
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Side-chain poly(phosphoramidate)s via acyclic diene metathesis polycondensation†
Alper
Cankaya
,
Mark
Steinmann
,
Yagmur
Bülbül
,
Ingo
Lieberwirth
and
Frederik R.
Wurm
 *
*
Max-Planck-Institut für Polymerforschung, Ackermannweg 10, 55128 Mainz, Germany. E-mail: wurm@mpip-mainz.mpg.de
First published on 18th July 2016
Abstract
Poly(phosphoester)s (PPEs) are interesting degradable multi-functional polymers. Here, we present the first synthesis of poly(phosphoramidate)s (PPAs) via acyclic diene metathesis (ADMET) polycondensation with amidate linkages in side chains. In contrast to conventional polyamides, the P–N-bond in phosphoramidates is more labile than the corresponding esters. Unsaturated PPAs were compared with structural analogues of PPEs: two novel α,ω-dienes, i.e. bis-(undecen-10-yl)-n-butyl-phosphoramidate (1) and bis-(undecen-10-yl)-n-butyl-phosphate (2) have been polymerized by Grubbs-type catalysts to polymers with molecular weights up to ca. 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1. After hydrogenation polyethylene-like structures were obtained with the phosphoramidate or -ester representing a precisely placed defect. PPAs were compared to their PPE analogues with respect to their thermal behavior and stability by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA), showing similar crystallization behavior for the saturated materials, but significant differences for unsaturated PPA vs. PPE. This synthesis of PPAs via ADMET polymerization offers an interesting approach to various PPAs. The hydrolytically labile pendant phosphoramidate further offers the possibility for the development of hydrolytically degradable materials or as processable intermediates for poly(phosphodiester)s which often show limited solubility.
000 g mol−1. After hydrogenation polyethylene-like structures were obtained with the phosphoramidate or -ester representing a precisely placed defect. PPAs were compared to their PPE analogues with respect to their thermal behavior and stability by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA), showing similar crystallization behavior for the saturated materials, but significant differences for unsaturated PPA vs. PPE. This synthesis of PPAs via ADMET polymerization offers an interesting approach to various PPAs. The hydrolytically labile pendant phosphoramidate further offers the possibility for the development of hydrolytically degradable materials or as processable intermediates for poly(phosphodiester)s which often show limited solubility.
Introduction
Biodegradable polymers with precise properties and structures are a field of research with increasing attention ranging from plastics to biomedical implants or drug delivery, for example. Polyesters and -amides lead the field in current literature due to established syntheses and have proven biocompatibility.1,2 However, as polyesters degrade, the pH may drop due to the release of carboxylic acids; also the chemical versatility of polyesters is limited or may demand several reaction steps to introduce functionality.3Poly(phosphoester)s (PPEs), with a backbone of repeating phosphoester bonds, are biodegradable and (typically) biocompatible polymers with a high degree of chemical versatility.4 Due to the formation of three ester-bonds, PPEs can be tailored along their main and side chains,5 offering a variety of possible polymer structures, making PPEs interesting for diverse applications like for drug6–8 and gene delivery9 and tissue engineering.10 The stability of the phosphorus-containing polymers is directly influenced by the first binding sphere around the central phosphorus: the development of poly(phosphonate)s with a stable P–C-bond as the main11 or side chain elements12 or poly(phosphoramidate)s (PPAs) with a hydrolysis-labile P–N bond in the side chain13 has a direct influence on the polymers’ degradation profile.
Side-chain PPAs have been only scarcely studied; their classical synthesis relies on the post-polymerization modification of poly(H-phosphonate)s by the versatile Atherton–Todd reaction.14–16 More recently, the Wooley lab presented an elegant approach by ring-opening polymerization13 of cyclic dioxaphospholane. PPAs have been studied for the delivery of nucleic acids or as flame-retardant polymers.2,14,17–19
Our group developed several protocols based on olefin metathesis polymerization (either as acyclic diene metathesis (ADMET) or ring-opening metathesis polymerization) for phosphorus-containing materials.20–22
Herein, we use the robust ADMET protocol23 to present the first strategy to aliphatic saturated and unsaturated PPAs with the phosphoramidate bond in the side chain. The labile P–N bond can be used to release the pendant chain (e.g. for pH-sensitive drug delivery) or can be regarded as a soluble precursor for poly(phosphodiester)s after acidic hydrolysis, which typically are hardly soluble and are interesting materials for bone targeting, for example.24 Such polymers may also find useful application as novel polymeric flame retardant additives.25 In addition, we compare the PPA to a structurally identical PPE (a single oxygen is exchanged by “NH”, cf. Scheme 1).
