
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmphiphilic hydroxyalkyl cellulose derivatives for amorphous solid dispersion prepared by olefin cross-metathesis†
Yifan
Dong
ab,
Laura I.
Mosquera-Giraldo
c,
Jacob
Troutman
d,
Brittny
Skogstad
e,
Lynne S.
Taylor
c and
Kevin J.
Edgar
*af
aDepartment of Sustainable Biomaterials, Virginia Tech, Blacksburg, VA 24061, USA. E-mail: kjedgar@vt.edu
bDepartment of Chemistry, Virginia Tech, Blacksburg, VA 24061, USA
cDepartment of Industrial and Physical Pharmacy, College of Pharmacy, Purdue University, West Lafayette, IN 47907, USA
dDepartment of Chemistry and Physics, Wingate University, Wingate, NC 28174, USA
eDepartment of Chemical Engineering, Virginia Tech, Blacksburg, VA 24061, USA
fMacromolecules Innovation Institute, Virginia Tech, Blacksburg, VA 24061, USA
First published on 7th July 2016
Abstract
Olefin cross-metathesis (CM) has enabled design and synthesis of diverse, amphiphilic cellulose ether derivatives (e.g. of ethyl and methyl cellulose). In this paper, hydroxyalkyl cellulose was selected as a hydrophilic starting material, with the additional advantage that it has DS (OH) 3.0 that allows targeting of a full range of DS of selected functional groups. Hydroxypropyl cellulose (HPC) was first etherified with 5-bromopent-1-ene to attach olefin “handles” for metathesis, whereby control of molar ratios of sodium hydride and 5-bromopent-1-ene permits full DS control of appended olefin. These olefin-terminated HPC ethers then were subjected to CM with acrylic acid and different acrylates, followed by diimide hydrogenation to reduce the resulting α,β-unsaturation. NMR and FT-IR spectroscopies were useful tools for following reaction progress. One of the product carboxyl-functionalized HPC derivatives, designated HPC-Pen106-AA-H, showed high promise as a crystallization inhibitor of the antiviral drug telaprevir. Its nucleation-induction inhibitory ability was compared to those of commercial controls, HPC and HPMCAS. All three polymers were very effective for inhibiting telaprevir crystallization, increasing induction time up to 8-fold. HPC did not effectively prevent amorphous particle growth, whereas the carboxyl-containing HPC-Pen106-AA-H and HPMCAS were able to prevent formation of agglomerates of amorphous drugs.
Introduction
Cellulose is a renewable, natural polysaccharide, readily available from plant and bacterial sources in almost inexhaustible quantities, and is a polymeric material with fascinating structure and properties.1–4 Commercial cellulose esters (e.g., acetate, propionate, butyrate, succinate and phthalate) and ethers (e.g., methyl, ethyl, carboxymethyl, hydroxyethyl and hydroxypropyl) are important for applications including automobile coatings, cleaning products, building materials and drug delivery systems.2–6 Compared to cellulose esters, where the ester linkages are relatively hydrolytically unstable and tend to hydrolyze in alkaline or acidic environments, cellulose ether derivatives are very stable even under extreme pH conditions. Cellulose ethers are frequently used in aqueous systems, for example as rheology modifiers, as part of drug delivery systems, and in waterborne coatings.7,8Hydroxyalkyl celluloses (e.g. hydroxyethyl and hydroxypropyl cellulose) are a commercial cellulose ether family prepared by reaction of deprotonated cellulose hydroxyl groups with epoxides (e.g. ethylene oxide or propylene oxide).9 Ring-opening of the epoxide by the cellulose alkoxide creates a new terminal hydroxyl group which is more distant from the backbone, and thus is more reactive than hydroxyls directly appended to the anhydroglucose rings. As a result, the side chains of hydroxyalkyl cellulose are typically oligo(hydroxyalkyl) groups, and each anhydroglucose unit (AGU) has a degree of substitution (DS) (OH) of 3.0. This provides a high degree of hydroxyl groups for further modifications, due to the broad available approach angle to the OH functionality because it is distant from the main cellulose chain. However, to date the scope of commercially available hydroxyalkyl cellulose derivatives is narrow, primarily due to the harsh conditions used in traditional alkaline etherification procedures.9 Given rising demand for high performance sustainable biomaterials, more efficient and powerful methods to greatly widen the range of hydroxyalkyl cellulose derivatives are needed.
It is estimated that up to 80% of current drugs and/or drug candidates10 suffer from poor bioavailability due to poor aqueous solubility.11 Several approaches have been developed to enhance drug solubility and improve bioavailability. Amorphous solid dispersions (ASDs) represent some of the most efficient formulations for this purpose.12,13 By creating an amorphous blend of polymer and drug, solution concentrations of released drug can be greatly enhanced. The energy barrier of the heat of fusion of the drug is eliminated by ASD, thereby leading to supersaturated solutions upon drug release.6 In order for polymers to be designed as efficient ASD matrices, certain structural criteria have been elucidated:14 (1) a terminal carboxyl group can interact with drug molecules and also function as a pH trigger to promote drug release;14 (2) moderate hydrophobicity can promote good miscibility with hydrophobic drugs; (3) some hydrophilicity can allow the polymer to release the drug in an aqueous environment and to maintain high supersaturations in solution prior to permeation through the gastrointestinal epithelium, and (4) a sufficiently high glass transition temperature (Tg), at least 50 °C higher than any possible ambient temperature, can immobilize the drug, prevent recrystallization, and ensure that the polymer dispersion remains in the glassy state.6
Cellulose ω-carboxyesters are ASD matrix polymers that have been shown to be miscible and effective at inhibiting crystallization for a wide variety of hydrophobic drugs.5,6 However the synthetic process of preparing discrete cellulose derivatives with terminal carboxylic acid and hydroxyl groups can be problematic, forcing synthetic chemists to adopt protection–deprotection strategies.14 In addition, one of the main limitations of the ω-carboxyesters produced to date is their hydrophobicity that impedes drug release from their ASDs. Building ASD polymer candidates upon polymer backbones that are amphiphilic, yet have greater water affinity may be an effective approach for solving this dilemma. Commercial hydroxypropylmethyl cellulose acetate succinate (HPMCAS)13,15,16 and hydroxypropyl cellulose (HPC)17–19 have been widely utilized as drug delivery matrices, including for ASD. Design and synthesis of amphiphilic hydroxyalkyl cellulose derivatives may thus be valuable for drug delivery applications including ASD matrices, as well as for other amphiphilic polymer applications. Such polymers may be effective at inhibiting drug-crystallization and at the same time able to release the drug into solution.5,6,20
Olefin cross-metathesis (CM) is a mild, efficient carbon–carbon bond forming transformation that has been extensively used in both organic and polymer chemistry.21–28 In 2014, Meng et al.29 reported the first successful CM of polysaccharide derivatives (CM reactions of mono- and oligosaccharides have been reported previously)30 by attaching an ester group possessing a terminal olefin onto a cellulose ester backbone as a metathesis handle. CM with an excess of acrylic acid (AA) and a Hoveyda–Grubbs’ 2nd generation catalyst provided discreet and soluble products with carboxyl functionality. Recent work by the Edgar group has shown that CM of cellulose esters (e.g. cellulose acetate)31,32 and ethers (e.g. ethyl cellulose and methyl cellulose)33,34 with various acrylates can be achieved, and hydrogenation of these CM products can provide saturated cellulose ester and ether derivatives with promising performance as amphiphiles, including as drug crystallization inhibitors.34
In this research, we explore CM modification of hydroxypropyl cellulose (HPC) derivatives, anticipating synthesis of amphiphilic CM products that possess useful features because: (1) HPC is more hydrophilic than most other cellulose esters and ethers and thus has good aqueous solubility, which may enable effective drug release; (2) hydroxyl groups of the oligo(hydroxypropyl) branches are separated from the backbone, thus less sterically hindered and therefore more reactive than backbone hydroxyl groups; (3) DS (OH) is 3.0, where all three hydroxyl groups in each AGU should be available for etherification and attaching terminal olefin metathesis “handles”.35 We hypothesize that olefin CM of ω-unsaturated HPC ethers could provide amphiphilic cellulose derivatives that display enhanced performance as ASD matrices. This would provide a range of derivatives with fully controllable DS, thereby enabling illumination of structure–property relationships relevant to ASD and other applications. We propose a three-step synthetic strategy: (1) etherification with an ω-unsaturated alkyl halide, (2) olefin CM, and (3) hydrogenation. We also carry out preliminary drug crystallization induction time experiments using these new amphiphilic HPC derivatives.
