
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVinyl monomers bearing a sulfonyl(trifluoromethane sulfonyl) imide group: synthesis and polymerization using nitroxide-mediated polymerization†
Trang N. T.
Phan
*a,
Adèle
Ferrand
a,
Hien The
Ho
a,
Livie
Liénafa
a,
Marion
Rollet
a,
Sébastien
Maria
a,
Renaud
Bouchet
b and
Didier
Gigmes
a
aAix-Marseille Université, CNRS, Institut de Chimie Radicalaire UMR 7273, 13397, Marseille, France. E-mail: trang.phan@univ-amu.fr
bLEPMI UMR-5279 CNRS—Grenoble INP—Univ. de Savoie—Univ. Joseph Fourier, 38402 St Martin d'Hères, France
First published on 7th September 2016
Abstract
A series of water-soluble styrene and propyl (meth)acrylate based monomers bearing a sulfonyl(trifluoromethane sulfonyl) imide (STFSI) group was prepared. These monomers were synthesized in good yield from the corresponding chloride sulfonyl monomer and trifluoromethanesulfonamide. Their chemical structures were further confirmed by HR-MS and NMR spectroscopy. Nitroxide-mediated polymerization of the prepared monomers was then performed in aqueous solution at 100 °C, 90 °C and 75 °C for acrylate, styrene and methacrylate derivatives, respectively. Controlled polymerization was successfully achieved as illustrated by the linearity of ln([M]0/[M]) vs. t2/3, the molar mass increase with conversion and the relatively low dispersity values for the resulting homopolymers. Compared to their neutral analogous polymers, the prepared anionic homopolymers exhibited a higher glass-transition temperature. This phenomenon was attributed to the electrostatic interactions between STFSI side groups along the polymer backbone.
Introduction
The bis(trifluoromethane sulfonyl) imide anion (TFSI or (CF3SO2)2N−) can be used in different applications depending on the nature of the associated cation, i.e. metallic or organic. When the TFSI anion is associated with a metal such as lithium, the blend of the corresponding salt with a polymer matrix was largely used as a solid or gel electrolyte for lithium–polymer batteries.1–4 Moreover, the association of this anion with cations such as imidazolium, pyrrolidonium, and pyridinium attached to a polymer backbone gave rise to a highly interesting family of polymeric ionic liquids (PILs).5,6 On the other hand, the neutral derivatives of TFSI have also been used in synthetic organic chemistry. For instance, (CF3SO2)2N–H is considered as a strong Brønsted acid and applied as a catalyst for organic reactions,7,8 while the N-fluoro derivative (CF3SO2)2N–F is a powerful reagent for electrophilic fluorination.9,10 The derivatives of TFSI have also been used as proton-exchange membranes for fuel-cell application.11,12 Interest in the TFSI salt and its derivatives is mainly due to their properties induced by the presence of the CF3SO2 group.13 Indeed, the strong electron-withdrawing effect of this group enhances the acidity of the hydrogen in (CF3SO2)2N–H and ensures a large delocalization of the negative charge. For these reasons, (CF3SO2)2N−M+ exhibits a high ionic conductivity and good resistance to oxidation.In the field of solid polymer electrolytes (SPEs) designed for lithium batteries, many polymer/lithium salt systems have been considered. However, to date the most widely studied and used system consists of a mixture of LiTFSI dissolved in an aprotic polymer matrix of poly(ethylene oxide) (PEO). In such binary salt–polymer systems, the ionic conductivity could be high above the melting temperature of PEO but the Li+ transference number, that gives the proportion of the current carried by Li+ ions, remains very low (t+ < 0.2),14 leading under current to the formation of an ionic concentration gradient that limits substantially the power performances. To avoid this problem, the anions were covalently attached to the polymer backbones to form single-ion conductor copolymers with a lithium transference number close to unity.15,16 However, these solid polymer electrolytes (SPEs) exhibit a low conductivity mostly owing to the low level of ionic dissociation of carboxylates15 or borates.16 Recently, we have developed a novel family of single-ion electrolytes (SIEL) based on BAB triblock copolymers having a PEO middle block and anionic polymers based on styrene and methacrylate derivatives bearing the lithium sulfonyl(trifluoromethane sulfonyl) imide group as outer blocks.17,18 The SIEL materials containing lithium poly(styrene sulfonyl(trifluoromethane sulfonyl) imide) (denoted as PSSTFSILi) have shown outstanding performances in terms of conductivity (1.3 × 10−5 S cm−1 at 60 °C), transport number (t+ > 0.85), mechanical properties and electrochemical stability window.17 These remarkable properties build on the combination of soft PEO-rich phases that enable the conduction of Li+ with rigid PSSTFSILi phases that provide mechanical strength and act as a reservoir of cations.
The use of block copolymers as SPEs to combine in the same material the two antagonistic properties (mechanical and conductivity) was firstly proposed by Sadoway et al.19 Since this study, interest in block copolymers as SPEs for lithium batteries has continuously grown. Block copolymers can be prepared by different methods, but the living/controlled polymerization is the only way, which allows the synthesis of block copolymers with a well-defined structure. The development of reversible-deactivation radical polymerization (RDRP) techniques in the past two decades has largely promoted the synthesis of block copolymers. Furthermore, RDRP is more tolerant to protic functional monomers than anionic or group transfer polymerization techniques, and can be run in many conventional solvents over a wide range of temperatures.20–24
Taking advantage of the charge delocalization character of STFSI and the very interesting electrolytes obtained with a monomer bearing STFSI anion, we were encouraged to develop new monomer structures containing a STFSI side group, namely methacrylate and acrylate based monomers. Prior to performing the preparation of block copolymers devoted to SPEs for lithium batteries, we first investigated the aqueous-polymerization of three monomer derivatives of styrene, methacrylate and acrylate (Scheme 1) using the Nitroxide-Mediated Polymerization (NMP) technique. This investigation is crucial to ensure that the polymerization reaction is well controlled and produces well-defined water-soluble polymers. All aqueous polymerizations were carried out using an SG1-based alkoxyamine bearing a carboxylic acid function (so-called MAMA-SG1 or BlocBuilder MA, Scheme 1). In its basic form, the carboxylate function confers water solubility to the alkoxyamine and thus allows various water-soluble monomers to be polymerized by NMP in homogeneous aqueous solution.
 | ||
| Scheme 1 Chemical formulae of nitroxide (SG1), alkoxyamine (MAMA-SG1) and vinyl monomers bearing sulfonyl(trifluoromethane sulfonyl) imide. | ||
It is well known now that the NMP of methacrylate monomers using MAMA-SG1 alkoxyamine can be performed in a controlled manner by adding a small amount of co-monomer such as styrene,25,26 sodium 4-styrenesulfonate,27,28 acrylonitrile,29,30N,N-dimethylacrylamide31 or vinylbenzyl carbazole.32 In the present work, potassium methacrylate-STFSI was copolymerized with potassium styrene-STFSI as a comonomer. To illustrate the features of a controlled polymerization process, kinetic studies were performed and the molar masses of the collected samples were analyzed by size exclusion chromatography with DMF/LiBr as the eluent. The thermodynamic behavior of the prepared water-soluble polymers was also characterized by using differential scanning calorimetry (DSC).