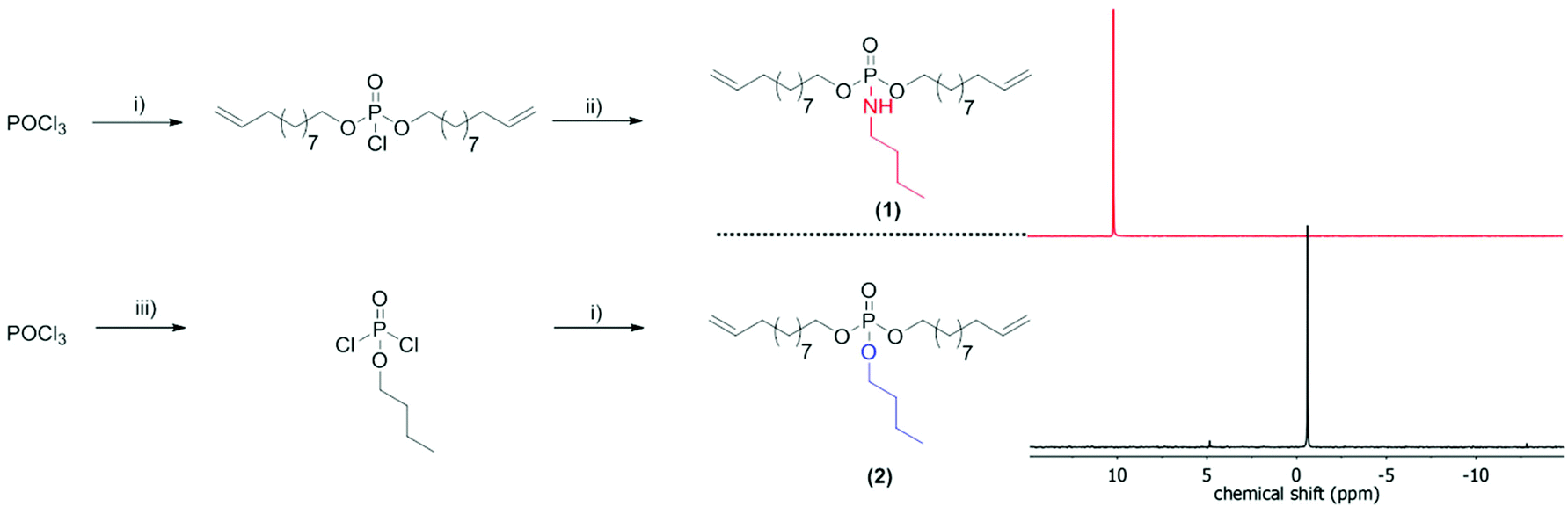 | ||
| Scheme 1 Synthesis of the ADMET monomers bis-(undecen-10-yl) butylphosphoramidate (1) and bis-(undec-10-en-1-yl)butylphosphate (2) and their 31P NMR spectra. (i) 10-undecen-1-ol, triethylamine, dichloromethane; (ii) butylamine, triethylamine, dichloromethane; (iii) butanol, triethylamine, dichloromethane. | ||
Experimental
Materials
Solvents were purchased from Acros Organics and Sigma Aldrich and used as received, unless otherwise stated. Phosphoroxychloride, 10-undecen-1-ol, triethylamine, tris(hydroxymethyl)phosphine and butan-1-ol were used as received from Sigma-Aldrich (Germany). The 1st generation Grubbs catalyst was purchased from Sigma-Aldrich and stored under an argon atmosphere. n-Butyl amine was purchased from TCI chemicals and used without further purification. Deuterated solvents were purchased from Acros Organics and Carl Roth Chemicals.Instrumentation
Gel-permeation chromatography (GPC) measurements were carried out in THF, with samples of the concentration of 1 g L−1. Sample injection was performed by using a 1260-ALS auto sampler (Waters) at 30 °C (THF). The flow rate was 1 mL min−1. In THF, three SDV columns (PSS) with dimensions of 300 × 80 mm, 10 μm particle size and pore sizes of 106, 104 und 500 Å were employed. Detection was accomplished with a DRI Shodex RI-101 detector (ERC) and UV-vis 1260-VWD detector (Agilent). Calibration was achieved vs. poly(styrene) standards provided by Polymer Standards Service. 1H-NMR spectra were recorded on a Bruker Avance 300 MHz spectrometer, 13C-NMR and 31P-spectra were recorded on a Bruker Avance III 500 MHz spectrometer. 1H15N-NMR spectra were recorded on a Bruker Avance 700 III MHz spectrometer. The 13C-NMR (176 MHz) and 31P-NMR (283 MHz) measurements were obtained with a 1H powergate decoupling method using a 30° degree flip angle.
1H-DOSY experiments were recorded with a 5 mm BBI 1H/X z-gradient on a 700 MHz spectrometer with a Bruker Avance III system. For a 1H NMR spectrum 64 transients were used with an 11 μs long 90° pulse and a 12600 Hz spectral width together with a recycling delay of 5 s. The temperature for all experiments was kept at 298.3 K. For the diffusion measurements a 2D sequence (DOSY1, dstebgp3s2) with a double stimulated echo for convection compensation and LED using a bipolar gradient pulse for diffusion was used additionally.26 The temperature was maintained at 298.3 K and regulated by using a standard 1H methanol NMR sample using the topspin 3.2 software (Bruker). The control of the temperature was realized with a VTU (variable temperature unit) and with an accuracy of ±0.1 K.
The proton, carbon and phosphorous spectra were measured in CDCl3 or DMSO-d6. The spectra were referenced to the residual proton signals of the deuterated solvent (CDCl3 (1H) = 7.26 ppm; DMSO-d6 (1H) = 2.50 ppm). All spectra were processed with MestReNova 6.1.1-6384 software. Differential Scanning Calorimetry (DSC) measurements were performed using a Perkin-Elmer 7 series thermal analysis system and a Perkin Elmer Thermal Analysis Controller TAC 7/DX in the temperature range from −150 to 130 °C. Three scanning cycles of heating–cooling were performed (in a N2 atmosphere 30 mL min−1) at a heating rate of 10 °C min−1.
Thermogravimetric analysis (TGA) was performed on Mettler Toledo ThermoSTAR TGA/SDTA 851-Thermowaage in the range of temperature between 25 °C and 600 °C, at a heating rate of 10 °C min−1. For wide-angle X-ray scattering (WAXS) and small-angle X-ray scattering (SAXS) experiments samples were prepared by hot pressing an approximately 200–400 μm thick film on a hot stage. A sufficient amount of the sample was placed on a preheated glass slide and allowed to melt. Subsequently, another hot glass slide was pressed on the melt. This sandwich was kept above the melting point for another 5 min before cooling it down to room temperature in order to eliminate any shear-induced orientation in the sample.