Results and discussion
Successful olefin CM of cellulose ester29,31,32 and ether derivatives33,34 has encouraged polysaccharide chemists to further explore this mild, efficient and modular strategy. In our previous study of olefin CM of commercial ethyl cellulose with DS (Et) 2.58 (EC2.58), the DS (OH) was limited to 0.42. Consequently, both the starting material EC2.58 and its metathesis products were quite hydrophobic; and the desired polymer amphiphilicity, crucial for success as an ASD polymer-matrix, could not be achieved. In order to overcome this limitation, we employed two different strategies: firstly, use of the more hydrophilic commercial methyl cellulose MC1.82; and secondly a one-pot synthesis of lower DS(Et) pentene-terminated EC2.30C5.34 Nevertheless, the DS(OH) was still limited, restricting the DS of terminal olefins and CM functional groups such as carboxylic acid. The maximum DS (OH) when using MC1.82 was 1.18, and 0.70 for the EC2.30 polymer.In the current research, we explored the possibility of CM modification of derivatives of hydrophilic HPC, with its DS (OH) 3.0. The DS(hydroxypropyl, HP) and MS(HP) of the commercial HPC and its phenyl carbamate derivative were measured by 1H NMR spectroscopy, using methods described in detail in the ESI (Fig. S1 and S2†). The results are in good agreement with previous reports of DS (HP) 2.2, and MS (HP) 4.4 for HPC, determined by 1H NMR spectroscopy.36,37
HPC was first etherified with 5-bromopent-1-ene (Fig. 1), as we have found that a chain length of C5 provides effective reaction with different CM partners.33 Compared to cellulose esters or ethers, HPC has not only significantly higher DS(OH), but most of the hydroxyl groups are also more reactive towards SN2 displacement due to enhanced approach angles resulting from their greater separation from the cellulose backbone. We started with a reduced molar equivalent ratio of NaH catalyst (5 equiv./–OH) and of 5-bromopent-1-ene (2 equiv./–OH) compared to those used in earlier work with commercial EC2.58 (10 equiv./–OH for both NaH and 5-bromopent-1-ene), due to the anticipated higher OH reactivity.33 Etherification results are briefly summarized in Table 1. These lower reagent ratios provided the olefin-terminated HPC-Pentene (HPC-Pen) derivative with pent-4-enyl DS of 2.82, which corresponds to 94% conversion of all the available hydroxyl groups. By further reducing the molar equivalents of NaH and 5-bromopent-1-ene, we were also able to prepare olefin-terminated HPC-Pen derivatives with predictably lower DS (Pen) (i.e. DS 1.06 and 0.69).
 | ||
| Fig. 1 Three-step synthetic route for HPC derivatives by etherification, CM, and hydrogenation. | ||
| Starting material | NaH equiv./–OH | 5-Bromopent-1-ene equiv./–OH | DS (olefin) | Product | OH conv.% |
|---|---|---|---|---|---|
| HPC | 5 | 2 | 2.82 | HPC-Pen282 | 94 |
| 3 | 1 | 1.06 | HPC-Pen106 | 35 | |
| 2 | 0.5 | 0.69 | HPC-Pen069 | 23 |
With these olefin-terminated HPC derivatives in hand, we then investigated the CM reaction of HPC-Pen with acrylic acid (AA). Here we adopted methodology that previously was successful in completely suppressing undesired self-metathesis (SM; crosslinking) in favor of the desired CM:29,33 (1) reacting olefins of two Grubbs types, the terminal olefin appended to cellulose (Type I olefin) and the small molecule acrylic acid (less SM-reactive Type II olefin); (2) using an excess of the Type II olefin (acrylic acid) to suppress SM of the Type I olefin; and (3) applying Hoveyda–Grubb's 2nd generation catalyst (HG II), with its high selectivity for CM vs. SM, and high activity.38,39 A minimal amount of the radical scavenger 3,5-di-tert-butylhydroxytoluene (BHT) was also applied to prevent radical-induced crosslinking during the CM reaction.29Table 2 summarizes CM results with HPC-Pen106. Initially, 30 equiv. of AA, 5 mol% of HG II catalyst, and 2 hours of reaction provided 83% CM conversion, which was an encouraging start. Conversion % and E/Z ratio were calculated based on proton NMR integration of olefin hydrogens vs. backbone hydrogens, as described in detail in Fig. S3.† By increasing the amount of CM partner and catalyst (50 equiv. AA and 10 mol% HG II), we achieved the desired 100% CM conversion. With these satisfying results with AA in hand, we explored the scope of the reaction with regard to Type II CM partner olefin. CM reactions with different acrylates including methyl acrylate (MA), 2-hydroxyethyl acrylate (HEA) and tert-butyl acrylate (tBuA) were carried out under conditions similar to those employed for AA. CM with MA using 30 equiv. of the acrylate ester and 5 mol% catalyst afforded 90% CM conversion; with 50 equivalents of MA and 8 mol% HG II the CM reaction reached 100%. We were also gratified to observe that under similar conditions (50 equivalents of the CM partner and 8 mol% HG II for 3 hours), CM with HEA and tBuA both achieved 100% conversion. Meanwhile the yields of these CM reactions were high to quantitative (>88%), supporting the high efficiency of CM modification of these polysaccharide ethers, and its tolerance for various functional groups. All metathesis products were predominantly in the E configuration (E/Z ratio >11.0), in accordance with previous reports.29,33
| Starting material | CM partnera | Olefin ratio (mol equiv.) | Cat. load (mol%) | Conv.b (%) | Yield (%) | E/Z |
|---|---|---|---|---|---|---|