Experimental section
Materials
Sodium 4-styrenesulfonate (SSNa, >90%), 3-sulfopropyl methacrylate potassium salt (MASPK, 98%), 3-sulfopropyl acrylate potassium salt (ASPK, >99%), oxalyl chloride (>99%), triethylamine (TEA, >99%), anhydrous acetonitrile (ACN) and anhydrous N,N-dimethylformamide (DMF) were purchased from Aldrich. Trifluoromethanesulfonamide (TFSA) was received from TCI Europe. SG1 (85%) and alkoxyamine based on SG1 derived from methacrylic acid (MAMA-SG1, >99%, trade name BlocBuilder MA) were kindly supplied by Arkema. All the above commercial chemicals were used as received. Sodium MAMA-SG1 was obtained after the deprotonation of the MAMA-SG1 alkoxyamine with sodium hydroxide, as described previously.33 All solvents and other reagents were synthesis grade and used without further purification.Analytical techniques
1H, 13C and 19F NMR spectra were recorded on a Bruker Advance 400 MHz spectrometer. Monomer conversion was determined by 1H NMR analysis using DMSO-d6 as the solvent.High-resolution mass spectroscopy (HR-MS) experiments were performed in the negative ion mode on a time-of-flight mass spectrometer (Synapt-G2 HDMS, Waters) equipped with a pneumatically assisted electrospray ion source. Samples were analyzed with the following conditions: capillary voltage 2.28 kV, cone voltage 10 V, and desolvation gas (nitrogen) flow rate 100 L h−1. Poly(ethylene glycol) oligomers with m/z 325.1868 and m/z 369.2130 were used for the calibration as internal references.
Differential scanning calorimetry (DSC) was carried out on a TA DSC Q20 using a heat/cool/heat cycle from −90 °C to 250 °C. The heating and cooling rates were 10 °C min−1. The glass-transition temperature, Tg, was determined from the second heating cycle of the DSC thermograms.
Size exclusion chromatography (SEC) experiments were performed on a Varian PL-GPC 120 apparatus, which was composed of a PL-AS-MT autosampler, an Agilent 1100 series pump, a degasser, an injection valve, a column oven and a refractive index (RI) detector. The following columns were used: one pre-column and two PL Resipore columns (300 mm × 7.8 mm). The injection loop, the columns and the RI detector were in the same oven thermostated at 70 °C. The eluent was a solution of 0.1 M LiBr in N,N-dimethylformamide filtered through a 0.45 μm nylon membrane and the flow rate was fixed at 0.7 mL min−1. The samples were prepared in a mixture of eluent and toluene (0.25 vol%) as the flowmarker, filtered through a 0.2 μm nylon filter (Interchim) and placed in an autosampler preheated at 50 °C. The sample concentration was 0.25 wt%. Calibration curves were established with either polystyrene (PS) standards or poly(methyl methacrylate) (PMMA) standards, both purchased from Agilent.
Synthesis of potassium 4-styrenesulfonyl(trifluoromethane sulfonyl) imide, SSTFSIK
SSTFSIK was prepared according to the literature.34 The monomer was obtained as a white powder in 50% yield.
1H NMR (400 MHz, DMSO-d6, δ ppm): 5.38 (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–, d, 1H), 5.95 (CH2CH–, d, 1H), 6.77 (CH2CH–, dd, 1H), 7.64 (C6H4–, dd, 4H). 19F NMR (DMSO-d6, δ ppm): −77.81. 7Li NMR (DMSO-d6, δ ppm): −1.00. 13C NMR (DMSO-d6, δ ppm): 116.78 (CH2CH–), 118.48 and 121.70 (–CF3), 126.07–126.61 (–CH2 aromatic), 135.74 (CH2CH–), 139.77 and 144.35 (–CH aromatic).
CH–, d, 1H), 5.95 (CH2CH–, d, 1H), 6.77 (CH2CH–, dd, 1H), 7.64 (C6H4–, dd, 4H). 19F NMR (DMSO-d6, δ ppm): −77.81. 7Li NMR (DMSO-d6, δ ppm): −1.00. 13C NMR (DMSO-d6, δ ppm): 116.78 (CH2CH–), 118.48 and 121.70 (–CF3), 126.07–126.61 (–CH2 aromatic), 135.74 (CH2CH–), 139.77 and 144.35 (–CH aromatic).
HR-MS (C9H7NO4S2F3K): m/z = [M − K+]found = 313.9775 g mol−1, [M − K+]cal = 313.9769 g mol−1.
Synthesis of potassium 3-sulfonyl(trifluoromethane sulfonyl) imide propyl acrylate, ASTFSIK
Oxalyl chloride (2.40 mL, 28.3 mmol) and N,N-dimethylformamide (0.50 mL) were added dropwise under argon to a three-necked round bottom flask containing anhydrous acetonitrile (30.0 mL) at 0 °C. The mixture was stirred for 1 h at room temperature. Then, the solution was chilled down to 0 °C and potassium 3-sulfopropyl acrylate (5.00 g, 21.6 mmol) was added into the solution under argon. The resulting solution was continuously stirred at 0 °C for 3 h and 2 h more at room temperature to achieve the formation of 3-(chlorosulfonyl) propyl acrylate. Then a solution of trifluoromethanesulfonamide (2.66 g, 1.79 × 10−2 mol) and triethylamine (6.80 mL, 4.91 × 10−2 mol) dissolved in acetonitrile (10.0 mL) at 0 °C under argon was added to the 3-(chlorosulfonyl) propyl acrylate solution. The reaction mixture was allowed to react at 0 °C for 2 h and then at room temperature for 16 h under argon. The reaction mixture was filtered off and the recovered white solid was washed with acetonitrile. The filtrate was concentrated under vacuum then dissolved in dichloromethane and washed successively with a saturated NaHCO3 solution, a solution of 1 M chloride acid and a saturated NaCl solution, dried over MgSO4, filtered and evaporated under reduced pressure. The product was obtained as amber oil (5.20 g, 68% yield). To the cold solution of triethylammonium-3-sulfonyl(trifluoromethane sulfonyl) imide propyl acrylate (3.00 g, 7.04 × 10−3 mol) in 30 mL of acetonitrile (30.0 mL) was slowly added potassium carbonate (1.94 g, 1.41 × 10−2 mol). After the complete addition of K2CO3, the mixture reaction was allowed to react at 0 °C to room temperature for 4 hours. The solution was then filtered and the filtrate was concentrated under vacuum. The yellowish solid was recrystallized from hot dichloromethane. The mixture solution was then kept at −20 °C for 3 h. The obtained solid was washed with cold n-pentane and dried under vacuum to obtain a white solid (60% yield).
1H NMR (400 MHz, DMSO-d6, δ ppm): 6.34 (CH2CH–, d, 1H), 6.18 (CH2CH–, dd, 1H), 5.95 (CH2CH–, d, 1H), 4.19 (–O–CH2–, t, 2H), 3.05 (–CH2–SO2–, t, 2H), 2.01 (–CH2–, m, 2H). 13C NMR (DMSO-d6, δ ppm): 165.87 (CO), 132.00 (CH2CH–), 128.71 (CH2CH–), 125.41–115.74 (–CF3), 63.01 (–COOCH2–), 51.72 (–CH2SO2–), 23.97 (–COOCH2–). 19F NMR (DMSO-d6, δ ppm): −77.54.
HR-MS (C7H9NO6S2F3K): m/z = [M − K+]found = 323.9832 g mol−1, [M − K+]cal = 323.9829 g mol−1.
Synthesis of potassium 3-sulfonyl(trifluoromethane sulfonyl) imide propyl methacrylate, MASTFSIK
A similar procedure to the synthesis of ASTFSIK was used to prepare MASTFSIK. In this case, 3-sulfopropyl methacrylate potassium salt was used instead of 3-sulfopropyl acrylate potassium salt. The product was obtained as a white powder (60% yield).
