of a high polymer molecular weight on the performance of polymer solar cells is well documented and generally ascribed to the suboptimal morphology that is formed when the molecular weight is low.46–48 In this respect, a fill factor (FF) of 0.63 for BDT-DTzBT:[70]PCBM cells is still relatively high, implying that highly efficient PSCs based on this polymer may be achievable once the molecular weight can be improved. This speculation is supported by IDT-DTzBT, which possesses a reasonable high Mn of 46.7 kDa and produces a Jsc and an FF that are comparable to IDT-DTBT, thereby offering a higher PCE due to its higher Voc. Both IDT-DTBT and IDT-DTzBT provide a relative low FF. We will discuss the possible reasons in more detail below. The EQE spectra (Fig. 3b) show virtually identical shapes for the two BDT and for the two IDT polymers. The main difference is the height of the EQE.
Morphology
It is well known that the morphology of the polymer:fullerene blend films plays a key role in exciton diffusion, exciton dissociation, and charge transport which determine the performance of bulk-heterojunction solar cells. The morphology was thus investigated by transmission electron microscopy (TEM). As shown in Fig. 4a and b, the BDT-DTBT:[70]PCBM bulk heterojunction film shows finely dispersed fibrillary structures and an intimately mixed film, while the BDT-DTzBT:[70]PCBM blend exhibits considerable phase separation with large (>100 nm), dark almost spherical fullerene domains and brighter fibrillary structures that we attribute to aggregated polymer chains. The large spherical domains in BDT-DTzBT:[70]PCBM are reminiscent of liquid–liquid phase separation during spin-coating. It is known that liquid–liquid phase separation during drying of spin coated polymer-fullerene films occurs when the polymer has an increased solubility and a decreased tendency to aggregate.50,51 The occurrence of liquid–liquid phase separation for BDT-DTzBT:[70]PCBM blends is therefore not surprising when we recall the low molecular weight and high solubility of BDT-DTzBT. With this morphology, the low Jsc and EQE are a consequence of a diminished polymer/fullerene interface for charge generation, while the relative high FF can be a consequence of high domain purity that we infer from the high contrast between the light (polymer rich) and dark (fullerene) regions in the TEM of the BDT-DTzBT:[70]PCBM blend (Fig. 4b). In addition, the larger domains in this blend (Fig. 4b) can provide unobstructed pathways for charge transport.
 |
| | Fig. 4 Bright field TEM images of the polymer:[70]PCBM blend films deposited with the same methods as those for PSC fabrication for BDT-DTBT (a), BDT-DTzBT (b), IDT-DTBT (c), and IDT-DTzBT (d). Image size: 1.5 × 1.5 μm2; scale bar: 200 nm. | |
As shown in Fig. 4c and d, both IDT-DTBT:[70]PCBM and IDT-DTzBT:[70]PCBM possess a relatively homogeneous film morphology without significant phase separation. In such an intimately mixed blend, charge separation is hindered because neither of the components crystallizes. Crystallization is known to be beneficial for the dissociation of photo-generated charges from the donor–acceptor interface,52 and without crystallization charges are more prone to geminate recombination. In agreement with this interpretation, the current density of illuminated IDT-DTBT and IDT-DTzBT based solar cells exhibits a considerable increase under reverse bias where the enhanced electric field promotes charge separation from the donor–acceptor interface (Fig. 3a). The fact that the dark current does not increase significantly under reverse bias (Fig. 3a) excludes the fact that the increased current density under illumination is due to a low shunt resistance. The morphology thus explains why both IDT-DTBT:[70]PCBM and IDT-DTzBT:[70]PCBM solar cells show a low FF that limits the PCE.