| a AA: acrylic acid, MA: methyl acrylate, HEA: 2-hydroxyethyl acrylate, tBuA: tert-butyl acrylate. b Analysis of CM conversion is based on the ratio of olefin peak integrations in 1H NMR spectra. c Not measured (measured only on complete conversion samples). | ||||||
| HPC-Pen106 |
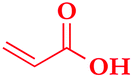
|
30 | 5 | 85 | 90 | NMc |
| AA | 50 | 10 | 100 | 92 | 14.3 | |

|
30 | 5 | 90 | 90 | NM | |
| MA | 50 | 8 | 100 | 88 | 11.0 | |
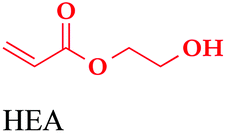
|
50 | 8 | 100 | 95 | 32.7 | |

|
50 | 8 | 100 | 93 | 14.3 | |
These α,β-unsaturated, ω-carboxy CM products are prone to H-atom abstraction from the γ-position, leading to radical reactions including radical coupling, which creates crosslinks. This chemistry, analogous to development of rancidity by natural unsaturated fatty acids, and similar to that observed by us and others29,33 for structurally related CM polysaccharide products, can be delayed by adding the radical scavenger BHT, and may be permanently addressed by hydrogenation to saturate the olefin.31,32,34 Therefore we performed homogeneous diimide hydrogenation to reduce the α,β-unsaturated CM products from HPC, thereby eliminating the propensity for radical-induced instability. Employing 6 equivalents of p-toluenesulfonyl hydrazide (pTSH) at reflux in DMF (135 °C) provided clean hydrogenation of all CM products with different functionalities, completed within 5 h. Similar CM and hydrogenation reactions were also performed for HPC-Pen282 and HPC-Pen069 with acrylic acid and various acrylates. For each combination of polymer and CM partner, 100% CM and complete hydrogenation were achieved under the conditions described above, providing a collection of saturated HPC derivatives with various functional groups (e.g. carboxylic acid and different esters) and controllable DS. Synthesis of HPC-Pen106-AA-H is used to exemplify the structural characterization methods used and results.
As shown in Fig. 2, the ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H stretch in the FT-IR spectrum at 3079 cm−1, resulting from the terminal olefin in the starting material HPC-Pen106, completely disappeared after CM. A distinct CO stretch from the carboxylic acid appeared at 1720 and 1697 cm−1, indicating successful CM and introduction of the carbonyl group. After hydrogenation, the CC stretch around 1654 cm−1 disappeared, leaving only the CO stretch at 1735 and 1710 cm−1, strongly supporting complete hydrogenation. 1H NMR spectra can be diagnostic and quantitative for successful CM and hydrogenation, simply by tracing the appearance, downfield migration with conjugation, and then disappearance upon saturation of the olefin peaks. As shown in Fig. 3, the two proton peaks from the terminal olefin in HPC-Pen106 at 4.96 and 5.82 ppm completely disappeared and two new peaks corresponding to the conjugated olefin protons emerged at 5.70 and 6.81 ppm. After hydrogenation, the olefinic peaks cleanly disappeared, indicating complete reduction of α,β-unsaturated HPC-Pen106-AA. 13C NMR spectra provided equally strong evidence of the proposed identity (Fig. 4). The starting material HPC-Pen106 has no carbonyl groups; after CM, a sharp and distinct signal at 167.1 ppm appeared, indicating successful introduction of the carbonyl group. Meanwhile the two carbon peaks at 114.8 and 138.5 ppm, assigned to the terminal olefin carbons in HPC-Pen106, also shifted downfield to 122.0 and 148.2 ppm respectively, with no evident residual carbon signals from the starting olefin, further supporting 100% CM and conjugation with the carbonyl groups. After hydrogenation, the carbonyl peak of the final product HPC-Pen106-AA-H shifted slightly downfield from 167.1 to 174.4 ppm, while the olefinic peaks disappeared completely, indicating quantitative hydrogenation.
C–H stretch in the FT-IR spectrum at 3079 cm−1, resulting from the terminal olefin in the starting material HPC-Pen106, completely disappeared after CM. A distinct CO stretch from the carboxylic acid appeared at 1720 and 1697 cm−1, indicating successful CM and introduction of the carbonyl group. After hydrogenation, the CC stretch around 1654 cm−1 disappeared, leaving only the CO stretch at 1735 and 1710 cm−1, strongly supporting complete hydrogenation. 1H NMR spectra can be diagnostic and quantitative for successful CM and hydrogenation, simply by tracing the appearance, downfield migration with conjugation, and then disappearance upon saturation of the olefin peaks. As shown in Fig. 3, the two proton peaks from the terminal olefin in HPC-Pen106 at 4.96 and 5.82 ppm completely disappeared and two new peaks corresponding to the conjugated olefin protons emerged at 5.70 and 6.81 ppm. After hydrogenation, the olefinic peaks cleanly disappeared, indicating complete reduction of α,β-unsaturated HPC-Pen106-AA. 13C NMR spectra provided equally strong evidence of the proposed identity (Fig. 4). The starting material HPC-Pen106 has no carbonyl groups; after CM, a sharp and distinct signal at 167.1 ppm appeared, indicating successful introduction of the carbonyl group. Meanwhile the two carbon peaks at 114.8 and 138.5 ppm, assigned to the terminal olefin carbons in HPC-Pen106, also shifted downfield to 122.0 and 148.2 ppm respectively, with no evident residual carbon signals from the starting olefin, further supporting 100% CM and conjugation with the carbonyl groups. After hydrogenation, the carbonyl peak of the final product HPC-Pen106-AA-H shifted slightly downfield from 167.1 to 174.4 ppm, while the olefinic peaks disappeared completely, indicating quantitative hydrogenation.
