
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo in one: use of azide functionality for controlled photo-crosslinking and click-modification of polymer microspheres†
Marco
Albuszis
a,
Peter J.
Roth
*b,
Werner
Pauer
*a and
Hans-Ulrich
Moritz
*a
aInstitute for Technical and Macromolecular Chemistry, University of Hamburg, Bundesstraße 45, 20146 Hamburg, Germany. E-mail: pauer@chemie.uni-hamburg.de; moritzhu@chemie.uni-hamburg.de
bDepartment of Chemistry, Faculty of Engineering and Physical Sciences, University of Surrey, Guildford, Surrey GU2 7XH, UK. E-mail: p.roth@surrey.ac.uk
First published on 11th August 2016
Abstract
Spherical, micrometer-sized, azide-functional particles were produced through dispersion copolymerization of styrene and vinylbenzyl azide (VBA, 1–100 wt% of monomer feed) in ethanol in the presence of stabilizers. The obtained microspheres were characterized by SEM, disc centrifuge, FT-IR and NMR spectroscopy, elemental analysis, DSC, and TGA, had measured azide loadings of up to 5.58 mmol g−1, and average diameters that decreased with increasing azide content from 2.8 to 0.8 μm. Microspheres were irradiated at a wavelength of 254 nm resulting in crosslinking based on azide-to-nitrene decomposition and subsequent C–H insertion and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C addition reactions. The conversion of azide functionality was monitored by FT-IR spectroscopy, elemental analysis, and DSC and was found to roughly follow first-order kinetics with increased rates found for microspheres with lower azide contents. Photo-crosslinking preserved shapes and size distributions and, above a crosslinking degree of 10%, prevented microsphere dissolution in good solvents. By controlling the irradiation time, the amount of azide consumed for photo-crosslinking could be precisely adjusted. Residual azide groups spared during the irradiation were shown to be amenable to highly efficient CuAAC click modification with a fluorescent dye, Rhodamine B propargyl ester. Given the demand for functional crosslinked microspheres and the inherent difficulties associated with common synthetic strategies in producing such materials, this methodology based on two orthogonal chemistries of the azide functionality provides simple access to well-defined microspheres with customizable degrees of crosslinking and functional group densities.
C addition reactions. The conversion of azide functionality was monitored by FT-IR spectroscopy, elemental analysis, and DSC and was found to roughly follow first-order kinetics with increased rates found for microspheres with lower azide contents. Photo-crosslinking preserved shapes and size distributions and, above a crosslinking degree of 10%, prevented microsphere dissolution in good solvents. By controlling the irradiation time, the amount of azide consumed for photo-crosslinking could be precisely adjusted. Residual azide groups spared during the irradiation were shown to be amenable to highly efficient CuAAC click modification with a fluorescent dye, Rhodamine B propargyl ester. Given the demand for functional crosslinked microspheres and the inherent difficulties associated with common synthetic strategies in producing such materials, this methodology based on two orthogonal chemistries of the azide functionality provides simple access to well-defined microspheres with customizable degrees of crosslinking and functional group densities.
Introduction
Spherical, polymer-based, micrometer-sized particles (microspheres) have many applications in materials science and technology including affinity bioseparation,1 liquid chromatography support materials,2,3 drug and enzyme carriers for medical applications,4 model systems to study rheology and crystallization of colloidal materials,5–7 and absorbents.8 Apart from size, type of polymer, and porosity, additional parameters that are crucial for applicability are crosslinking (which improves mechanical stability and prevents disintegration in solvents for the base polymer)6,9–11 and chemical surface functionality (which improves compatibility with solvents/host materials and is essential for immobilizing labels or payloads).12A range of protocols for microsphere synthesis is available and includes the classical dispersion, emulsion, and suspension techniques,13–15 polymerization-induced self-assembly (PISA),16–18 as well as multistep seed–swelling procedures.19–22 Most of these methods, however, are not well suited for the direct synthesis of crosslinked and functionalized microspheres. Dispersion polymerization, during which a homogeneous solution of monomer, initiator, and stabilizers produces insoluble microspheres, is a well-established and simple method for the preparation of micrometer-sized, non-porous, monodisperse particles.23–29 However, this method has limitations in the preparation of crosslinked particles, with the presence of crosslinkers during the nucleation step typically resulting in non-spherical or coagulated particles while step-wise addition of crosslinkers after nucleation allows only for relatively low crosslinking densities.11,30–34 Other methods, seed–swelling suspension polymerizations, for example, offer excellent control over the crosslinking density (formulations with 100% crosslinkers are possible),19–22 but the incorporation of functional or reactive comonomers can result in the formation of irregular, hollow, or collapsed particles.35 Crosslinking and/or chemical modification of microspheres are thus often performed in additional post-polymerization modification steps.12,26,36–42
An expedient functional group for such post-modification is the azide functionality. Azide-functional particles (whether produced through dispersion polymerization or other methods) are gaining increased attention35,43–46 due to the versatility and scope of the Cu-catalyzed alkyne–azide cycloaddition (CuAAC) click reaction.47 It is worth noting that the azide functionality is usually installed into pre-made particles (typically through substitution with sodium azide on chloromethyl groups) and that this reaction often suffers from poor conversions, due, in part, to solubility limitations. Only very few published reports have used a direct copolymerization of azide-functional comonomers (such as 4-vinylbenzyl azide) for the preparation of azide-functional microspheres and, to the best of our knowledge, dispersion polymerization based on 4-vinylbenzyl azide has not yet been described.35,46,48
The azide functionality also allows for post-synthesis photo-crosslinking,42,49 which has been demonstrated on soluble polymers50 and polymer films.51,52 This reaction is based on the decomposition of the azide moiety upon UV-irradiation, loss of a nitrogen molecule, and formation of a nitrene intermediate. Being highly reactive, the nitrene species can undergo several reactions with the surrounding material (often a combination of reaction pathways is found) predominantly through C–H insertion and CC addition reactions, which result in crosslinks. To the best of our knowledge, the azide photosensitivity has not yet been used for the crosslinking of polymer microspheres prepared through heterogeneous polymerization methods.