Charge transport and recombination
To further understand the photovoltaic properties of the polymers, the charge carrier transport and recombination of the polymer:[70]PCBM blend films were investigated. Hole mobilities were determined from hole-only polymer:[70]PCBM blend devices by fitting the data using the space-charge-limited current (SCLC) model. The current density versus voltage characteristics are shown in Fig. S6 (ESI†), and the mobility values acquired from the data fitting are listed in Tables 2 and S7 (ESI†). Table 2 shows that the hole mobilities of the bulk heterojunction films based on thiazole-containing polymers are one order of magnitude lower than those of their thiophene-containing analogues.
Bimolecular charge recombination kinetics of the PSCs were qualitatively investigated by analysing the ratio between the EQE measured with light bias to afford a short-circuit current equivalent to AM1.5G illumination and the EQE measured without light bias (denoted as ρ = EQEbias/EQEno bias). Since the bimolecular recombination efficiency can be approximated as ηBR = 1 − ρ,53 a high ρ indicates lower bimolecular recombination. Fig. 5 shows the ρ average values in the wavelength range from 400 to 700 nm of the PSCs. The original biased and unbiased EQEs of the corresponding solar cells are provided in the ESI (Fig. S7†). In all cases the bimolecular recombination efficiency is estimated to be less than 10%. PSCs based on the thiazole-containing polymers produce lower average ρ values than the corresponding thiophene-containing polymers, indicating more bimolecular recombination losses. These results are consistent with the hole mobilities of the corresponding polymer:[70]PCBM blends.
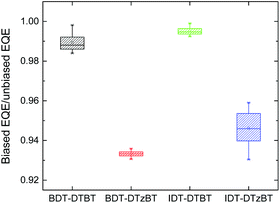 |
| | Fig. 5 Average EQEbias/EQEno bias values of polymer:[70]PCBM solar cells based on BDT-DTBT, BDT-DTzBT, IDT-DTBT, and IDT-DTzBT. | |
Considering its lower hole mobility and higher bimolecular recombination, the higher FF of the BDT-DTzBT:[70]PCBM cell (FF = 0.63) compared to the BDT-DTBT:[70]PCBM cell (FF = 0.60) may seem surprising. Likewise, the FF of the IDT-DTzBT:[70]PCBM cells (FF = 0.46) is only slightly lower than that of the IDT-DTBT:[70]PCBM devices (FF = 0.49). This shows that the devices operate in a range where the hole mobility and bimolecular recombination are not the main factors that determine the FF and Jsc. We propose that rather the efficiency of charge separation from the donor–acceptor interface and the effect of the electric field on this process determine the FF. In agreement, the highest FF is found for the most phase-separated blend (BDT-DTzBT:[70]PCBM), where the pure phases enhance the separation of photo-generated electrons and holes from the donor–acceptor interface.
Conclusions
In conclusion, four wide bandgap conjugated polymers with BT as an acceptor unit, BDT or IDT as a donor, and thiazole or thiophene as a π-spacer were synthesized and applied in polymer–fullerene solar cells. The results show that the replacement of thiophene by thiazole can effectively downshift the frontier orbital levels of the polymers without significantly changing the optical bandgap. Consequently, the Voc of the PSCs is enhanced by about 0.2 V. The PCE, however, is only significantly improved for IDT-DTzBT compared to IDT-DTBT, but not for BDT-DTzBT with respect to BDT-DTBT. In the latter case, the low Mn and higher solubility cause a sub-optimal morphology for BDT-DTzBT:[70]PCBM blends. Also for the corresponding IDT polymers the Mn is significantly reduced when introducing thiazole, but in this case the absolute value remains high and the blend morphologies are similar such that the increased Voc indeed increases the PCE. The reason for the reduced Mn seems to be a lower intrinsic reactivity of the thiazole-flanked BT (DTzBT) monomer (M4) in the Stille cross-coupling polymerization reaction, or a possible chelation of the palladium catalyst by the DTzBT unit. As a result, the real potential of the thiazole-containing wide bandgap polymers in PSCs is obscured by the low molecular weights. Nevertheless, the improved device performance for the thiazole-containing polymer (IDT-DTzBT), in which reasonably high molecular weight was achieved, suggests that the thiazole-implementing strategy can be a fruitful approach.