 | ||
| Fig. 2 FT-IR spectra of HPC-Pen106, the CM product with acrylic acid HPC-Pen106-AA, and the hydrogenated product HPC-Pen106-AA-H. | ||
 | ||
| Fig. 3 1H NMR spectra of HPC-Pen106, the CM product with acrylic acid HPC-Pen106-AA, and the hydrogenated product HPC-Pen106-AA-H. | ||
 | ||
| Fig. 4 13C NMR spectra of HPC-Pen106, the CM product with acrylic acid HPC-Pen106-AA, and the hydrogenated product HPC-Pen106-AA-H. | ||
We applied SEC to evaluate the molecular weight of the starting material and final product. As we can see from Fig. 5, the starting material HPC has a molecular weight about 112.0 kDa (DP = 268), which is close to the 100 kDa reported by the supplier. The HPC-Pen106 has much lower molecular weight (20.5 kDa) and DP decreased to 42, which is a result of the strongly alkaline conditions during the four-day etherification reaction. We expect based on our previous studies that the olefin metathesis and hydrogenation reactions would not cause significant change in DP. However, from the SEC results, the final product HPC-Pen106-AA-H has a much higher molecular weight (69.0 kDa) and increased DP (129). We also noticed that the SEC in THF system is providing a much lower dispersity (Đ) and dn/dc value for the carboxy-containing final product. We assume that the carboxyl groups at the ends of polymer branches are promoting some self-association in solution and this was also confirmed by dynamic light scattering (DLS) of the polymer under the same condition (0.1 mg mL−1, THF, sonication for 20 min). The polymer HPC-Pen106-AA-H was showing an average diameter as 240 nm and PDI about 0.28, suggesting polymer aggregation with a relatively uniform particle size rather than entering the column as single chains.
 | ||
| Fig. 5 SEC curves of HPC-Pen106 and the final product HPC-Pen106-AA-H after CM and hydrogenation. | ||
Useful ASD polymer matrices must have Tg values that are sufficiently high so that after mixing with drug, and even in very high ambient temperature and humidity conditions, the ASD will still remain in the glassy state, immobilizing the drug and helping to prevent crystallization. While cellulose derivatives tend to be high Tg polymers, the addition of multiple hydrophobic and hydrophilic substituents is certainly likely to result in some degree of internal plasticization, so it was important to see whether the products still had sufficiently high Tg values. In this study, modulated DSC (MDSC) was applied to determine product Tg values. Starting HPC comprises a large number of different monosaccharides, due to the many possible chain lengths of oligo(hydroxypropyl) group, the possibility of occasional nucleophilic attack at the more substituted end of the polysaccharide, and the different possible positions of substitution. For this reason the heterogeneous polymer has a very broad Tg, which can only be readily observed by MDSC, and a Tm around 205 °C, as reported by other investigators using either thermal or dynamic mechanical analysis.40,41 Glass transition temperatures of our CM products, which are synthesized from this heterogeneous polymer, would also be expected to be broad and indistinct, as was indeed observed; thus MDSC was required to measure them. As shown in Fig. 6, the olefin terminated HPC-Pen106 has a broad Tg around 91 °C, while the CM product with AA has a Tg around 96 °C. The hydrogenated product HPC-Pen106-AA-H has a very broad Tg around 94 °C, which is barely detectable. By analogy to our previous studies,31,34 the hydrogenated products would be expected to have a Tg value similar to that of the CM product prior to hydrogenation, due to their similar chemical structures. Overall, the final product HPC-Pen106-AA-H with its Tg above 90 °C shows promise for maintaining the polymer–drug dispersion in the glassy state, thereby preventing recrystallization.
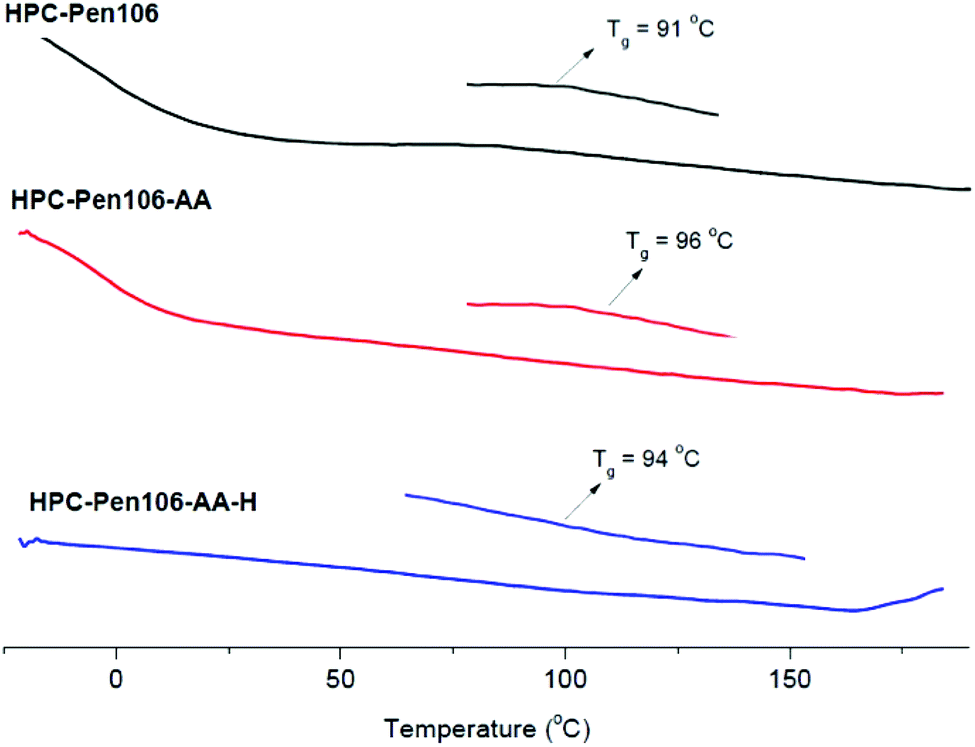 | ||
| Fig. 6 MDSC curves of HPC-Pen106 and the final product HPC-Pen106-AA-H after CM and hydrogenation. | ||
Crystallization induction experiments measure the time required for drug molecules to crystallize and achieve a detectable size of crystal nuclei. This is a first screening test to determine the effectiveness of polymers at inhibiting nucleation, an important requirement for effective ASDs. The experiments were conducted as previously described, using an in situ UV-vis spectrometer.34 The poorly water soluble (4.6 μg mL−1) antiviral drug telaprevir42,43 was chosen as the model drug compound. Supersaturated drug solutions corresponding to 150 μg mL−1 of telaprevir were prepared in pH 6.8 100 mM sodium phosphate buffer solution (with or without predissolved polymer). The concentration of drug in solution was monitored by UV-vis spectroscopy, and the time when the apparent solution concentration of the drug started to drop was selected as the induction time.