SAXS was recorded using CuKα radiation (wavelength 1.54 Å) from a rotating anode source (Rigaku MicroMax 007 X-ray generator) with curved multilayer optics (Osmic Confocal Max-Flux). The scattered intensity was recorded on a 2D detector (Mar345 image plate) with a sample–detector distance of 2 m. For WAXS measurements the sample–detector distance was set to 20 cm. For Transmission Electron Microscopy (TEM), a FEI Tecnai F20 transmission electron microscope operating at an acceleration voltage of 200 kV was used to determine the crystal morphology, thickness, and crystal structure.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C
C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) –), δ 4.96–4.82 (m, 4H, C2CH–), δ 4.20–3.90 (m, 4H, –O–C2–), δ 2.03 (q, J = 6 Hz, 4H, CH2CH–C2–), δ 1.81–1.71 (m, 4H, –O–CH2–C2–), δ 1.44–1.28 (m, 24H, –C2–).
:acetone (8:1) as an eluent (Rf: 0.58, yield: 63%, 3.5 g).
–), δ 4.96–4.82 (m, 4H, C2CH–), δ 4.20–3.90 (m, 4H, –O–C2–), δ 2.03 (q, J = 6 Hz, 4H, CH2CH–C2–), δ 1.81–1.71 (m, 4H, –O–CH2–C2–), δ 1.44–1.28 (m, 24H, –C2–).
:acetone (8:1) as an eluent (Rf: 0.58, yield: 63%, 3.5 g).
1H-NMR (298 K, 300 MHz, DMSO-d6, ppm): δ 5.81 (ddt, J1 = 12 Hz, J2 = 6 Hz, J3 = 3 Hz 2H, CH2C–), δ 5.02–4.90 (m, 4H, C2CH–), δ 4.79–4.74 (dt, 1H, –N–), δ 3.97 (qd, 4H, –O–C2–), δ 2.92–2.84 (m, 2H, –NH–C2–), δ 2.03 (m, CH–C2–), δ 1.65 (q, J = 9 Hz, 4H, –O–CH2–C2–), δ 1.31 (m, 28H, –C2–, –C2–CH3), δ 0.91 (t, J = 6 Hz, 3H, –CH2–C3). 13C-NMR (298 K, 75 MHz, DMSO-d6, ppm): δ 139.06 (CH2![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H–), 114.04 (H2CH–), 66.21 (–O–H2–), 41.06 (–NH–H2–), 33.71 (CH–H2–), 30.31 (–O–CH2–H2–), 29.01 (–H2–), 25.44 (–NH–CH2–H2–), 19.56 (–H2–CH3), 13.69 (–H3). 31P-NMR (298 K, 125 MHz DMSO-d6, ppm): δ 10.00.
H–), 114.04 (H2CH–), 66.21 (–O–H2–), 41.06 (–NH–H2–), 33.71 (CH–H2–), 30.31 (–O–CH2–H2–), 29.01 (–H2–), 25.44 (–NH–CH2–H2–), 19.56 (–H2–CH3), 13.69 (–H3). 31P-NMR (298 K, 125 MHz DMSO-d6, ppm): δ 10.00.
:ethylacetate (8:1) as an eluent (Rf: 0.8, yield: 37%, 7.4 g).
1H-NMR (298 K, 300 MHz, CDCl3, ppm): δ 5.74 (ddt, J1 = 12 Hz, J2 = 9 Hz, J3 = 3 Hz, 2H, CH2C–), δ 4.96–4.84 (m, 4H, C2CH–), δ 4.00–3.92 (m, 6H, –O–C2–), δ 2.01–1.93 (m, 4H, CH2CH–C2–), δ 1.65–1.55 (m, 6H, –O–C2–), δ 1.36–1.17 (m, 26H, –C2–), δ 0.87 (t, J = 6 Hz, 3H, –C3). 13C-NMR (298 K, 75 MHz, CDCl3, ppm): δ 139.09 (CH2H–), 113.93 (H2CH–), 67.45 (–O–H2–), 33.66 (CH–H2–), 30.35 (–O–CH2–H2–), 29.15 (–H2–), 25.32 (–H2–H2–CH3), 18.49 (–H2–CH3), 13.38 (–H3). 31P-NMR (298 K, 125 MHz, CDCl3, ppm): δ −0.65.
P1 . 1H-NMR (298 K, 300 MHz, CDCl3, ppm): δ 5.33–5.25 (m, 2H, –C
C–), δ 3.90 (qq, 4H, –O–C2–), δ 2.81 (dt, J1 = 9 Hz, J2 = 6 Hz, 2H, –NH–C2–), δ 1.89 (q, J = 6 Hz, 4H, CH–C2–), δ 1.58 (q, J = 6 Hz, 4H, –O–CH2–C2–), δ 1.42–1.20 (m, 28H, –C2–), δ 0.84 (t, J = 6 Hz 3H, –C3). 13C-NMR (298 K, 75 MHz, CDCl3, ppm): δ 130.21 (–CC–), 66.14 (–O–C2–), 41.00 (–NH–H2–), 33.88 (–CHCH–H2–), 32.61 (–O–CH2–H2–), 30.43 (–H2–), 25.60 (–NH–CH2–H2–), 19.74 (–H2–CH3), 13.69 (–H3). 31P-NMR (298 K, 125 MHz, CDCl3, ppm): δ 10.02.