Experimental section
Synthesis
All synthetic procedures were performed under an argon atmosphere. All reactants and reagents are commercially available and used as received, unless otherwise specified. Solvents were dried over 4 Å molecular sieves. [60]PCBM (purity of 99%) and [70]PCBM (purity of 99%) were purchased from Solenne BV. 2,6-Bis(trimethyltin)-4,8-di(2,3-didecylthiophen-5-yl)-benzo[1,2-b:4,5-b′]dithiophene (M1) was purchased from SunaTech Inc. 2,7-Bis(trimethyltin)-4,4,9,9-tetrakis(4-hexylphenyl)-4,9-dihydro-s-indaceno[1,2-b:5,6-b′]dithiophene (M2),54 4,7-bis(5-bromothiophen-2-yl)benzo[2,1,3]thiadiazole (M3),55 and 4,7-bis(5-bromothiazol-2-yl)benzo[2,1,3]thiadiazole (M4)56 were synthesized according to the literature procedures.
Poly{(4,8-di(2,3-didecylthiophen-5-yl)-benzo[1,2-b:4,5-b′]dithiophen-2,6-diyl)-alt-((4,7-di(thiophen-2-yl)benzo[2,1,3]thiadiazole)-5,5′-diyl)} (BDT-DTBT).
To a degassed solution of monomer M1 (99.30 mg, 0.080 mmol) and M2 (36.66 mg, 0.080 mmol) in anhydrous toluene (1.8 mL) and anhydrous N,N-dimethylformamide (DMF) (0.2 mL), tris(dibenzylideneacetone)-dipalladium(0) (2.19 mg, 0.0024 mmol) and tri(o-tolylphosphine) (5.84 mg, 0.0192 mmol) were added. The mixture was stirred at 115 °C for 18 hours, after which 2-(tributylstannyl)thiophene and 2-bromothiophene were sequentially added to the reaction with a 2 hour interval. After another 2 hours, the reaction mixture was diluted with o-DCB, and refluxed with ethylenediaminetetraacetic acid dipotassium salt dihydrate (EDTA) (100 mg) and water (2 mL) for 2 hours. Upon cooling, the reaction mixture was precipitated in methanol and filtered through a Soxhlet thimble. The polymer was subjected to sequential Soxhlet extraction with acetone, hexane, dichloromethane, and chloroform under argon protection. The residue in the thimble was dissolved in o-DCB at 140 °C for 2 hours, filtered hot, concentrated and precipitated in methanol to obtain BDT-BT-2T as a dark shiny solid (80 mg, yield 82%). Mn = 32.6 kg mol−1, PDI = 2.8.
Poly{(4,8-di(2,3-didecylthiophen-5-yl)-benzo[1,2-b:4,5-b′]dithiophen-2,6-diyl)-alt-((4,7-di(thiazol-2-yl)benzo[2,1,3]thiadiazole)-5,5′-diyl)} (BDT-DTzBT).
BDT-DTzBT was synthesized as a dark solid with the yield of 81% according to the method of BDT-DTBT described above by collecting the chloroform fraction in Soxhlet extraction. Mn = 18.6 kDa, PDI = 3.4.
Poly{(4,4,9,9-tetrakis(4-hexylphenyl)-4,9-dihydro-s-indaceno[1,2-b:5,6-b′]dithiophen-2,7-diyl)-alt-((4,7-di(thiophen-2-yl)benzo[2,1,3]thiadiazole)-5,5′-diyl)} (IDT-DTBT).
IDT-DTBT was synthesized as a dark solid with the yield of 56% according to the method of BDT-DTBT described above. Mn = 93.7 kDa, PDI = 6.5.