1H NMR (400 MHz, DMSO-d6, δ ppm): 6.04 (CH2C, s, 1H), 5.68 (CH2C, s, 1H), 4.17 (–O–CH2–, t, 2H), 3.06 (–CH2–SO2–, t, 2H), 2.01 (–CH2–, m, 2H), 1.88 (–CH3, s, 3H). 13C NMR (DMSO-d6, δ ppm): 166.42 (CO), 135.81 (CH2CH–), 125.75 (CH2CH–), 123.10–115.26 (–CF3), 62.07 (–COOCH2–), 51.25 (–CH2SO2–), 23.53 (–COOCH2–), 17.94 (–CH3). 19F NMR (DMSO-d6, δ ppm): −77.53.
HR-MS (C8H11NO6S2F3): m/z = [M − K+]found = 337.9985 g mol−1, [M − K+]cal = 337.9985 g mol−1.
General procedure for the NMP of different anionic monomers
Anionic monomer, SG1 and distilled water were placed in a three-neck flask equipped with a reflux condenser and a magnetic stir bar. The mixture was purged for 30 min with argon at 40 °C to remove oxygen. In parallel, the water-soluble alkoxyamine, sodium MAMA-SG1 was dissolved in 2 mL of distilled water and degassed by bubbling argon for 20 min at room temperature. This alkoxyamine solution was transferred, via a cannula, to the degassed monomer solution. The mixture was heated to appropriate temperature according to the monomer nature. The [SG1]/[MAMA-SG1] ratio was between 5% and 10%, and the monomer concentration in the polymerization mixture was varied from 10 to 30 wt%. For kinetic studies, samples were collected from the reaction mixture at given time intervals. Conversion and molecular weights were determined by 1H NMR spectroscopy and LiBr-DMF SEC, respectively. Table 1 summarizes the experimental conditions of the performed polymerization experiments.| Entry | Monomer | T (°C) | m monomer (g) | molMAMA-SG1 (mmol) | r (%) | [Monomer] wt% in water | Target Mn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b at full conversion (g mol−1) b at full conversion (g mol−1) |
|---|---|---|---|---|---|---|---|
| a r = 100 × [SG1]0/[MAMA-SG1]0. b Theoretical number-average molar mass, calculated according to Mn = MM(MAMA-SG1) + conversion × initial weight of the monomer/initial mol number of MAMA-SG1, with conversion = 1 and molar mass of the initiator, MM(MAMA-SG1) = 381 g mol−1. | |||||||
| 1 | SSTFSILi | 90 | 0.975 | 26.2 | 5 | 10 | 40000 |
| 2 | MASTFSIK/SSTFSIK | 75 | 0.81/0.19 | 47.2 | 10 | 20 | 30000 |
| 3 | ASTFSIK | 100 | 1.3 | 47.2 | 7 | 30 | 30000 |
Because of the poor solubility of SSTFSIK in water, this monomer was converted to the lithium salt and then polymerized in diluted solution at 10 wt%. For the polymerizations of acrylate-STFSIK and methacrylate-STFSIK, targeted molar masses (Mn) of anionic polymers at 100% conversion were targeted at 30000 g mol−1, while that of styrene-STFSILi was targeted at 40000 g mol−1.
Results and discussion
Monomers design and synthesis
Three vinyl monomers based on styrene and methacrylate and acrylate monomers functionalized with a sulfonyl(trifluoromethane sulfonyl) imide group were prepared (Scheme 1). These monomers were selected with the aim to elaborate thereafter single-ion block copolymer electrolytes for lithium batteries. By varying the composition and the structure of the investigated block copolymers, our motivation was to tune their corresponding electrolyte properties such as thermal stability, chain mobility and conductivity performance.The synthesis of SSTFSIK was first described by Armand35 and readapted by other authors.8,34,36 4-Vinylbenzenesulfonyl chloride, as an intermediate compound, was synthesized and reacted in situ with trifluoromethanesulfonamide in the presence of triethylamine to yield a viscous liquid. Cation exchange of the liquid with an excess of LiOH in methanol yields the lithium styrene-STFSI derivative. A high purity product can be obtained after washing the solid with n-pentane and cold dichloromethane. The formation of the lithium styrene-STFSI monomer was proven by 19F NMR with a singlet at −77.81 ppm, to be compared with the signal of the starting compound trifluoromethanesulfonamide CF3SO2NH2, which is at −79.32 ppm. 1H, 7Li and 13C NMR were also used to characterize the structure of SSTFSILi (Fig. S1–S3†).
The synthetic route for potassium methacrylate-STFSI and potassium acrylate-STFSI is depicted in Scheme 2. The corresponding 3-sulfopropyl (meth)acrylate potassium salts are commercially available and were converted into sulfonyl chloride derivatives upon reaction with oxalyl chloride in the presence of DMF as the catalyst (Vilsmeier–Haack reaction). The intermediate chloride products were then reacted with trifluoromethanesulfonamide in the presence of an excess of triethylamine. The use of an excess of triethylamine ensures the obtention of all monomers in their triethylammonium salt form avoiding the obtention of a mixture of salt and acid forms. The last step consisted of an ionic exchange of the triethylammonium ion with the potassium ion. This step was performed by using K2CO3 instead of Li2CO3 or LiOH used in the synthesis of SSTFSILi. Indeed, the basicity of the latter is too strong and may induce the hydrolysis of the (meth)acrylate ester bond. The global yield was typically close to 60%. Each compound was characterized by 1H, 19F, 13C NMR and HRMS. The 19F NMR spectra of the purified monomers (Fig. S4†) showed a shift of the characteristic signal of the trifluoromethyl group before and after the reaction. The combination of this result and those of other analyses indicates the formation of the right products. 1H and 13C NMR spectra are shown in the ESI (Fig. S5–S8†).