Telaprevir induction times were measured in the absence and presence of 5 μg mL−1 of HPMCAS, HPC and HPC-Pen106-AA-H. Fig. 7a shows an example of a concentration versus time profile for the diverse polymers. Fig. 7b summarizes average induction time values for each polymer. Our previous study34 showed the effectiveness of the relatively hydrophobic 5-carboxypentyl cellulose ether derivatives made from commercial EC, commercial MC, and lab-made EC-C5 (EC2.58C5-AA-H, MC1.82C5-AA-H and EC2.30C5-AA-H). The most efficient polymer, EC2.30C5-AA-H, delayed the crystallization induction times of telaprevir to more than 90 minutes. In the present study, the induction times for HPC and HPMCAS were measured as negative and positive controls, respectively, where carboxylic acid groups are present in HPMCAS and absent in HPC. Fig. 7a shows that in the absence of polymer or in the presence of HPC, there are two drops in the apparent concentration of telaprevir. The first drop in the apparent concentration is assigned to a change in the optical properties of amorphous aggregates due to agglomeration/particle growth, and the second drop is attributed to crystallization with a subsequent decrease of molecularly dissolved drug. We also observed similar results for systems containing HPMC, PVP and PVP-VA, which do not contain carboxylic acid groups.
 | ||
| Fig. 7 (a) Change in apparent concentration as a function of time for supersaturated solutions containing 5 μg ml−1 polymer and 150 μg ml−1 telaprevir. (b) Nucleation induction times in the absence and presence of different predissolved polymers (n = 3). | ||
Although the induction time results for HPC, HPMCAS and HPC-Pen106-AA-H are similar (Fig. 7b), the concentration versus time profiles suggest that the addition of substituents that include a terminal carboxylic acid helps to prevent the formation of agglomerates of amorphous drug. Addition of the 5-carboxypentyl groups to the HPC polymer (HPC-Pen106-AA-H) generates concentration versus time profiles more similar to HPMCAS than to HPC. These polymers allow an increase in the induction time of telaprevir to around 160 minutes, which is an 8-fold increase vs. that of the drug itself. We speculate that the excellent performance of these polymers is due to the moderate balance of hydrophobicity and hydrophilicity, and in the case of HPMCAS and HPC-Pen106-AA-H is also due to the pendent carboxyl groups.
Modification of commercial HPC by this CM synthetic method permits full control of DS of functional groups, including carboxylic acid. Organic solubility of ASD polymers is important not only because spray drying from organic solvents is an important method of making drug–polymer ASDs, but also because good organic solubility is a predictor of broad polymer–drug miscibility. Solubility properties of the CM products are summarized in Table 3, where they are compared with those of the important cellulosic ASD polymers HPC and HPMCAS. All HPC CM derivatives have some organic solubility, and those from moderate DS(pentenyl) substrate (DS 0.69 or 1.06) are soluble in all organic solvents tested. The water-soluble starting material HPC allows synthesis of more hydrophilic products compared to previously reported methyl and ethyl cellulose derivatives: aqueous solubility of HPC-Pen106-AA-H is 43.5 mg mL−1 (for comparison, HPMCAS solubility is 23.4 mg mL−1,44 while that of 5-carboxypentyl ethyl cellulose (EC2.30C5-COOH) is 1.0 mg mL−1 (ref. 34)), which is a very promising sign for fast dissolution of polymer/drug solid dispersions. In future work, we plan to study the impact of DS (carboxylic acid) on the crystallization behavior of different drugs to determine the optimal content of carboxylic acid groups for effective ASD performance, and performance in other applications.
| CH2Cl2 | CHCl3 | THF | Acetone | DMSO | DMF | H2O | |
|---|---|---|---|---|---|---|---|
| (+), Soluble; (−), insoluble; (P), partially soluble. | |||||||
| HPC | + | + | + | + | + | + | + |
| HPMCAS | + | + | + | + | + | + | − |
| HPC-Pen069-AA-H | + | + | + | + | + | + | P |
| HPC-Pen106-AA-H | + | + | + | + | + | + | P |
| HPC-Pen282-AA-H | − | − | − | − | + | + | + |
Given that a transition metal is being applied for the synthesis of HPC derivatives, we also did an initial ICP-MS test to check the ruthenium content in HPC-Pen106-AA-H as an exemplary and promising ASD polymer. We were pleased to find the residual ruthenium is only about 67 ppm (i.e. 67 μg per 1 gram polymer) compared to the U.S. Food and Drug Administration's limit for ruthenium oral consumption from pharmaceutical formulations of 100 μg per day. This is overall a quite low concentration of ruthenium even without any special measures to remove and/or recover the catalyst.45 Further studies on target ruthenium level and methods to minimize the level in the product will await completion of our structure–property studies to identify the optimum polymer structure for ASD.
Experimental
Materials
Hydroxypropyl cellulose (HPC, Mw ∼ 100 kg mol−1 as reported, MS and DS(HP) measured as described below) and potassium phosphate dibasic (K2HPO4) were purchased from Acros Organics. Sodium hydride (95%, NaH), 5-bromopent-1-ene, anhydrous tetrahydrofuran (THF), acrylic acid (AA), methyl acrylate (MA), 2-hydroxyethyl acrylate (HEA), tert-butyl acrylate (tBuA), 3,5-di-tert-butylhydroxytoluene (BHT), Hoveyda–Grubbs’ 2nd generation catalyst, and p-toluenesulfonyl hydrazide (pTSH) were from Sigma-Aldrich. Monopotassium phosphate (KH2PO4), dichloromethane (DCM), isopropanol, and N,N-dimethylformamide (DMF) were purchased from Fisher Scientific. Ethyl vinyl ether was purchased from Fluka Analytical. All reagents were used as received without further purification. Dialysis tubing (MWCO 3.5 kDa) was from Fisher Scientific. Telaprevir was purchased from Attix Pharmaceuticals.Measurements