Poly{(4,4,9,9-tetrakis(4-hexylphenyl)-4,9-dihydro-s-indaceno[1,2-b:5,6-b′]dithiophen-2,7-diyl)-alt-((4,7-di(thiazol-2-yl)benzo[2,1,3]thiadiazole)-5,5′-diyl)} (IDT-DTzBT).
IDT-DTzBT was synthesized as a dark solid with the yield of 64% according to the method of BDT-DTBT described above. Mn = 46.7 kDa, PDI = 2.7.
Measurements and characterization
1H nuclear magnetic resonance (NMR) spectroscopy spectra were recorded at 400 MHz on a VARIAN mercury spectrometer at 25 °C unless otherwise noted. 1H NMR spectra are referenced to tetramethylsilane (TMS) using the residual solvent peak impurity of the given solvent. The molecular weights and polydispersity index (PDI) were determined with gel permeation chromatography (GPC) at 140 °C on a PL-GPC 120 system using a PL-GEL 10 μm MIXED-B column and o-DCB as the eluent against polystyrene standards. All the polymer samples were dissolved in o-DCB at 140 °C overnight and the solutions were filtered through PTFE filters (0.2 μm) prior to injection. UV-visible spectroscopy was recorded on a Perkin Elmer Lambda 900 UV/vis/near IR spectrophotometer at room temperature unless noted otherwise. All solution UV-vis experiments were performed in o-DCB with a sample concentration of 0.05 mg mL−1. Films were prepared by spin coating o-DCB solutions on glass substrates. Cyclic voltammetry (CV) studies were performed at a scan rate of 0.1 V s−1 under an inert atmosphere with 1 M tetrabutylammonium hexafluorophosphate in acetonitrile as the electrolyte. The working electrode was an ITO bar and the counter electrode was a silver electrode. A silver wire coated with silver chloride (Ag/AgCl) was used as a quasi-reference electrode in combination with Fc/Fc+ as an internal standard. The samples were spin coated on top of the ITO work electrode to form ∼10 nm thick films. TEM was performed on a Tecnai G2 Sphera transmission electron microscope operated at 200 kV. Space-charge-limited-current hole mobility was acquired through the hole-only devices with a configuration of ITO/MoOx (10 nm)/polymer:[70]PCBM/MoOx (10 nm)/Ag (100 nm). The thickness of the polymer:[70]PCBM blend films is similar to those of the corresponding solar cells. The dark current densities of the polymer:[70]PCBM blends were measured by applying a voltage between 0 and 4 V using a computer-controlled Keithley 2400 source meter under an N2 atmosphere. These data were analysed according to the Mott–Gurney laws that includes a Poole–Frenkel-type dependence of mobility on the electric field, given by 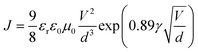 , where ε0 is the permittivity of free space, εr is the dielectric constant of the polymer which is assumed to be around 3 for the conjugated polymers, μ0 is the zero-field mobility, V is the voltage drop across the device, d is the film thickness of the active layer, and γ is a parameter that describes the strength of the field-dependence effect. The applied voltage is used without correcting for series resistance or built-in voltage, which offers the best fitting of the experimental data following the protocol reported in the literature.57 The hole mobilities are extracted with the fit parameters at an electric field (E) of 2 × 105 V cm−1 (corresponding to an applied voltage of 2 V across the bulk of a 100 nm device) by the Murgatroyd equation
, where ε0 is the permittivity of free space, εr is the dielectric constant of the polymer which is assumed to be around 3 for the conjugated polymers, μ0 is the zero-field mobility, V is the voltage drop across the device, d is the film thickness of the active layer, and γ is a parameter that describes the strength of the field-dependence effect. The applied voltage is used without correcting for series resistance or built-in voltage, which offers the best fitting of the experimental data following the protocol reported in the literature.57 The hole mobilities are extracted with the fit parameters at an electric field (E) of 2 × 105 V cm−1 (corresponding to an applied voltage of 2 V across the bulk of a 100 nm device) by the Murgatroyd equation 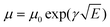 .
.