 | ||
| Scheme 2 Synthetic route for the preparation of potassium 3-sulfonyl(trifluoromethane sulfonyl) imide propyl methacrylate (MASTFSIK) and potassium 3-sulfonyl(trifluoromethane sulfonyl) imide propyl acrylate (ASTFSIK). | ||
Aqueous NMP of different anionic monomers bearing a sulfonyl(trifluoromethane sulfonyl) imide group
The NMP of hydrophilic monomers has been very seldom performed in aqueous solution. We found only a few examples available in the literature28,30,33,37–40 and among them only five articles28,33,38–40 that describe the use of SG1 nitroxide. In these studies, the considered monomers were either neutral monomers (acrylamide, N,N-dimethylacrylamide, poly(ethylene glycol) methyl ether methacrylate)33,38,39 or anionic monomers (sodium 4-styrenesulfonate, methacrylic acid).28,33,40 Anionic monomers with a sulfonyl(trifluoromethane sulfonyl) imide group have never been investigated in aqueous medium using RDRP and we barely know if this kind of functional monomer could be polymerized in a controlled manner. Then the first aim of this work was to follow the polymerization kinetics and to determine the most appropriate conditions for achieving polymerization with all the controlled features. All characteristics of the obtained polymers are summarized in Table 2.| Polymer | Target Mna at full conversion (g mol−1) |
Polymerization time (min) | Total conversion (NMR) (%) |
M
nb (th) (g mol−1) |
M n (SEC) (g mol−1) | Dispersity |
|---|---|---|---|---|---|---|
| M n, the number-average molar mass, calculated according to the equation Mn = MM(MAMA-SG1) + conversion × initial weight of monomer/initial mol number of MAMA-SG1, MM(MAMA-SG1) = 381 g mol−1.a Values calculated with conversion of 100%.b Values calculated with conversion determined by 1H NMR. | ||||||
| PSSTFSILi | 40000 |
300 | 55 | 22000 |
45000 |
1.09 |
| P(MASTFSIK-SSTFSIK) | 30000 |
80 | 59 | 17700 |
23500 |
1.19 |
| PASTFSIK | 30000 |
380 | 57 | 17100 |
24000 |
1.17 |
Aqueous NMP of lithium 4-styrenesulfonyl(trifluoromethane sulfonyl) imide
Conventional radical polymerization of potassium or sodium 4-styrene sulfonyl(trifluoromethane sulfonyl) imide using K2S2O8 as the initiator34,36 as well as those using Reversible Addition–Fragmentation chain Transfer (RAFT) polymerization in DMF solution41,42 were already reported. Cloutet et al. have recently demonstrated that SSTFSIK could be polymerized with DMF as the solvent by the RAFT method to yield a homopolymer with low dispersity (Đ = 1.02) at high monomer conversion (98%) for a polymerization time of 14 hours. In the present study, lithium styrene-STFSI was polymerized at 90 °C in dilute aqueous solution (10 wt% of monomer) using sodium MAMA-SG1 as the initiator in the presence of 5 mol% of free SG1 nitroxide compared to alkoxyamine. It was demonstrated that the addition of free SG1 allows one to obtain better control of hydrophilic monomers polymerization in terms of kinetic evolution and dispersities.33 To support that the NMP of Li styrene-STFSI was well-controlled, a complete kinetic study for a target Mn of 40000 g mol−1 at full conversion was performed. Fig. 1 gives the kinetic plots of ln([M]0/[M]) vs. reaction time (t and t2/3). The linearity of the kinetic plots when plotted as ln([M]0/[M]) vs. t2/3 rather than t lies in the radical persistent effect. According to Fischer's43 and Fukuda's44 work, the minimization of the irreversible termination in the NMP process is governed by the persistent radical effect. Considering an alkoxyamine compound (RY) that decomposes into a transient radical (R˙) and a persistent radical (Y˙), this persistent radical effect leads to two unusual rate laws for R˙ and Y˙ during the intermediate regimes:  and
and  , where kd, kc and kt are the rate constants of the decomposition reaction, the combination reaction of RY and the termination reaction between two R˙, respectively. Consequently, the kinetic law of monomer concentration is thus given by
, where kd, kc and kt are the rate constants of the decomposition reaction, the combination reaction of RY and the termination reaction between two R˙, respectively. Consequently, the kinetic law of monomer concentration is thus given by  ; hence, the plots of ln([M]0/[M]) versus time should show a downward curvature while the plots of ln([M]0/[M]) versus t2/3 should be linear. This persistent radical effect was experimentally validated in the NMP of styrene and acrylates by different groups.45–47 On the other hand, in the case of high propagation rate constant monomers, an initial excess of persistent radicals is added at the beginning of the polymerization process to slow down its rate and improve the polymerization control.33,48 Under these conditions, the concentration of the transient radicals reaches the stationary value [R˙]stationary = K[RY]0/[Y˙]0 (with K = kd/kc) and the linear evolution of the polymerization index ln([M]0/[M]) with time is generally observed.49,50 However, since the self-termination of the propagating radicals increases with the polymerization time and causes an additional Y˙ amount, when this released nitroxide concentration [Y˙] exceeds the initial one [Y˙]0, the behavior of the system turns to that observed without the initial persistent species. Fischer et al.50 theoretically demonstrated that if [Y˙]0 > (3[ln(10)]K[RY]0kt/kp)1/2, the polymerization index ln([M]0/[M]) varies linearly with time. On the contrary, if [Y˙]0 < (3K[RY]0kt/kp)1/2, the initial presence of the persistent species has no influence, and the polymerization index ln([M]0/[M]) increases linearly with t2/3.
; hence, the plots of ln([M]0/[M]) versus time should show a downward curvature while the plots of ln([M]0/[M]) versus t2/3 should be linear. This persistent radical effect was experimentally validated in the NMP of styrene and acrylates by different groups.45–47 On the other hand, in the case of high propagation rate constant monomers, an initial excess of persistent radicals is added at the beginning of the polymerization process to slow down its rate and improve the polymerization control.33,48 Under these conditions, the concentration of the transient radicals reaches the stationary value [R˙]stationary = K[RY]0/[Y˙]0 (with K = kd/kc) and the linear evolution of the polymerization index ln([M]0/[M]) with time is generally observed.49,50 However, since the self-termination of the propagating radicals increases with the polymerization time and causes an additional Y˙ amount, when this released nitroxide concentration [Y˙] exceeds the initial one [Y˙]0, the behavior of the system turns to that observed without the initial persistent species. Fischer et al.50 theoretically demonstrated that if [Y˙]0 > (3[ln(10)]K[RY]0kt/kp)1/2, the polymerization index ln([M]0/[M]) varies linearly with time. On the contrary, if [Y˙]0 < (3K[RY]0kt/kp)1/2, the initial presence of the persistent species has no influence, and the polymerization index ln([M]0/[M]) increases linearly with t2/3.
 | ||
| Fig. 1 Evolution of ln([M]0/[M]) versus reaction time (t and t2/3) for the NMP of lithium styrene-STFSI in aqueous solution at 90 °C using sodium MAMA-SG1 as the initiator in the presence of 5 mol% of free SG1. | ||
Depending on the polymerization system, the propagation can take place in the first mode or could happen in both modes and in certain cases; the presence of the persistent species has no influence and the kinetics vary with time as t2/3. There are relatively few experimental studies that have investigated the effect of free nitroxide concentration on the NMP. Becer et al.51 in a systematic kinetic screening have studied the influence of the concentration of free nitroxide SG1 (0–10 mol% with respect to the initiator) on the NMP of styrene and tert-butyl acrylate. They found no significant effect of the introduction of a slight excess of free nitroxide for the styrene polymerization; however, the polymerization rate of tert-butyl acrylate was reduced with the increasing SG1 concentration. For an initial concentration of SG1 introduced in the polymerization medium ≥1 mol% with respect to the initiator, the polymerization index ln([M]0/[M]) varies linearly with time for both monomers.
In our case, as no values of kp and kt are available in the literature for lithium styrene-STFSI and the two other monomers studied in the present work, it is difficult for us to estimate precisely if the initial SG1 concentration introduced in the polymerization medium was higher than (3[ln(10)]K[MAMA-SG1]0kt/kp)1/2 or lower than (3K[MAMA-SG1]0kt/kp)1/2. For the NMP of lithium styrene-STFSI in the presence of 5 mol% of SG1 with respect to MAMA-SG1, we found a linear fit of ln([M]0/[M]) with t2/3 rather than with time. This suggests that the initial concentration of SG1 used was not enough to suppress the persistent radical effect conditions due probably to the fast activation step in the polar medium. The similar phenomenon was also observed in the Atom Transfer Radical Polymerization (ATRP) system by Zhang et al.52 They demonstrated through a full kinetic analysis of the CuBr-mediated ATRP of methyl methacrylate that when [Cu(II)]0/[Cu(I)]0 ≤ 0.1, the reaction kinetics fits quite well Fischer's equation with ln([M]0/[M]) as a function of t2/3 and when [Cu(II)]0/[Cu(I)]0 ≥ 0.1, the reaction kinetics fits quite well with ln([M]0/[M]) as a function of t. In this case, the addition of at least 10 mol% of Cu(II) in the initial mixture is needed to neglect the persistent radical effect.