P2 . 1H-NMR (298 K, 300 MHz, CDCl3, ppm): δ 5.33–5.26 (m, 2H, –C
C–), δ 3.96 (qd, 6H, –O–C2–), δ 1.88 (q, J = 6 Hz, 4H, CH–C2–), δ 1.59 (dtt, 6H, –O–CH2–C2–), δ 1.40–1.20 (m, 26H, –C2–), δ 0.87 (t, J = 6 Hz, 3H, –C3). 13C-NMR (298 K, 75 MHz, CDCl3, ppm): δ 130.70 (–CC–), 68.15 (–O–C2–), 33.14 (–CHCH–H2–), 30.78 (–O–CH2–H2–), 30.01 (–H2–), 25.95 (–H2–CH3), 19.15 (–H2–CH3), 14.05 (–H3). 31P-NMR (298 K, 125 MHz, CDCl3, ppm): δ −0.84.
P1-H . 1H-NMR (298 K, 300 MHz, CDCl3, ppm): δ 3.93 (qq, 4H, –O–C
2–), δ 2.84 (dt, J1 = 9 Hz, J2 = 6 Hz, 2H, –NH–C2–), δ 1.58 (q, J = 7 Hz, 4H, –O–CH2–C2–), δ 1.42–1.20 (m, 28H, –C2–), δ 0.84 (t, J = 5 Hz 3H, –C3). 13C-NMR (298 K, 75 MHz, CDCl3, ppm): δ 66.14 (–O–C2–), 41.00 (–NH–H2–), 32.01 (–O–CH2–H2–), 30.43 (–H2–), 26.20 (–NH–CH2–H2–), 19.53 (–H2–CH3), 13.71 (–H3). 31P-NMR (298 K, 125 MHz, CDCl3, ppm): δ 10.02.
P2-H . 1H-NMR (298 K, 300 MHz, CDCl3, ppm): δ 3.96 (qd, 6H, –O–C
2–), δ 1.66 (dtt, 6H, –O–CH2–C2–), δ 1.40–1.20 (m, 26H, –C2–), δ 0.93 (t, J = 6 Hz, 3H, –C3). 13C-NMR (298 K, 75 MHz, CDCl3, ppm): δ 67.15 (–O–C2–), 38.18 (–O–CH2–H2–), 29.91 (–H2–), 25.95 (–H2–CH3), 19.15 (–H2–CH3), 14.31 (–H3). 31P-NMR (298 K, 125 MHz, CDCl3, ppm): δ −0.84.
:4). The degradation was measured every day over the duration of 1.5 weeks by 1H NMR and 31P NMR spectroscopy.
Results and discussion
For the comparison of PPEs with PPAs, we designed two novel α,ω-dienes, i.e. bis-(undecen-10-yl) butylphosphoramidate (1) and its phosphoester equivalent (2) (characterization data can be found in the ESI†). Both monomers can be synthesized starting from phosphoroxychloride with sequential substitution of the chlorides with alcohols (10-undecen-1-ol and/or n-butanol) and/or the amine (n-butylamine) (Scheme 1). Due to its high nucleophilicity, the amine is attached after the incorporation of the polymerizable groups, while for the phosphoester side chain (2) butanol can be also added in the first step. Both monomers were purified by column chromatography and are stable at room temperature over a period of at least several months (1H, 13C, 31P, 1H15N HMBC NMR spectra can be found in the ESI†). The 15N NMR chemical shift for the amidate-N is detected at 42.3 ppm (Fig. S4†), while the 31P NMR spectra show distinct resonances for the phosphoramidate at ca. 10 ppm and the phosphoester at −0.65 ppm (Scheme 1).Both α,ω-dienes (phosphoramidate/-ester) monomers were polymerized by ADMET polycondensation using the 1st generation Grubbs catalyst or the 2nd generation Hoveyda–Grubbs catalyst. The reaction was carried out in bulk at 80 °C for 12 h at reduced pressure in order to remove the ethylene evolved during this reaction (Scheme 2). For the poly(phosphoramidate)s polymerization by the 1st generation Grubbs catalyst (3 mol%) produced polymers with apparent Mn's of up to 4800 g mol−1. The polymerization of 1 was also investigated with the typically more active 2nd generation Grubbs–Hoveyda catalyst. 1, 3, 5, and 10 mol% were used and the apparent Mn values were increased to ca. 9000 g mol−1 for 5 mol% catalyst loading. 10 mol% of catalyst loading changed the molar mass only slightly (Table 1 & Fig. S14†). In addition to SEC, apparent molecular weights have been determined by 1H DOSY NMR spectroscopy for two examples: 1H-DOSY NMR. In order to determine Mw of the polymers, poly(styrene) samples with known absolute molecular weights (GPC standards) were measured via DOSY and used as a calibration.27,28 The determined Mws from the H-DOSY NMR spectra are in a range between 9400 g mol−1 and 11000 g mol−1 for the PPAs P1-b and P1-d, respectively.