Fabrication and characterization of solar cells
Photovoltaic devices were made by vacuum (∼3 × 10−7 mbar) evaporating 10 nm of MoOx onto pre-cleaned, patterned ITO substrates. The polymer–fullerene photoactive layers were deposited by spin coating in a nitrogen-filled glovebox from the solutions containing the corresponding polymers and [70]PCBM with different content ratios at room temperature. The IDT-DTzBT:[70]PCBM film was annealed at 130 °C for 10 min before depositing an electron-collecting layer. No thermal annealing was applied to the other blend films. The thickness of active layer films was controlled by spin speed. All active layer films possess a similar thickness of 100 ± 10 nm. Before the evaporation of the Al electrode, a thin PFN layer was spin-coated on top of the active layer from its methanol solution (0.2 mg mL−1) at 3000 rpm. Finally, Al (100 nm) was deposited by vacuum evaporation at ∼3 × 10−7 mbar as the back electrode. The active area of the cells was 0.09 or 0.16 cm2, which provided similar results. Current density–voltage (J–V) characteristics were measured under simulated solar light (100 mW cm−2) from a tungsten–halogen lamp filtered by a Hoya LB100 daylight using a Keithley 2400 source meter. No mismatch correction was done. All measurements were conducted in a nitrogen-filled glove box. The accurate Jsc was determined from the EQE by integration with the AM1.5G solar spectrum. EQE measurements were performed in a homebuilt set-up, with the devices kept in a nitrogen filled box with a quartz window and illuminated through an aperture of 2 mm. Mechanically modulated (Stanford Research, SR 540) monochromatic (Oriel, Cornerstone 130) light from a 50 W tungsten halogen lamp (Osram 64610) was used as probe light, in combination with continuous bias light from a solid state laser (B&W Tek Inc. 532 nm, 30 mW). The intensity of the bias laser light was adjusted using a variable-neutral density filter. The response was recorded as the voltage over a 50 Ω resistance, using a lock-in amplifier (Stanford Research Systems SR 830). For all devices, measurement was carried out under representative illumination intensity (AM1.5G equivalent, provided by the 532 nm laser).
Acknowledgements
We thank Ralf Bovee and Gaël Heintges for GPC analysis and Fei Huang and Zhicheng Hu of the South China University of Technology for providing PFN. The work was performed in the framework of the Mujulima project that received funding from the European Commission's Seventh Framework Programme (Grant Agreement No. 604148). The work of JJVF forms part of the research programme of the Dutch Polymer Institute (DPI), project #734. The research leading to these results has also received funding from the European Research Council under the European Union's Seventh Framework Programme (FP/2007–2013)/ERC Grant Agreement No. 339031. The research forms part of the Solliance OPV program and has received funding from the Ministry of Education, Culture and Science (Gravity program 024.001.035).
Notes and references
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666 CrossRef CAS PubMed.
- F. C. Krebs, J. Fyenbo and M. Jorgensen, J. Mater. Chem., 2010, 20, 8994 RSC.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CAS.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef.
- C.-C. Chen, W.-H. Chang, K. Yoshimura, K. Ohya, J. You, J. Gao, Z. Hong and Y. Yang, Adv. Mater., 2014, 26, 5670 CrossRef CAS PubMed.
- A. R. B. M. Yusoff, D. Kim, H. P. Kim, F. K. Shneider, W. J. da Silva and J. Jang, Energy Environ. Sci., 2015, 8, 303 CAS.
- Y. Liang, Z. Xu, J. Xia, S.-T. Tsai, Y. Wu, G. Li, C. Ray and L. Yu, Adv. Mater., 2010, 22, E135 CrossRef CAS PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- V. Vohra, K. Kawashima, T. Kakara, T. Koganezawa, I. Osaka, K. Takimiya and H. Murata, Nat. Photonics, 2015, 9, 403 CrossRef CAS.
- H. X. Zhou, L. Q. Yang and W. You, Macromolecules, 2012, 45, 607 CrossRef CAS.