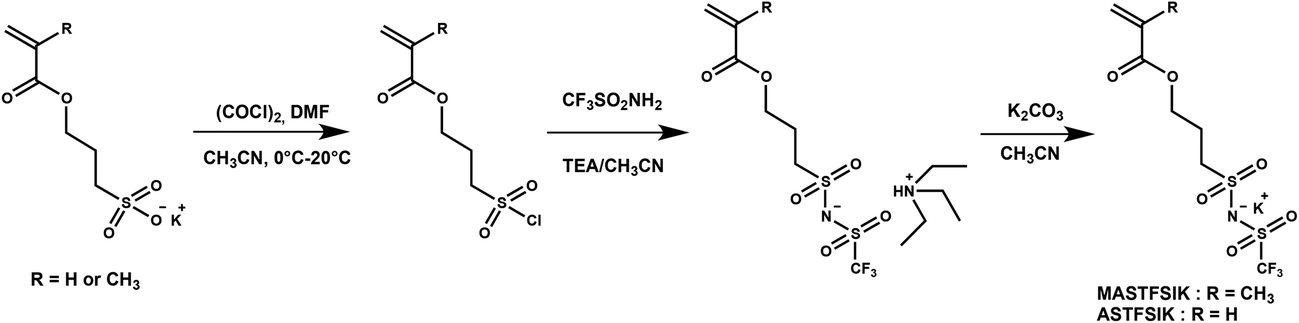
To further characterize the control of the chain growth polymerization, evolution of the theoretical and experimental Mn values was plotted (Fig. 2a). The experimental Mn values increased linearly with monomer conversion and the molar mass distributions were narrow as attested by the dispersity values. However, the experimental Mn values evaluated from the SEC calibration curve established with polystyrene standards were considerably higher than the expected ones. According to the principle of the SEC technique, the determination of the polymer molecular weight is based on the hydrodynamic volume of the polymer in the mobile phase. DMF used for the SEC mobile phase is considered as a “poor” solvent for PS but as a “good” solvent for poly(styrene-STFSI), it means that PS is poorly solvated and poly(styrene-STFSI) is well solvated in DMF. Considering the two polymers with a similar length, the hydrodynamic volume of PS will be smaller than that of poly(styrene-STFSI). This explains why we obtained the experimental Mn values for poly(styrene-STFSI) which were higher than the expected values. The SEC traces (Fig. 2b) show the continuous shift to smaller retention volumes with the increasing reaction time and are all unimodal with no evidence of the presence of high molecular weight polymers that may be indicative of uncontrolled polymerization and/or termination reactions. These data show clearly the controlled nature of the aqueous NMP of lithium styrene-STFSI using MAMA-SG1 as the initiator. However, some tailing is observed at high elution volume of the SEC chromatograms, probably coming from the interactions between the polymer chains and stationary phase.
 | ||
| Fig. 2 (a) Evolution of theoretical and experimental Mn and dispersity with conversion; experimental Mn estimated by using the calibration curve established with polystyrene standards. (b) Size exclusion chromatograms (DMF solution with 0.1 mol L−1 of LiBr) for poly(lithium 4-styrene-STFSI) homopolymer at various polymerization times. | ||
Aqueous NMP of potassium 3-sulfonyl(trifluoromethane sulfonyl) imide propyl methacrylate
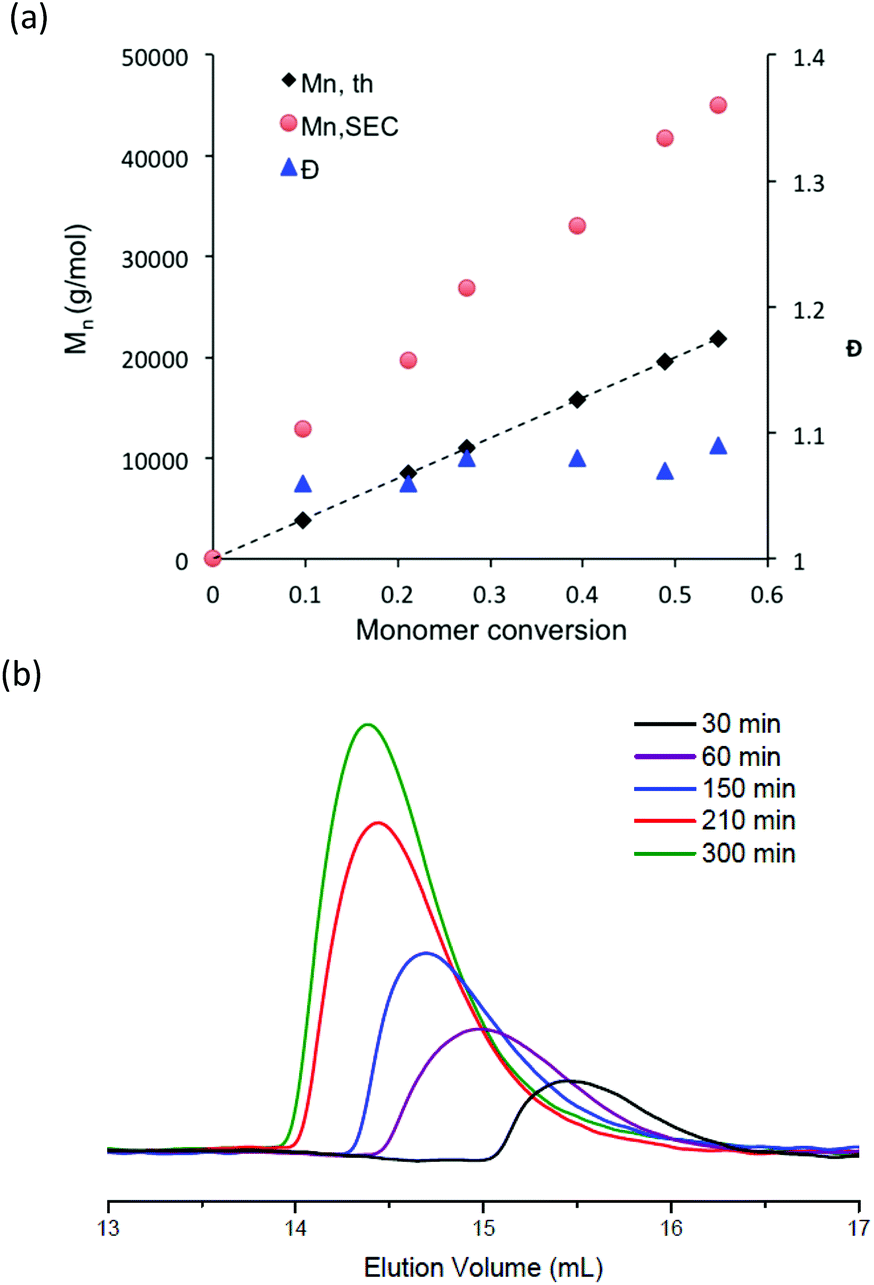
Apart from the synthesis of a specific nitroxide exclusively devoted to methacrylate monomers,53 the NMP of methacrylate monomers using nitroxide agents is difficult to achieve due to a too high equilibrium constant for the propagation step and/or undesired side reaction.20,54,55 To favor the formation of stable chain-end alkoxyamine and obtain a controlled/living system, methacrylate monomers are usually copolymerized with a comonomer, which is well controlled by NMP. The NMP of hydrophilic methacrylate monomers was generally carried out in organic solvent solution such as dioxane, dimethylsulfoxide or ethanol. There is only one paper that describes the nitroxide-mediated copolymerization of hydrophilic methacrylate monomers in water solution.28 In this study, the authors succeeded to control the SG1-mediated polymerization of methacrylic acid performed in water solution at 76 °C and in the presence of less than 10 mol% of sodium 4-styrenesulfonate. In the present work, potassium methacrylate-STFSI was copolymerized with potassium styrene-STFSI in water solution with a monomer content of 20 wt%, using sodium MAMA-SG1 as an alkoxyamine initiator and in the presence of 10 mol% of free-SG1 nitroxide. The initial molar fraction of potassium styrene-STFSI was chosen at 0.2 and polymerization reactions were performed at 65 °C and 75 °C. For the copolymerization performed at 65°, we observed polymerization kinetics to be extremely slow with a retardation period up to 80 min (Fig. S9†). During this period, neither of the two monomers was polymerized. From 80 min to 300 min, we observed nevertheless the linearity of ln([M]0/[M]) when plotted versus t2/3, however, SEC chromatograms (not shown here) of these samples did not exhibit any shifts toward higher molar masses. When the polymerization was performed at 75 °C for a target Mn of 30000 g mol−1 at full conversion, we observed that the reaction was fast. Indeed, conversions close to 100% were reached within 100 min (Fig. 3a). ln([M]0/[M]) of individual monomers was also plotted versus t2/3 with the aim to check the controlled