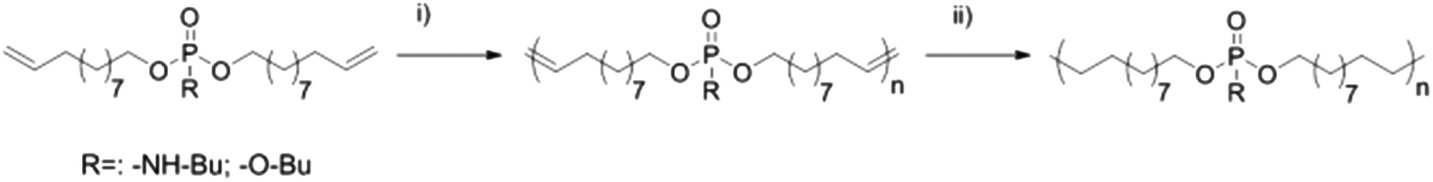 | ||
| Scheme 2 ADMET polymerization of 1 and 2 and subsequent hydrogenation (i) Grubbs catalyst, vacuum, 60–80 °C; (ii) Pd/H2, toluene). | ||
| Code | Conditionsa |
M
nb |
M
w/Mnb |
T
gc/°C |
T
mc/°C |
|---|---|---|---|---|---|
| a A: 1st generation Grubbs, 3 mol%, 24 h. 2nd gen. Grubbs–Hoveyda. 1 mol%, 24 h = B; 3 mol%, 24 h = C; 5 mol%, 24 h = D; 10 mol%, 24 h = E; 5 mol%, 48 h = F. b Determined via SEC in THF (vs. PS standards). c Determined by differential scanning calorimetry (10 K min−1). n.d. = not determined. | |||||
| P1 -a | A | 3100 | 1.25 | n.d. | n.d. |
| P1 -b | A | 4800 | 2.19 | −72 | −8 |
| P1 -c | B | 4400 | 1.82 | n.d. | n.d. |
| P1 -d | C | 6500 | 1.70 | −71 | 8 |
| P1 -e | D | 9000 | 1.72 | n.d. | n.d. |
| P1 -f | E | 9500 | 1.66 | n.d. | n.d. |
| P1 -g | F | 14000 |
2.01 | n.d | n.d. |
| P2 -a | A | 9000 | 2.70 | −84 | 0 |
| P2 -b | A | 9600 | 2.71 | n.d. | n.d. |
| P2 -c | D | 19600 |
1.81 | n.d. | n.d. |
| P1 -d-H | — | 7000 | 1.60 | −50 | 51 |
| P2 -b-H | — | 10000 |
2.55 | −80 | 47 |
The poly(phosphoester) based on 2 was not reported in the literature, however similar polymers with phenoxy side chains have been prepared and GPC indicates for P2s a Mn of ca. 9000 g mol−1 in this case (which is probably not the upper limit). Successful polymerization of both monomers can be easily detected from the 1H NMR spectra, as the terminal olefin resonances change into internal alkenes after the polymerization (end groups may be detected, but isomerization is known to shift the end groups also to internal alkenes, spectra can be found in the ESI†). These unsaturated PPAs/PPEs converted by catalytic hydrogenation with Pd/C into their saturated counterparts. The 1H NMR spectra show the disappearance of any alkene resonances after successful hydrogenation (Fig. S11 & S12†) and SEC proves the stability of the polymer under these conditions (Fig. S16†).
As possible flame-retardant materials phosphoramidates typically exhibit different thermal behavior compared to their ester analogues.25 The thermal properties of the unsaturated and saturated poly(phosphoramidate)s and poly(phosphoester)s were studied by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) (Fig. S17 & S18†). P2 was studied by TGA and single thermal decomposition at ca. 310 °C (10% weight loss: T10% = 311 °C) with a char residue at 450 °C of 9% was observed. The thermal degradation of phosphoramidates is different compared to phosphoesters and was already studied for various flame retardant additives or modified polymers (typically with low molecular weight additives or modification of a common polymer matrix).29,30P1 shows a more complex degradation profile than the corresponding polyester: a first mass loss of ca. 23% mass up to ca. 298 °C, followed by a second process until ca. 325 °C and 60% weight loss. This char slowly degrades under further heating to ca. 15% remaining at 440 °C. Compared to low molecular weight flame retardant phosphoramidates, this TGA profile also makes PPAs interesting candidates for flame retardants additives.25
The hydrogenated samples (P1-H and P2-H) can also be regarded as a defective poly(ethylene) (PE) with the phosphate/-amidate acting as defects, spaced by 20 methylene groups. The unsaturated PPE (P2) shows a low Tg of ca. −84 °C and a melting endotherm at ca. 0 °C with a melting enthalpy of ΔHm = −31 J g−1. After hydrogenation (P2-H), the Tg remains rather unchanged (−80 °C) and the melting temperature is increased to ca. 47 °C (ΔHm = −59 J g−1), similar to previously reported PPEs.21,31 Interestingly, a second melting endotherm is detected after hydrogenation at ca. −1 °C with a very low ΔHm = −8 J g−1, indicating another crystalline structure, which may be due to side chain crystallinity (Fig. S18†). For the unsaturated PPAs P1 a different behavior is detected: the Tg is −72 °C higher than that for the corresponding PPE; in addition above Tg the polymer crystallizes at Tc = −40 °C, before the melting is detected at −8 °C. Also in the cooling curve of the P1-series, no recrystallization is observed under these conditions (10 °C min−1), leaving a completely amorphous polymer after cooling. In contrast, P2 recrystallizes during the same cooling procedure. The hydrogenated PPA (P1-H) exhibits a similar thermal behavior as its PPE-analog with a melting endotherm at ca. 51 °C with a similar melting enthalpy of ΔHm = −45 J g−1.