1H NMR spectra were acquired on a Bruker Avance II spectrometer operating at 500 MHz or an Agilent MR4 operating at 400 MHz. Samples were analyzed as solutions in CDCl3 (δ 7.26) or DMSO-d6 (δ 2.50) (ca. 10 mg mL−1) at 25 °C in standard 5 mm o.d. tubes. Three drops of trifluoroacetic acid were added to shift the water peak in DMSO-d6 downfield from the spectral region of interest. 13C NMR spectra were acquired using a Bruker Avance II 500 MHz NMR with samples in solutions of CDCl3 or DMSO-d6 (ca. 40 mg mL−1) and with a minimum of 5000 scans. FT-IR spectra were obtained using a Nicolet 8700 instrument with 1 mg of each polymer and 99 mg of potassium bromide (KBr) matrix compressed into transparent pellets. Glass transition temperatures (Tg) were measured by a TA Instruments Discovery differential scanning calorimeter using modulated differential scanning calorimetry (MDSC). Each polymer sample (ca. 5 mg dry powder in a Tzero aluminum pan) was first equilibrated at −25 °C and then heated to 190 °C at 3 °C min−1 with a modulation amplitude of 0.5 °C and an oscillation period of 60 s. Molecular weights (Mw) were measured by size exclusion chromatography (SEC) in HPLC grade THF at 30 °C with a 1 mL min−1 flow rate, applying two connected Agilent PLgel Mixed-B columns with a Wyatt Dawn Heleos light scattering detector and a Wyatt Optilab Rex refractive index (RI) detector. The dn/dc values were obtained by assuming 100% mass elution from the columns and no calibration standards were used. Ruthenium concentration was determined using a Thermo Electron X-Series inductively coupled plasma mass spectrometer (ICP-MS) using Standard Method 3125-B.46 Polymer solubility was tested by adding 1 mL of solvent into a 4 mL vial with ∼10 mg sample. The mixture was subjected to vortex mixing for 10 min and then allowed to stand overnight at room temperature. Solubility was judged by visual examination.Determination of molar substitution (MS) and DS hydroxypropyl (HP)
DS(HP) of HPC was determined by 1H NMR spectroscopy in CDCl3 solution (ESI S1†) as 2.2. The MS of HPC was determined by the carbanilation of HPC with phenylisocyanate and subsequent 1H NMR analysis. The ratio of the integral of the internal HP methyl groups and that of the terminal methyl group (the one closest to the electron-withdrawing carbamate moiety, ESI S2†) enabled MS determination.36,37 The HPC used in this work had MS(HP) 4.4.Preparation of hydroxypropyl pent-4-enyl cellulose (HPC-Pen106)
HPC etherification was modeled on an alkyl cellulose etherification procedure from our previous paper.34 HPC (1.0 g, 2.40 mmol AGU, 7.19 mmol –OH) was dispersed in 15 mL of anhydrous THF using a mechanical stirrer and with a nitrogen purge. Polymer gradually dissolved to form a clear and transparent solution after overnight stirring. NaH (95%, 0.28 g, 21.6 mmol, 3 equiv./–OH) was added with N2 protection. The reaction system was cooled to 0 °C in an ice bath and then 5-bromopent-1-ene (1.13 g, 7.2 mmol, 1.0 equiv./–OH) was added dropwise under vigorous stirring. The solution was stirred at room temperature for 1 d and then at 50 °C for 3 d. After cooling to room temperature, isopropanol was gradually added to deactivate any residual catalyst. The solution was added to 300 mL of pH 7.4 buffer solution (3.54 g KH2PO4 and 7.14 g K2HPO4 in 1000 mL deionized water) to precipitate the product, which was then recovered by filtration. The precipitate was redissolved in THF, then reprecipitated into hexanes and recovered by filtration, before vacuum drying at 40 °C overnight. DS determined by 1H NMR: DS (HP) = 2.2, DS (Pen) = 1.06; yield: 1.03 g, 89%. 1H NMR (CDCl3): 1.12 (br, s, OCH2CHC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3OH), 1.63 (br, s, OCH2C2CH2CHCH2), 2.10 (br, s, OCH2CH2C2CHCH2), 2.78–4.58 (m, cellulose backbone, OC2CHCH3OH, OC2CH2CH2CHCH2), 4.88–5.07 (dd, OCH2CH2CH2CHC2), 5.82 (m, OCH2CH2CH2CCH2). 13C NMR (CDCl3): 17.6 (OCH2CH
3OH), 1.63 (br, s, OCH2C2CH2CHCH2), 2.10 (br, s, OCH2CH2C2CHCH2), 2.78–4.58 (m, cellulose backbone, OC2CHCH3OH, OC2CH2CH2CHCH2), 4.88–5.07 (dd, OCH2CH2CH2CHC2), 5.82 (m, OCH2CH2CH2CCH2). 13C NMR (CDCl3): 17.6 (OCH2CH![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3OH), 29.4 (OCH2H2CH2CHCH2), 30.4 (OCH2CH2H2CHCH2), 65.8 (C-6), 68.5 (OCH2HCH3OH), 73.4–77.4 (C-2, C-3, C-5, OH2CHCH3OH), 83.8 (C-4), 102.6 (C-1), 114.8 (OCH2CH2CH2CHH2), 138.5 (OCH2CH2CH2HCH2).
H3OH), 29.4 (OCH2H2CH2CHCH2), 30.4 (OCH2CH2H2CHCH2), 65.8 (C-6), 68.5 (OCH2HCH3OH), 73.4–77.4 (C-2, C-3, C-5, OH2CHCH3OH), 83.8 (C-4), 102.6 (C-1), 114.8 (OCH2CH2CH2CHH2), 138.5 (OCH2CH2CH2HCH2).
General procedure for olefin cross-metathesis reaction
The olefin CM reactions were performed according to a previously reported method.33,34 In brief, BHT (5 mg) and 3 mL of THF were added to a flask containing HPC-Pen106 (100 mg; 0.20 mmol AGU; 0.22 mmol terminal olefin). The mixture was stirred under N2 protection at 37 °C until all reagents were completely dissolved. Next, acrylic acid (0.82 g; 10.8 mmol; 50 equiv./terminal olefin) was added, followed by the Hoveyda–Grubbs’ 2nd generation catalyst (12.8 mg; 8 mol%, dissolved in 4 mL THF). After stirring at 37 °C for 2 h, the reaction was stopped by adding two drops of ethyl vinyl ether. The product solution was concentrated by evaporating the THF solvent with a nitrogen purge and was then added to water to precipitate the product, which was collected by vacuum filtration, then vacuum dried overnight at 40 °C. DS by 1H NMR (500 MHz, DMSO-d6): DS(HP) 2.2, DS (carboxylic acid) 1.06. Percent conversion by 1H NMR: 100%; yield: 108 mg, 92%. 1H NMR (DMSO-d6): 1.02 (br, s, OCH2CHC3OH), 1.58 (br, s, OCH2C2CH2CHCHCOOH), 2.16 (br, s, OCH2CH2C2CHCHCOOH), 2.70–4.14 (m, cellulose backbone, OC2CHCH3OH, OC2CH2CH2CHCHCOOH), 5.70 (d, OCH2CH2CH2CHCCOOH), 6.81 (m, OCH2CH2CH2CCHCOOH). 13C NMR (DMSO-d6): 17.3 (OCH2CHH3OH), 28.2 (OCH2H2CH2CHCHCOOH), 28.3 (OCH2CH2H2CHCHCOOH), 67.4 (C-6), 72.2–77.4 (OCH2HCH3OH, C-2, C-3, C-5, OH2CHCH3OH), 82.5 (C-4), 101.3 (C-1), 122.0 (OCH2CH2CH2CHHCOOH), 148.2 (OCH2CH2CH2HCHCOOH), 167.1 (OCH2CH2CH2CHCHOOH).