character of the polymerization (Fig. 3b). As can be seen in Fig. 3b, ln([M]0/[M]) versus t2/3 was almost linear for potassium styrene-STFSI and potassium methacrylate-STFSI at least until 70 min. However, at this time, the conversion of SSTFSI-K was quasi-complete (>92%) and about 60% for the overall monomer conversions. After the complete disappearance of the styrene monomer in the polymerization mixture, the control of methacrylate polymerization was lost. Indeed, to ensure good control of the methacrylate monomer the polymerization mixture must always contain at least 7–10 mol% of comonomer.25,56 Below 60% overall monomer conversions, the Mn increased linearly with monomer conversion (Fig. 4a) and dispersity values were low (Đ < 1.2). Like for the polymerization of SSTFSI-Li discussed above, the experimental Mn values, derived from the calibration curve established with PMMA standards, were above the expected data. However, the difference between the experimental Mn values and the expected ones is now considerably reduced compared to the case of poly(styrene-STFSI) since DMF is considered as a “theta” (quite good) solvent of PMMA. The controlled character of the polymerization was further attested by the shift of the SEC chromatograms with the polymerization time (Fig. 4b). The SEC peaks corresponding to the polymerization time of 100 min and 130 min were superimposed with the large distributions of molar masses indicating the loss of polymerization control.
 | ||
| Fig. 3 Evolution of (a) monomer conversion vs. t and (b) ln([M]0/[M]) vs. t2/3 for the nitroxide-mediated copolymerization of potassium methacrylate-STFSI and potassium styrene-STFSI in water solution at 75 °C using sodium MAMA-SG1 as the initiator in the presence of 10 mol% of free SG1. | ||
 | ||
| Fig. 4 (a) Evolution of theoretical and experimental Mn and dispersity with conversion; experimental Mn estimated by using the calibration curve established with PMMA standards. (b) Size exclusion chromatograms (DMF solution with 0.1 mol L−1 of LiBr) for the poly(SSTFSI-K-co-MASSTFSI-K) copolymer at various polymerization times. | ||
From these results, it undoubtedly appeared that the SG1-mediated copolymerization of potassium methacrylate-STFSI and potassium styrene-STFSI under the employed conditions was fast and ill-controlled above 60% overall monomer conversion.
Aqueous NMP of potassium 3-sulfonyl(trifluoromethane sulfonyl) imide propyl acrylate
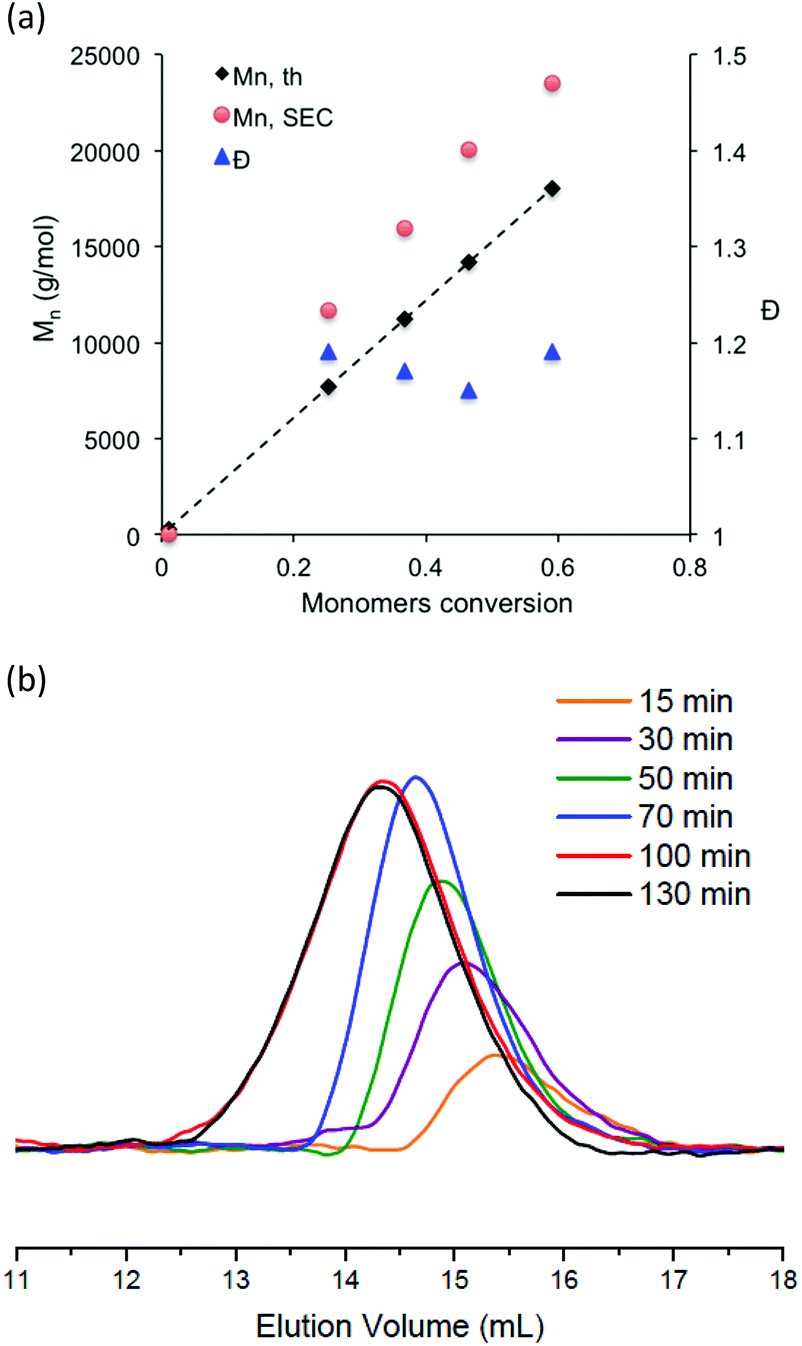
In comparison with hydrophilic styrene or methacrylate monomers, hydrophilic acrylate monomers and anionic acrylate derivatives have been the subject of a limited number of studies. As far we are aware, there have been only a few reports of the polymerization of anionic acrylates by living radical polymerization in aqueous media and they were mainly from RAFT polymerization.57,58 In the present work, potassium 3-sulfonyl(trifluoromethane sulfonyl) imide propyl acrylate was polymerized at 100 °C in water solution using sodium MAMA-SG1 as sodium MAMA-SG1 alkoxyamine. The reaction temperature was chosen based on our previous work on the NMP of the cationic acrylate monomer.33 Polymerization was performed with a target Mn of 30000 g mol−1, a monomer concentration of 30 wt% in water and in the presence of 7 mol% of free SG1 to initiator. In all cases, the polymerization reactions proceeded smoothly with reaction kinetics following ln([M]0/[M]) as a function of t2/3 (Fig. 5b). Monomer conversion was lower compared to styrene and methacrylate derivatives although the reaction temperature and monomer content were higher (Fig. 5a). The SEC chromatograms of the produced polymer shifted steadily to a lower elution volume with increasing monomer conversion (Fig. 6a). The evaluation of the molar masses from the calibration established with PMMA standards indicated low dispersity values (Đ < 1.2) and molar masses increased linearly with conversion (Fig. 6b). However, the apparent Mn values are higher than the theoretical values based on conversion, as the polymer standard is not appropriate and also because of the difference of solvation between the polymer standard and the analyzed polymer in the mobile phase as well as the possible interactions between the polymer chain and the stationary phase.