The solid state properties of P1-H and P2-H were analyzed by WAXS and SAXS measurements. The small angle X-ray scattering diagram (Fig. 1) shows a dominant peak at q = 0.2368 Å−1, corresponding to a long period of 2.7 nm. This peak is found in the wide angle X-ray scattering (WAXS) as well at 2Θ = 3.6°. Accordingly, the lamellar thickness of P1-H crystals corresponds to the length of the 20 CH2 groups in all-trans conformations. P1-H and P2-H were dissolved in hot n-octane and crystallized slowly during the cooling process from solution. Drop-cast TEM (cf. Fig. S19 & S20†) shows the resulting crystals for both polymers. The electron diffraction pattern (inset Fig. S19 and S20†)) indicates that the crystal packing is similar to the pseudohexagonal crystal phase of polyethylene.32 For P1-H a lattice spacing of 4.1 Å was measured from TEM diffraction and also the WAXS measurement yields a lattice spacing of 4.1 Å. Interestingly, the structural analogue of PPE, i.e.P2-H, behaves similarly in the bulk phase: the SAXS diagram (Fig. S21†) shows a major signal at q = 0.2808 Å−1, corresponding to a long period of 2.2 nm, similar to P1-H. The WAXS shows a small peak at 2Θ = 6.8° which corresponds to a spacing of 1.3 nm. The dominant peak in the WAXS data corresponds to a lattice spacing of 4.2 Å, indicating again the pseudo-hexagonal crystal structure. Fig. S20† shows a TEM micrograph of P2-H crystals with the corresponding diffraction pattern yielding a lattice spacing of 4.2 Å (compare above 4.1 Å for P1-H). From these structural examinations the P2-H crystallizes in a pseudo-hexagonal crystal structure with a lamellar long period of 2.2 nm as P1-H. As reported previously for other defect-PEs, the pendant groups are located outside the crystalline lamellae33 and thus for P1-H the amidate side chains are located outside the crystals. Due to the easy cleavage of P–N bonds (see below), this might be a handle for future tuning of crystal surfaces and currently studied in our department.
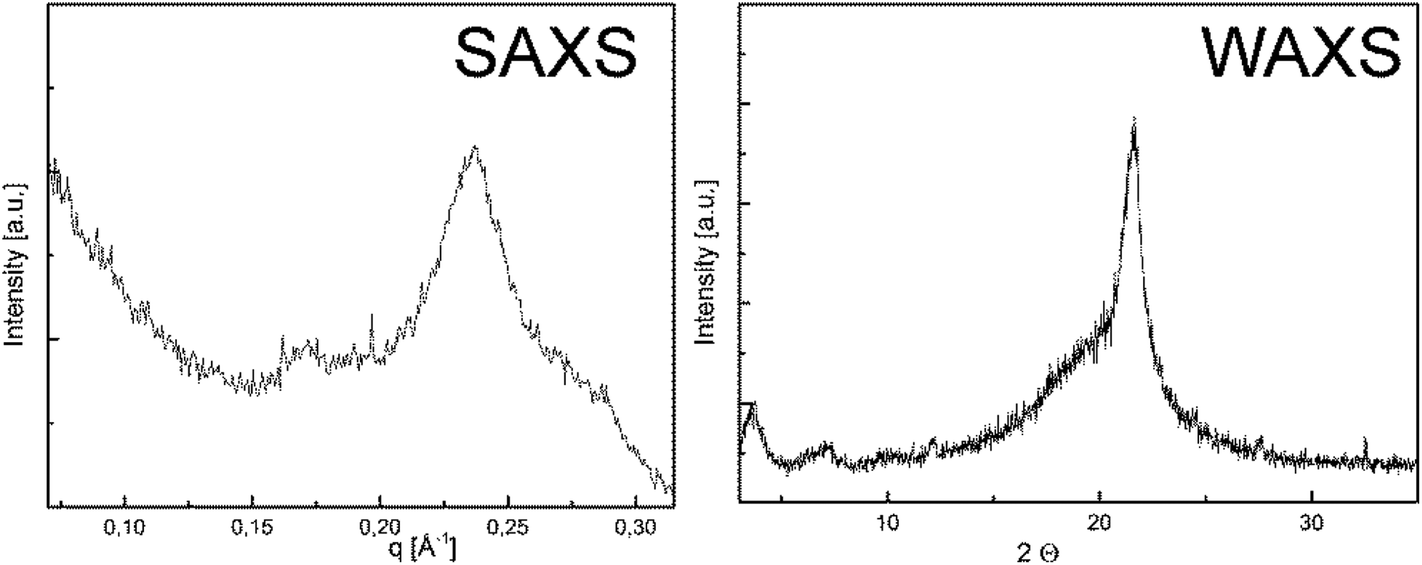 | ||
| Fig. 1 Small- and wide angle X-ray scattering of P1-H. Prior to the X-ray measurement the sample was annealed at 42 °C for 24 hours. | ||
The P–N-bond in phosphoramidates is known to be hydrolytically labile in contrast to the stronger P–O bond.13 Thus, PPAs can be used to release the pendant groups in an acidic environment; while at neutral pH PPAs are stable. This behavior makes PPAs advantageous for using in a low pH-triggered release of the side chain-bound material, e.g. for drug release.
The cleavability of the phosphoramidate bond with p-toluenesulfonic acid was investigated by 1H NMR (Fig. 2) and 31P NMR (Fig. S13†): the complete conversion of the phosphoramidate (9.78 ppm) into a phosphate group (0.40 ppm) was detected after 7 days. No scission of the main chain was observed under these conditions from the NMR spectra. It has to be noted that the polyphosphodiester exhibits low solubility in common solvents (either methanol/chloroform mixtures or basic conditions, e.g. pyridine, can be used), and thus SEC analysis was not possible. However, literature reports show that phosphodiesters have a very high stability under acidic conditions.34 This makes the pendant amidate also a potential protective group for the P–OH group, which allows easy handling due to higher solubility as the resulting polyphosphodiester.