Hydrogenation of the CM product HPC-Pen106-AA by p-toluenesulfonyl hydrazide (pTSH)
BHT (8 mg; 10 wt%) and 4 mL DMF were added to a flask containing HPC-Pen106-AA (80 mg; 0.15 mmol AGU; 0.16 mmol olefin). The mixture was stirred under N2 protection at room temperature until all reagents were completely dissolved. Next, pTSH (0.18 g; 0.95 mmol; 6 equiv./olefin) was added. After refluxing at 135 °C for 5 h, the product was dialyzed against acetone for 3 d and then against water for 2 d, with the acetone and water being changed daily, and then freeze dried. 1H NMR (DMSO-d6): 0.86 (s, OCH2CHC3OH), 1.02 (s, OCH2CH2C2CH2CH2COOH), 1.26 (s, OCH2CHC3OCH2CH2CH2CH2CH2COOH), 1.45 (m, OCH2C2CH2C2CH2COOH), 2.17 (br, s, OCH2CH2CH2CH2C2COOH), 2.63–4.76 (cellulose backbone, OC2CHCH3OH, OC2CH2CH2CH2CH2COOH). 13C NMR (DMSO-d6): 17.4 (OCH2CHH3OH), 24.4 (OCH2CH2CH2H2CH2COOH), 25.4 (OCH2CH2H2CH2CH2COOH), 29.5 (OCH2H2CH2CH2CH2COOH), 33.7 (OCH2CH2CH2CH2H2COOH), 65.3 (C-6), 68.2 (OH2CH2CH2CH2CH2COOH), 72.1–76.5 (OH2HCH3OH, C-2, C-3, C-5), 82.9 (C-4), 101.6 (C-1), 174.4 (OCH2CH2CH2CH2CH2OOH). Yield: 55 mg, 69%.
Nucleation-induction time
The experimental nucleation time (Tind) is defined as the sum of the time for the critical nucleus formation (Tn) and the growth to a detectable size (Tg).47| Tind = Tn + Tg |
The crystallization induction time from unseeded-desupersaturation experiments was measured using an SI Photonics UV/vis spectrometer (Tucson, Arizona) coupled to a fiber optic probe (path length 5 mm). Polymers were dissolved in sodium phosphate buffer pH 6.8 100 mM and then sonicated until no undissolved material was observed. Supersaturated solutions were created by adding 150 μg mL−1 of a telaprevir methanolic stock solution (7 mg mL−1) to 47 mL buffer solution with predissolved polymer, held at 37 °C using a 50 mL jacketed flask coupled to a water bath, and 300 rpm. Measurements were recorded every 1 minute at two wavelengths: the maximum UV absorbance wavelength of telaprevir (270 nm) and a non-absorbing wavelength (370 nm) to account for changes in the scattering due to the presence of a second phase. The point at which the apparent concentration drops was assumed to be the induction time. The methodology has been described by Dong et al.34
Water solubility test
300 mg dried polymer was dispersed in 6 mL pH 6.8 potassium phosphate buffer and the suspension was stirred at room temperature for 24 h. After centrifuging at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 10 min, 1 mL of the supernatant liquid was transferred into a small vial and vacuum dried at 40 °C for 12 h to remove water. The weight of the dissolved polymer was calculated by subtracting the weight of salt (6.8 mg mL−1) in the buffer and concentration was determined by dividing the weight by volume (1 mL).
000g for 10 min, 1 mL of the supernatant liquid was transferred into a small vial and vacuum dried at 40 °C for 12 h to remove water. The weight of the dissolved polymer was calculated by subtracting the weight of salt (6.8 mg mL−1) in the buffer and concentration was determined by dividing the weight by volume (1 mL).
Ruthenium concentration measurement
Polymer (10 mg) was digested with 200 μL nitric acid and 2.8 mL deionized water, placed on a shaker at room temperature for three days. The solution was then diluted to 10 mL with deionized water and filtered before ICP-MS measurement. Ruthenium concentration was determined as 67 ppb in 10 mL solution, which is 0.67 μg per 10 mg polymer (67 ppm).Conclusions
We have demonstrated that olefin CM is an efficient method for synthesizing amphiphilic derivatives of hydroxypropyl cellulose. Etherification with 5-bromopent-1-ene created olefin-terminated HPC derivatives with controllable DS. CM with acrylic acid and various acrylates provided different functionalities and hydrogenation by pTSH afforded stable, saturated products. This is the first application of CM to hydroxyalkyl cellulose ethers; the starting material is significantly more hydrophilic than cellulose esters or ethers previously used in CM studies, which enables higher aqueous solubility of the final products than our previously reported cellulose ester/ether derivatives. The 5-carboxypentyl HPC derivative HPC-Pen106-AA-H has shown excellent ability to inhibit crystallization of telaprevir from supersaturated solution. This is a highly promising in vitro indication of future success for these polysaccharide derivatives in ASD and other drug delivery applications. Furthermore, a series of HPC-based derivatives with the same DP, different terminal functional groups, and controllable DS of functional group can be prepared through this procedure, which enables a valuable collection of derivatives for detailed structure–property relationship investigations. These versatile amphiphiles, efficiently prepared by a mild, tolerant, and rapid process, have substantial promise not only for drug delivery but for many other delivery applications and for other applications in which amphiphilicity is valuable.Acknowledgements
This research was supported in part by a Graduate Student Fellowship Award to LIMG from the American Association of Pharmaceutical Scientists. We would like to thank the AAPS for their support. We thank the National Science Foundation for funding this work through award DMR-1308276. We thank the Institute of Critical Technologies and Applied Science at Virginia Tech for facility support, and the Department of Chemistry and the Macromolecules Innovation Institute for educational and facility support. We also thank Scott Radzinski and Ami Jo of Virginia Tech for performing SEC and DLS analyses. We also thank Jeffrey Parks of Virginia Tech for ICP-MS testing.Notes and references
- K. Makuuchi, Radiat. Phys. Chem., 2010, 79, 267–271 CrossRef CAS.
- S. Kamel, N. Ali, K. Jahangir, S. M. Shah and A. A. El-Gendy, eXPRESS Polym. Lett., 2008, 2, 758–778 CrossRef CAS.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- M. Jonoobi, R. Oladi, Y. Davoudpour, K. Oksman, A. Dufresne, Y. Hamzeh and R. Davoodi, Cellulose, 2015, 22, 935–969 CrossRef CAS.
- G. A. Ilevbare, H. Liu, K. J. Edgar and L. S. Taylor, Mol. Pharmaceutics, 2013, 10, 2381–2393 CrossRef CAS PubMed.
- G. A. Ilevbare, H. Liu, K. J. Edgar and L. S. Taylor, Cryst. Growth Des., 2012, 13, 740–751 Search PubMed.