 | ||
| Fig. 5 Evolution of (a) monomer conversion vs. t and (b) ln([M]0/[M]) vs. t2/3 for the NMP of potassium propyl acrylate-STFSI in water solution at 100 °C using sodium MAMA-SG1 as the initiator in the presence of 7 mol% of free SG1. | ||
 | ||
| Fig. 6 (a) Size exclusion chromatograms (DMF solution with 0.1 mol L−1 of LiBr) for poly(ASTFSI-K) at various polymerization times. (b) Evolution of theoretical and experimental Mn and dispersity with conversion; experimental Mn estimated by using the calibration curve established with PMMA standards. | ||
Thermodynamic behavior of water-soluble polymers bearing a sulfonyl(trifluoromethane sulfonyl) imide group
The thermodynamic behavior of the prepared polymers was studied by differential scanning calorimetry (DSC). For this purpose, samples were heated between 40 °C and 250 °C for styrene and methacrylate based polymers and between −90 °C and 150 °C for acrylate based polymers. DSC traces of these polymers are shown in Fig. S10 and S11.† We found that the Tg of the homopolymer lithium PSSTFSI was 172 °C, which is in good agreement with those reported in the literature. For example, the lithium PSSTFSI synthesized by free-radical polymerization presents a Tg value of about 152 °C,34,41 however Long et al. have noted that Tg varied significantly depending on the choice of the counterion; in particular, voluminous cations or divalent cations lead to polymer networks with a higher Tg.41 Recently, Hadziioannou et al. showed that PSSTFSIK samples obtained by RAFT polymerization exhibited also high Tg values which increased from 179 °C to 239 °C as the polymer molar masses varied from 6000 g mol−1 to 80000 g mol−1.42 The poly(SSTFSIK-co-MASTFSIK) copolymer exhibits two Tg values at 132 °C and 174 °C with a high heat flow amplitude for the first Tg. The observation of two Tg values suggests that the copolymer is not random but rather a gradient copolymer with a rich part of methacrylate and a rich part of styrene. Thus, the first Tg was attributed to PMASTFSIK and the second, close to the Tg value observed for PSSTFSILi previously, to PSSTFSIK. As expected for the polyacrylate backbone, the Tg value of the PASTFSIK homopolymer was much lower than those observed for styrene and methacrylate based polymers, which was 17 °C. However, one can note that thermal transition took place on a very large temperature domain. By comparison with the corresponding polystyrene and poly(propyl (meth)acrylate), all the studied water-soluble polymers bearing a sulfonyl(trifluoromethane sulfonyl) imide group exhibit higher Tg values (50 °C to 80 °C) due to strong electrostatic interactions between the potassium or lithium STFSI groups. These anionic polymers with their interesting thermodynamic behavior will probably give rise to the good block copolymer electrolytes for lithium battery when they associate with a ionic conductor block.
Conclusion
Besides higher performance batteries, safer battery technologies are the next major issues in the battery industry. For this purpose, we have recently developed single-ion solid block copolymer electrolytes.17 In order to gain more information on such kinds of electrolytes and improve their performances, other anionic monomers bearing a sulfonyl(trifluoromethane sulfonyl) imide group, namely potassium propylmethacrylate-STFSI and potassium propylacrylate-STFSI, were successfully synthesized with good yield (60%) and fully characterized by HR-MS and 1H, 13C and 19F NMR spectroscopy. Then, nitroxide-mediated polymerization of these hydrophilic monomers bearing an STFSI anion (SSTFSILi, MASTFSIK and ASTFSIK) in homogeneous aqueous medium was achieved using a SG1-based alkoxyamine bearing a carboxylic acid group. These polymerizations present the typical features of controlled polymerization, such as the linearity of ln([M]0/[M]) as a function of t2/3 up to high conversions, a linear increase of the molecular weight with conversion and low dispersity values (Đ < 1.2). In the case of the methacrylate-based monomer, 20 mol% of the styrene based monomer was required to reach good polymerization control. The NMP in water yields well-defined polymers with methacrylate, styrene and acrylate based water-soluble monomers at 75, 90, and 100 °C, respectively. The obtained protocol will be usefully applied in the preparation of block copolymers devoted to SPE for lithium batteries. Furthermore, these anionic polymers show a higher glass transition temperature compared to their neutral analogous polymers. The thermodynamic behavior of these anionic polymers once associated with the PEO block should lead to interesting solid electrolytes for lithium batteries.Acknowledgements
The present work was undertaken within the French ANR program under the contract COPOLIBAT 2, no. ANR-13-PRGE-0002-02. The authors wish to thank Arkema for kindly providing the MAMA-SG1 initiator and the nitroxide SG1. They are grateful to Jean-Pierre Bonnet for his help in MASTFSIK synthesis.References
- W. H. Meyer, Adv. Mater., 1998, 10, 439–448 CrossRef CAS PubMed.
- K. Murata, S. Izuchi and Y. Yoshihisa, Electrochim. Acta, 2000, 45, 1501–1508 CrossRef CAS.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- R. Younesi, G. M. Veith, P. Johansson, K. Edströmbe and T. Veggea, Energy Environ. Sci., 2015, 8, 1905–1922 CAS.
- D. Mecerreyes, Prog. Polym. Sci., 2011, 36, 1629–1648 CrossRef CAS.
- J. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009–1036 CrossRef CAS.
- T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed.
- Z.-H. Ma, H.-B. Han, Z.-B. Zhou and J. Nie, J. Mol. Catal. A: Chem., 2009, 311, 46–53 CrossRef CAS.
- J. Zhang and D. D. DesMarteau, J. Fluorine Chem., 2001, 111, 253–257 CrossRef CAS.
- G. S. Lal, G. P. Pez and R. G. Syvret, Chem. Rev., 1996, 96, 1737–1756 CrossRef CAS PubMed.
- C. G. Cho, Y. S. Kim, X. Yu, M. Hill and J. E. McGrath, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6007–6014 CrossRef CAS.