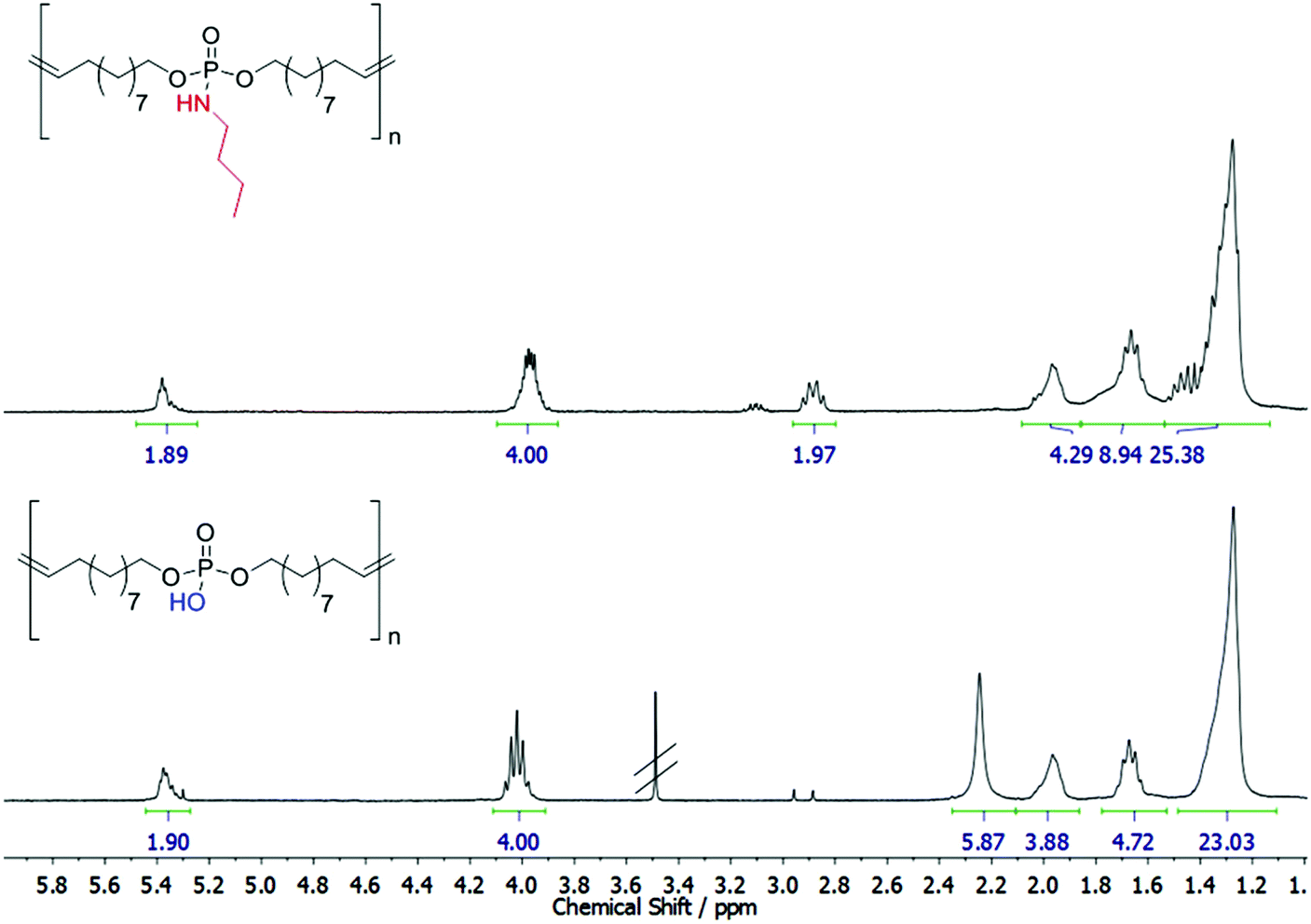 | ||
| Fig. 2 Degradation of P1 and preparation of a poly(phosphodiester). Top: 1H NMR of P1. Bottom: 1H NMR of degraded P1. | ||
Conclusions
In summary, this work presented the first synthesis of poly(phosphoramidate)s via acyclic diene metathesis polycondensation. In addition the comparison of structural analogues of saturated and unsaturated poly(phosphoester)s was performed. Distinct differences for both classes of materials have been identified based on their thermal behavior, thermal stability, and hydrolytic stability. Their crystallization behavior was found to be very similar and rather independent of the binding motif of the pendant chain. The pendant amidate was selectively hydrolyzed under acidic conditions, producing a poly(phosphodiester) with a very high stability towards acids. This selective degradation profile makes phosphoramidates a powerful protective group for P–OH groups, which in general exhibit strong hydrogen bonding and low solubility. These materials extend the class of degradable P-containing polymers that may find applications ranging from fire retardant additives to biodegradable scaffolds for tissue engineering or drug delivery.Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (WU 750/5-1) for funding. The authors thank Prof. Dr Katharina Landfester (MPIP) for continuous support. We thank Dr Manfred Wagner (MPIP) for NMR measurements.References
- J. Wang, P. Zhang, H. Mao and K. Leong, Gene Ther., 2002, 9, 1254–1261 CrossRef CAS PubMed.
- S.-W. Huang and R.-X. Zhuo, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 340–348 CrossRef CAS.
- Y. Ikada and H. Tsuji, Macromol. Rapid Commun., 2000, 21, 117–132 CrossRef CAS.
- T. Steinbach and F. R. Wurm, Angew. Chem., Int. Ed., 2015, 54, 6098–6108 CrossRef CAS PubMed.
- Y. H. Lim, G. S. Heo, Y. H. Rezenom, S. Pollack, J. E. Raymond, M. Elsabahy and K. L. Wooley, Macromolecules, 2014, 47, 4634–4644 CrossRef CAS PubMed.