- Y. Li, Acc. Chem. Res., 2012, 45, 723 CrossRef CAS PubMed.
- C. H. Duan, F. Huang and Y. Cao, J. Mater. Chem., 2012, 22, 10416 RSC.
- Z.-G. Zhang and Y. Li, Sci. China: Chem., 2015, 58, 192 CrossRef CAS.
- C. Duan, F. Huang and Y. Cao, Polym. Chem., 2015, 6, 8081 RSC.
- S. C. Price, A. C. Stuart, L. Q. Yang, H. X. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 4625 CrossRef CAS PubMed.
- Y. Dong, X. Hu, C. Duan, P. Liu, S. Liu, L. Lan, D. Chen, L. Ying, S. Su, X. Gong, F. Huang and Y. Cao, Adv. Mater., 2013, 25, 3683 CrossRef CAS PubMed.
- K. Li, Z. Li, K. Feng, X. Xu, L. Wang and Q. Peng, J. Am. Chem. Soc., 2013, 135, 13549 CrossRef CAS PubMed.
- J.-H. Kim, J. B. Park, F. Xu, D. Kim, J. Kwak, A. C. Grimsdale and D.-H. Hwang, Energy Environ. Sci., 2014, 7, 4118 CAS.
- C. Duan, A. Furlan, J. J. van Franeker, R. E. M. Willems, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2015, 27, 4461 CrossRef CAS PubMed.
- M. Zhang, X. Guo, W. Ma, H. Ade and J. Hou, Adv. Mater., 2015, 27, 4655 CrossRef CAS PubMed.
- L. Huo, T. Liu, X. Sun, Y. Cai, A. J. Heeger and Y. Sun, Adv. Mater., 2015, 27, 2938 CrossRef CAS PubMed.
- Z. Li, H. Lin, K. Jiang, J. Carpenter, Y. Li, Y. Liu, H. Hu, J. Zhao, W. Ma, H. Ade and H. Yan, Nano Energy, 2015, 15, 607 CrossRef CAS.
- L. Gao, Z.-G. Zhang, L. Xue, J. Min, J. Zhang, Z. Wei and Y. Li, Adv. Mater., 2016, 28, 1884 CrossRef CAS PubMed.
- H. Bin, Z.-G. Zhang, L. Gao, S. Chen, L. Zhong, L. Xue, C. Yang and Y. Li, J. Am. Chem. Soc., 2016, 138, 4657 CrossRef CAS PubMed.
- R. Steim, T. Ameri, P. Schilinsky, C. Waldauf, G. Dennler, M. Scharber and C. J. Brabec, Sol. Energy Mater. Sol. Cells, 2011, 95, 3256 CrossRef CAS.
- D. Veldman, S. C. J. Meskers and R. A. J. Janssen, Adv. Funct. Mater., 2009, 19, 1939 CrossRef CAS.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789 CrossRef CAS.
- N. Blouin, A. Michaud and M. Leclerc, Adv. Mater., 2007, 19, 2295 CrossRef CAS.
- E. G. Wang, L. Wang, L. F. Lan, C. Luo, W. L. Zhuang, J. B. Peng and Y. Cao, Appl. Phys. Lett., 2008, 92, 033307 CrossRef.
- H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew. Chem., Int. Ed., 2011, 123, 3051 CrossRef.
- M. Zhang, X. Guo, X. Wang, H. Wang and Y. Li, Chem. Mater., 2011, 23, 4264 CrossRef CAS.
- Y. Lin, H. Fan, Y. Li and X. Zhan, Adv. Mater., 2012, 24, 3087 CrossRef CAS PubMed.
- I. Bulut, P. Chavez, A. Mirloup, Q. Huaulme, A. Hebraud, B. Heinrich, S. Fall, S. Mery, R. Ziessel, T. Heiser, P. Leveque and N. Leclerc, J. Mater. Chem. C, 2016, 4, 4296 RSC.