- L. Landoll, J. Polym. Sci., Polym. Chem., 1982, 20, 443–455 CrossRef CAS.
- J. Posey-Dowty, K. Seo, K. Walker and A. Wilson, Surf. Coat. Int. PT B–C, 2002, 85, 203–208 CrossRef CAS.
- K. E. D, US3278521A, 1966 Search PubMed.
- A. Fahr and X. Liu, Expert Opin. Drug Delivery, 2007, 4, 403–416 CrossRef CAS PubMed.
- S. N. Pace, G. W. Pace, I. Parikh and A. K. Mishra, Pharm. Technol., 1999, 23, 116–134 CAS.
- Y. Bao, J. Ma and N. Li, Carbohydr. Polym., 2011, 84, 76–82 CrossRef CAS.
- W. K. Miller, D. K. Lyon, D. T. Friesen, W. B. Caldwell, D. T. Vodak and D. E. Dobry, Hydroxypropyl methyl cellulose acetate succinate with enhanced acetate and succinate substitution, U.S. Patent, 0102691, 2013 Search PubMed.
- H. Liu, G. A. Ilevbare, B. P. Cherniawski, E. T. Ritchie, L. S. Taylor and K. J. Edgar, Carbohydr. Polym., 2014, 100, 116–125 CrossRef CAS PubMed.
- C. L. Li, L. G. Martini, J. L. Ford and M. Roberts, J. Pharm. Pharmacol., 2005, 57, 533–546 CrossRef CAS PubMed.
- D. T. Friesen, R. Shanker, M. Crew, D. T. Smithey, W. Curatolo and J. Nightingale, Mol. Pharmaceutics, 2008, 5, 1003–1019 CrossRef CAS PubMed.
- A. L. Sarode, S. A. Malekar, C. Cote and D. R. Worthen, Carbohydr. Polym., 2014, 112, 512–519 CrossRef CAS PubMed.
- H. Häbel, H. Andersson, A. Olsson, E. Olsson, A. Larsson and A. Särkkä, J. Controlled Release, 2016, 222, 151–158 CrossRef PubMed.
- M. Pozzoli, P. Rogueda, B. Zhu, T. Smith, P. M. Young, D. Traini and F. Sonvico, Drug Dev. Ind. Pharm., 2016, 1–9 CrossRef PubMed.
- T. Vasconcelos, B. Sarmento and P. Costa, Drug Discovery Today, 2007, 12, 1068–1075 CrossRef CAS PubMed.
- A. K. Chatterjee, T. L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360–11370 CrossRef CAS PubMed.
- R. H. Grubbs, Tetrahedron, 2004, 60, 7117–7140 CrossRef CAS.
- O. Brummer, A. Ruckert and S. Blechert, Chem. – Eur. J., 1997, 3, 441–446 CrossRef CAS.
- J. J. Van Veldhuizen, S. B. Garber, J. S. Kingsbury and A. H. Hoveyda, J. Am. Chem. Soc., 2002, 124, 4954–4955 CrossRef CAS PubMed.
- S. J. Connon and S. Blechert, Angew. Chem., Int. Ed., 2003, 42, 1900–1923 CrossRef CAS PubMed.
- S. J. Meek, R. V. O'Brien, J. Llaveria, R. R. Schrock and A. H. Hoveyda, Nature, 2011, 471, 461–466 CrossRef CAS PubMed.
- R. Smith, S. Boothroyd, R. Thompson and E. Khosravi, Green Chem., 2016, 18, 3448–3455 RSC.
- L. Fournier, C. Robert, S. Pourchet, A. Gonzalez, L. Williams, J. Prunet and C. M. Thomas, Polym. Chem., 2016, 7, 3700–3704 RSC.
- X. Meng, J. B. Matson and K. J. Edgar, Biomacromolecules, 2014, 15, 177–187 CrossRef CAS PubMed.
- A. Aljarilla, J. C. Lopez and J. Plumet, Eur. J. Org. Chem., 2010, 6123–6143, DOI:10.1002/Ejoc.201000570.
- X. Meng, J. B. Matson and K. J. Edgar, Polym. Chem., 2014, 5, 7021–7033 RSC.
- X. Meng and K. J. Edgar, Carbohydr. Polym., 2015, 565–573 CrossRef PubMed.
- Y. Dong and K. J. Edgar, Polym. Chem., 2015, 6, 3816–3827 RSC.
- Y. Dong, L. I. Mosquera-Giraldo, L. S. Taylor and K. J. Edgar, Biomacromolecules, 2015, 17, 454–465 CrossRef PubMed.
- M. Donbrow and Y. Samuelov, J. Pharm. Pharmacol., 1980, 32, 463–470 CrossRef CAS PubMed.
- C. J. Malm, L. Tanghe, B. C. Laird and G. D. Smith, Anal. Chem., 1954, 26, 188–190 CrossRef CAS.
- F. F. Ho, R. R. Kohler and G. A. Ward, Anal. Chem., 1972, 44, 178–181 CrossRef.
- A. K. Chatterjee, J. P. Morgan, M. Scholl and R. H. Grubbs, J. Am. Chem. Soc., 2000, 122, 3783–3784 CrossRef CAS.
- S. Gessler, S. Randl and S. Blechert, Tetrahedron Lett., 2000, 41, 9973–9976 CrossRef CAS.
- T. T. Kararli, J. B. Hurlbut and T. E. Needham, J. Pharm. Sci., 1990, 79, 845–848 CrossRef CAS PubMed.
- J. Siepmann and N. Peppas, Adv. Drug Delivery Rev., 2012, 64, 163–174 CrossRef.
- A. D. Kwong, R. S. Kauffman, P. Hurter and P. Mueller, Nat. Biotechnol., 2011, 29, 993–1003 CrossRef CAS PubMed.
- L. I. Mosquera-Giraldo and L. S. Taylor, Mol. Pharmaceutics, 2015, 12, 496–503 CrossRef CAS PubMed.
- B. Li, K. Harich, L. Wegiel, L. S. Taylor and K. J. Edgar, Carbohydr. Polym., 2013, 92, 1443–1450 CrossRef CAS PubMed.
- G. C. Vougioukalakis, Chem. – Eur. J., 2012, 18, 8868–8880 CrossRef CAS PubMed.
- APHA, AWWA, and WEF, Standard Methods for Examination of Water and Wastewater, APHA, AWWA, and WEF (American Public Health Association, American Water Works Association, and Water Environment Federation), Washington, D.C., 20th edn, 1998 Search PubMed.
- O. Söhnel and J. W. Mullin, J. Colloid Interface Sci., 1988, 123, 43–50 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py00960c |
| This journal is © The Royal Society of Chemistry 2016 |