- Z. Zhao, J. Wang, H.-Q. Mao and K. W. Leong, Adv. Drug Delivery Rev., 2003, 55, 483–499 CrossRef CAS PubMed.
- F. Zhang, S. Zhang, S. F. Pollack, R. Li, A. M. Gonzalez, J. Fan, J. Zou, S. E. Leininger, A. Pavía-Sanders, R. Johnson, L. D. Nelson, J. E. Raymond, M. Elsabahy, D. M. P. Hughes, M. W. Lenox, T. P. Gustafson and K. L. Wooley, J. Am. Chem. Soc., 2015, 137, 2056–2066 CrossRef CAS PubMed.
- F. Zhang, J. A. Smolen, S. Zhang, R. Li, P. N. Shah, S. Cho, H. Wang, J. E. Raymond, C. L. Cannon and K. L. Wooley, Nanoscale, 2015, 7, 2265–2270 RSC.
- J. Wang, H.-Q. Mao and K. W. Leong, J. Am. Chem. Soc., 2001, 123, 9480–9481 CrossRef CAS PubMed.
- J.-J. Qiu, C.-M. Liu, F. Hu, X.-D. Guo and Q.-X. Zheng, J. Appl. Polym. Sci., 2006, 102, 3095–3101 CrossRef CAS.
- K. N. Bauer, H. T. Tee, I. Lieberwirth and F. R. Wurm, Macromolecules, 2016, 49, 3761–3768 CrossRef CAS.
- T. Wolf, T. Steinbach and F. R. Wurm, Macromolecules, 2015, 48, 3853–3863 CrossRef CAS.
- S. Zhang, H. Wang, Y. Shen, F. Zhang, K. Seetho, J. Zou, J.-S. A. Taylor, A. P. Dove and K. L. Wooley, Macromolecules, 2013, 46, 5141–5149 CrossRef CAS PubMed.
- J. Wang, S. J. Gao, P. C. Zhang, S. Wang, H. Q. Mao and K. W. Leong, Gene Ther., 2004, 11, 1001–1010 CrossRef CAS PubMed.
- K. Troev, I. Tsatcheva, N. Koseva, R. Georgieva and I. Gitsov, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1349–1363 CrossRef CAS.
- V. Mitova, N. Koseva and K. Troev, RSC Adv., 2014, 4, 64733–64736 RSC.
- X.-Q. Zhang, X.-L. Wang, S.-W. Huang, R.-X. Zhuo, Z.-L. Liu, H.-Q. Mao and K. W. Leong, Biomacromolecules, 2005, 6, 341–350 CrossRef CAS PubMed.
- X. Jiang, W. Qu, D. Pan, Y. Ren, J.-M. Williford, H. Cui, E. Luijten and H.-Q. Mao, Adv. Mater., 2013, 25, 227–232 CrossRef CAS PubMed.
- W. Batorewicz, Uniroyal Inc., US Pat., US3932565-A, 1976 Search PubMed.
- T. Steinbach, C. Wahlen and F. R. Wurm, Polym. Chem., 2015, 6, 1192–1202 RSC.
- T. Steinbach, E. M. Alexandrino, C. Wahlen, K. Landfester and F. R. Wurm, Macromolecules, 2014, 47, 4884–4893 CrossRef CAS.
- T. Steinbach, E. M. Alexandrino and F. R. Wurm, Polym. Chem., 2013, 4, 3800–3806 RSC.
- H. Mutlu, L. M. de Espinosa and M. A. R. Meier, Chem. Soc. Rev., 2011, 40, 1404–1445 RSC.
- S. A. Low and J. Kopeček, Adv. Drug Delivery Rev., 2012, 64, 1189–1204 CrossRef CAS PubMed.
- M. Neisius, S. Liang, H. Mispreuve and S. Gaan, Ind. Eng. Chem. Res., 2013, 52, 9752–9762 CrossRef CAS.
- A. Jerschow and N. Müller, J. Magn. Reson., Ser. A, 1996, 123, 222–225 CrossRef.
- W. Li, H. Chung, C. Daeffler, J. A. Johnson and R. H. Grubbs, Macromolecules, 2012, 45, 9595–9603 CrossRef CAS PubMed.
- M. Mazarin, S. Viel, B. Allard-Breton, A. Thévand and L. Charles, Anal. Chem., 2006, 78, 2758–2764 CrossRef CAS PubMed.
- T.-M. Nguyen, S. Chang, B. Condon, R. Slopek, E. Graves and M. Yoshioka-Tarver, Ind. Eng. Chem. Res., 2013, 52, 4715–4724 CrossRef CAS.
- S. Gaan, P. Rupper, V. Salimova, M. Heuberger, S. Rabe and F. Vogel, Polym. Degrad. Stab., 2009, 94, 1125–1134 CrossRef CAS.
- F. Marsico, M. Wagner, K. Landfester and F. R. Wurm, Macromolecules, 2012, 45, 8511–8518 CrossRef CAS.
- W. Qiu, J. Sworen, M. Pyda, E. Nowak-Pyda, A. Habenschuss, K. B. Wagener and B. Wunderlich, Macromolecules, 2006, 39, 204–217 CrossRef CAS.
- Y. R. Zheng, H. T. Tee, Y. Wei, X. L. Wu, M. Mezger, S. Yan, K. Landfester, K. Wagener, F. R. Wurm and I. Lieberwirth, Macromolecules, 2016, 49, 1321–1330 CrossRef CAS.
- A. J. Kirby and M. Younas, J. Chem. Soc. B, 1970, 510–513 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py00999a |
| This journal is © The Royal Society of Chemistry 2016 |