- F. Caffy, N. Delbosc, P. Chavez, P. Leveque, J. Faure-Vincent, J. P. Travers, D. Djurado, J. Pecaut, B. Grevin, N. Lemaitre, N. Leclerc and R. Demadrille, Polym. Chem., 2016, 7, 4160 RSC.
- J.-L. Wang, Z. Wu, J.-S. Miao, K.-K. Liu, Z.-F. Chang, R.-B. Zhang, H.-B. Wu and Y. Cao, Chem. Mater., 2015, 27, 4338 CrossRef CAS.
- H. Bronstein, E. Collado-Fregoso, A. Hadipour, Y. W. Soon, Z. Huang, S. D. Dimitrov, R. S. Ashraf, B. P. Rand, S. E. Watkins, P. S. Tuladhar, I. Meager, J. R. Durrant and I. McCulloch, Adv. Funct. Mater., 2013, 23, 5647 CrossRef CAS.
- E. Zaborova, P. Chavez, R. Bechara, P. Leveque, T. Heiser, S. Mery and N. Leclerc, Chem. Commun., 2013, 49, 9938 RSC.
- W. Li, K. H. Hendriks, A. Furlan, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2015, 137, 2231 CrossRef CAS PubMed.
- M. Akhtaruzzaman, N. Kamata, J.-I. Nishida, S. Ando, H. Tada, M. Tomura and Y. Yamashita, Chem. Commun., 2005, 3183 RSC.
- A. Garcia, G. C. Welch, E. L. Ratcliff, D. S. Ginley, G. C. Bazan and D. C. Olson, Adv. Mater., 2012, 24, 5368 CrossRef CAS PubMed.
- F. Huang, H. B. Wu, D. L. Wang, W. Yang and Y. Cao, Chem. Mater., 2004, 16, 708 CrossRef CAS.
- C. H. Duan, C. D. Wang, S. J. Liu, F. Huang, C. H. W. Choy and Y. Cao, Sci. China: Chem., 2011, 54, 685 CrossRef CAS.
- J. Lee, M. Kim, B. Kang, S. B. Jo, H. G. Kim, J. Shin and K. Cho, Adv. Energy Mater., 2014, 4, 1400087 CrossRef.
- Y. Yang, R. Wu, X. Wang, X. Xu, Z. Li, K. Li and Q. Peng, Chem. Commun., 2014, 50, 439 RSC.
- C. Duan, R. E. M. Willems, J. J. van Franeker, B. J. Bruijnaers, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem. A, 2016, 4, 1855 CAS.
- W. Li, L. Yang, J. R. Tumbleston, L. Yan, H. Ade and W. You, Adv. Mater., 2014, 26, 4456 CrossRef CAS PubMed.
- J. Subbiah, B. Purushothaman, M. Chen, T. Qin, M. Gao, D. Vak, F. H. Scholes, X. Chen, S. E. Watkins, G. J. Wilson, A. B. Holmes, W. W. H. Wong and D. J. Jones, Adv. Mater., 2015, 27, 702 CrossRef CAS PubMed.
- J. J. van Franeker, G. H. L. Heintges, C. Schaefer, G. Portale, W. Li, M. M. Wienk, P. van der Schoot and R. A. J. Janssen, J. Am. Chem. Soc., 2015, 137, 11783 CrossRef CAS PubMed.
- M. M. Wienk, J. M. Kroon, W. J. H. Verhees, J. Knol, J. C. Hummelen, P. A. van Hal and R. A. J. Janssen, Angew. Chem., Int. Ed., 2003, 42, 3371 CrossRef CAS PubMed.
- S. Kouijzer, J. J. Michels, M. van den Berg, V. S. Gevaerts, M. Turbiez, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2013, 135, 12057 CrossRef CAS PubMed.
- J. J. van Franeker, M. Turbiez, W. Li, M. M. Wienk and R. A. J. Janssen, Nat. Commun., 2015, 6, 6229 CrossRef CAS PubMed.