- J. M. Alsheheri, H. Ghassemi and D. A. Schiraldi, J. Power Sources, 2014, 267, 316–322 CrossRef CAS.
- J. Foropoulos Jr. and D. D. DesMarteau, Inorg. Chem., 1984, 23, 3720–3723 CrossRef.
- D. Devaux, R. Bouchet, D. Glé and R. Denoyel, Solid State Ionics, 2012, 227, 119–127 CrossRef CAS.
- D. R. Sadoway, B. Y. Huang, P. E. Trapa, P. P. Soo, P. Bannerjee and A. M. Mayes, J. Power Sources, 2001, 97–98, 621–623 CrossRef CAS.
- X.-G. Sun and J. B. Kerr, Macromolecules, 2006, 39, 362–372 CrossRef CAS.
- R. Bouchet, S. Maria, R. Meziane, A. Aboulaich, L. Lienafa, J. P. Bonnet, T. N. T. Phan, D. Bertin, D. Gigmes, D. Devaux, R. Denoyel and M. Armand, Nat. Mater., 2013, 12, 452–457 CrossRef CAS PubMed.
- R. Bouchet, A. Aboulaich, S. Maria, T. Phan, D. Gigmes, D. Bertin, R. Meziane, J.-P. Bonnet and M. Armand, US Pat., US20140272600A1, 2014 Search PubMed.
- P. P. Soo, B. Huang, Y. I. Jang, Y. M. Chiang, D. R. Sadoway and A. M. Mayes, J. Electrochem. Soc., 1999, 146, 32 CrossRef CAS.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef CAS.
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, P. Wilson, K. Kempe, J. F. Quinn, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Rev., 2015, 116, 835–877 CrossRef PubMed.
- A. Anastasaki, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2016, 7, 1002–1026 RSC.
- J. Nicolas, C. Dire, L. Mueller, J. Belleney, B. Charleux, S. R. Marque, D. Bertin, S. Magnet and L. Couvreur, Macromolecules, 2006, 39, 8274–8282 CrossRef CAS.
- J. Nicolas, P. Couvreur and B. Charleux, Macromolecules, 2008, 41, 3758–3761 CrossRef CAS.
- S. Brusseau, J. Belleney, S. Magnet, L. Couvreur and B. Charleux, Polym. Chem., 2010, 1, 720–729 RSC.
- S. Brusseau, F. D'Agosto, S. Magnet, L. Couvreur, C. Chamignon and B. Charleux, Macromolecules, 2011, 44, 5590–5598 CrossRef CAS.
- J. Nicolas, S. Brusseau and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 34–47 CrossRef CAS.
- M. Chenal, S. Mura, C. Marchal, D. Gigmes, B. Charleux, E. Fattal, P. Couvreur and J. Nicolas, Macromolecules, 2010, 43, 9291–9303 CrossRef CAS.
- T. N. T. Phan, S. Maiez-Tribut, J. P. Pascault, A. Bonnet, P. Gerard, O. Guerret and D. Bertin, Macromolecules, 2007, 40, 4516–4523 CrossRef CAS.
- B. Lessard, E. J. Y. Ling, M. S. T. Morin and M. Marić, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1033–1045 CrossRef CAS.
- T. N. T. Phan and D. Bertin, Macromolecules, 2008, 41, 1886–1895 CrossRef CAS.
- R. Meziane, J.-P. Bonnet, M. Courty, K. Djellab and M. Armand, Electrochim. Acta, 2011, 57, 14–19 CrossRef CAS.
- M. Armand, Y. Choquette, M. Gauthier and C. Michot, France Pat., EP0850920A2, 1997 Search PubMed.
- J. Li, H. Zhu, X. Wang, M. Armand, D. R. MacFarlane and M. Forsyth, Electrochim. Acta, 2015, 175, 232–239 CrossRef CAS.
- W. Huang, B. Charleux, R. Chiarelli, L. Marx, A. Rassat and J.-P. Vairon, Macromol. Chem. Phys., 2002, 203, 1715–1723 CrossRef CAS.
- B. Grassl, G. Clisson, A. Khoukh and L. Billon, Eur. Polym. J., 2008, 44, 50–58 CrossRef CAS.
- J. Rigolini, B. Grassl, S. Reynaud and L. Billon, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5775–5782 CrossRef CAS.
- R. Nicolaÿ, L. Marx, P. Hémery and K. Matyjaszewski, Macromolecules, 2007, 40, 6067–6075 CrossRef.
- C. Jangu, A. M. Savage, Z. Zhang, A. R. Schultz, L. A. Madsen, F. L. Beyer and T. E. Long, Macromolecules, 2015, 48, 4520–4528 CrossRef CAS.
- M. Mumtaz, K. Aissou, D. Katsigiannopoulos, C. Brochon, E. Cloutet and G. Hadziioannou, RSC Adv., 2015, 5, 98559–98565 RSC.
- H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS PubMed.
- A. Goto and T. Fukuda, Prog. Polym. Sci., 2004, 29, 329–385 CrossRef CAS.
- J.-F. Lutz, P. Lacroix-Desmazes and B. Boutevin, Macromol. Rapid Commun., 2001, 22, 189–193 CrossRef CAS.
- E. Drockenmuller, J.-P. Lamps and J.-M. Catala, Macromolecules, 2004, 37, 2076–2083 CrossRef CAS.
- F. Chauvin, P. E. Dufils, D. Gigmes, Y. Guillaneuf, S. R. A. Marque, P. Tordo and D. Bertin, Macromolecules, 2006, 39, 5238–5250 CrossRef CAS.
- D. Benoit, G. Grimaldi, S. Robin, J.-P. Finet, P. Tordo and Y. Gnanou, J. Am. Chem. Soc., 2000, 122, 5929–5939 CrossRef CAS.
- T. Fukuda, A. Goto and K. Ohno, Macromol. Rapid Commun., 2000, 21, 151–165 CrossRef CAS.
- M. Souaille and H. Fischer, Macromolecules, 2002, 35, 248–261 CrossRef CAS.
- C. R. Becer, R. M. Paulus, R. Hoogenboom and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6202–6213 CrossRef CAS.
- H. Zhang, B. Klumperman, W. Ming, H. Fischer and R. van der Linde, Macromolecules, 2001, 34, 6169–6173 CrossRef CAS.
- Y. Guillaneuf, D. Gigmes, S. R. A. Marque, P. Astolfi, L. Greci, P. Tordo and D. Bertin, Macromolecules, 2007, 40, 3108–3114 CrossRef CAS.
- R. Mchale, F. Aldabbagh and P. B. Zetterlund, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2194–2203 CrossRef CAS.
- C. Dire, J. Belleney, J. Nicolas, D. Bertin, S. Magnet and B. Charleux, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6333–6345 CrossRef CAS.
- B. Charleux, J. Nicolas and O. Guerret, Macromolecules, 2005, 38, 5485–5492 CrossRef CAS.
- J. T. Lai, D. Filla and R. Shea, Macromolecules, 2002, 35, 6754–6756 CrossRef CAS.
- M. Mertoglu, A. Laschewsky, K. Skrabania and C. Wieland, Macromolecules, 2005, 38, 3601–3614 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The 1H, 19F, 13C NMR spectra of hydrophilic monomers bearing the STFSI anion and the DSC thermograms of their corresponding homopolymers. See DOI: 10.1039/c6py01004k |
| This journal is © The Royal Society of Chemistry 2016 |