- D. Veldman, O. Z. İpek, S. C. J. Meskers, J. R. Sweelssen, M. M. Koetse, S. C. Veenstra, J. M. Kroon, S. S. V. Bavel, J. Loos and R. A. J. Janssen, J. Am. Chem. Soc., 2008, 130, 7721 CrossRef CAS PubMed.
- L. J. A. Koster, M. Kemerink, M. M. Wienk, K. Maturová and R. A. J. Janssen, Adv. Mater., 2011, 23, 1670 CrossRef CAS PubMed.
- Y. Zhang, J. Y. Zou, H. L. Yip, K. S. Chen, D. F. Zeigler, Y. Sun and A. K. Y. Jen, Chem. Mater., 2011, 23, 2289 CrossRef CAS.
- Q. Hou, Y. S. Xu, W. Yang, M. Yuan, J. B. Peng and Y. Cao, J. Mater. Chem., 2002, 12, 2887 RSC.
- J. Y. Lee, K. W. Song, H. J. Song and D. K. Moon, Synth. Met., 2011, 161, 2434 CrossRef CAS.
- J. C. Blakesley, F. A. Castro, W. Kylberg, G. F. A. Dibb, C. Arantes, R. Valaski, M. Cremona, J. S. Kim and J.-S. Kim, Org. Electron., 2014, 15, 1263 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py01083k |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a 





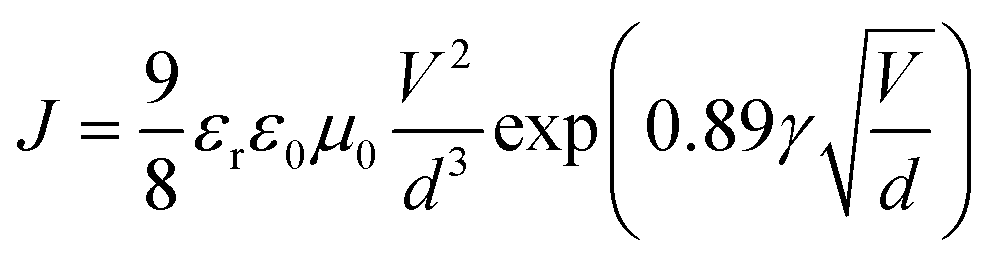 , where ε0 is the permittivity of free space, εr is the dielectric constant of the polymer which is assumed to be around 3 for the conjugated polymers, μ0 is the zero-field mobility, V is the voltage drop across the device, d is the film thickness of the active layer, and γ is a parameter that describes the strength of the field-dependence effect. The applied voltage is used without correcting for series resistance or built-in voltage, which offers the best fitting of the experimental data following the protocol reported in the literature.57 The hole mobilities are extracted with the fit parameters at an electric field (E) of 2 × 105 V cm−1 (corresponding to an applied voltage of 2 V across the bulk of a 100 nm device) by the Murgatroyd equation
, where ε0 is the permittivity of free space, εr is the dielectric constant of the polymer which is assumed to be around 3 for the conjugated polymers, μ0 is the zero-field mobility, V is the voltage drop across the device, d is the film thickness of the active layer, and γ is a parameter that describes the strength of the field-dependence effect. The applied voltage is used without correcting for series resistance or built-in voltage, which offers the best fitting of the experimental data following the protocol reported in the literature.57 The hole mobilities are extracted with the fit parameters at an electric field (E) of 2 × 105 V cm−1 (corresponding to an applied voltage of 2 V across the bulk of a 100 nm device) by the Murgatroyd equation 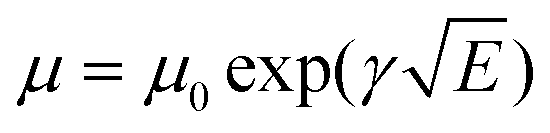 .
.