The purpose of the present study is twofold. Firstly, successful dispersion copolymerization of 4-vinylbenzyl azide (VBA) with styrene in ratios ranging from 1–100 wt% VBA is demonstrated to produce microspheres with a tunable azide content. Compared to our previous work in which azide functionality was installed into microspheres through postpolymerization modification of residual vinyl groups of divinylbenzene-based particles43 or through polymerization of VBA inside PS template spheres,35 the current method leads to much higher measured azide loadings of up to 5.58 mmol g−1. It is further shown that this direct copolymerization is favorable over the post-synthesis installation of azide into 4-vinylbenzyl chloride comonomers, because the chloromethyl groups were found to be susceptible to unwanted nucleophilic substitution by the solvent ethanol resulting in a loss of reactive groups during the polymerization step. Secondly, photo-crosslinking of azide-functional microspheres is demonstrated to yield well-defined particles that can be dispersed without disintegration in good solvents. Importantly, we found that the amount of azide groups consumed for photo-crosslinking can be precisely tuned through controlling the irradiation time. As a result, the crosslinking density and the amount of residual azide groups can be controlled. This residual azide functionality is demonstrated to be amenable to chemical modification through a near-quantitative CuAAC click modification with an alkyne-functional fluorescent dye, Rhodamine B. This study emphasizes how the azide functionality, as a single functional group, can be installed, without loss, into well-defined microspheres and can be exploited for both a controllable degree of crosslinking and for surface modification, providing simple access to well-defined functional materials expected to facilitate many of the aforementioned applications.
Experimental section
Materials
Unless otherwise noted, chemicals were purchased from Sigma Aldrich, Merck Chemicals, or Acros Organics in analytical grade and were used without further purification. Styrene (S) was distilled in vacuum to remove inhibitors and impurities directly before use. 2,2′-Azobis-(2-isobutyronitrile) (AIBN) was recrystallized from methanol. 4-Vinylbenzyl chloride was of technical grade (90%) and distilled in vacuum directly before use. Rhodamine B propargyl ester and 4-vinylbenzyl azide (VBA) were prepared as published elsewhere.35Dispersion polymerization synthesis of poly(styrene-co-4-vinylbenzyl azide), P(S-co-VBA) microspheres
Reactive, azide containing microspheres were produced following a typical literature procedure.43 Briefly, reactions were performed in 3-necked 250 ml double walled glass reactors connected to a thermostat and placed on a shaking plate without additional stirrers. A typical polymerization procedure is given as follows: solvent ethanol (135 g) and stabilizers polyvinylpyrrolidone (PVP-K30, Mn = 40 kg mol−1, 1.5 g) and Aliquat 336 (0.6 g) were mixed in a flask overnight and transferred into a reactor followed by purging with argon for 30 minutes while shaking at 150 rpm. The temperature was increased to 70 °C. A monomer mixture (15 g total) consisting of AIBN (0.3 g), styrene, and 4-vinylbenzyl azide (in variable mass ratios) was purged with argon for 15 minutes in a separate flask and transferred into the preheated reactor. The polymerization was run for 24 h with shaking at 150 rpm. The mixture was cooled to room temperature and the microspheres were purified by repeated washing–centrifugation steps using ethanol, hot water, and ethanol again for redispersing before drying in vacuum. 1H NMR (CDCl3) δ/ppm = 7.00, 6.50 (bm, Ph); 4.15 (–CH2N3); 2.28–0.72 (bm, backbone). Molar copolymer compositions were determined by comparison of the integrals of the benzylic protons (2 H) of VBA to those of the phenylic side chains (5 H for S, 4 H for VBA) and the backbone (3 H). Results from FT-IR, SEM, DSC, TGA, and elemental analysis are presented below.Poly(styrene-co-4-vinylbenzyl chloride) microspheres were prepared in analogy by substituting VBA for 4-vinylbenzyl chloride.
Microsphere photo-crosslinking
Azide-functional microspheres (0.2 g) were suspended in water (200 mL) using sonication and stirred mechanically in a narrow glass cylinder. A Heraeus UV-2 laboratory reactor system consisting of a cooling tube and dip tube made of quartz equipped with a TQ 150 immersion emitter (output 150 W, emission maxima at 254, 313, and 366 nm) was dipped directly into the stirred dispersion. Irradiation was done at room temperature for a predetermined amount of time before particles were isolated by centrifugation and washing.Click modification of partially photo-crosslinked P(S-co-VBA) microspheres
Partially photo-crosslinked microspheres containing residual azide functionality (100 mg) were dispersed in a mixture of DMF (5 mL), N,N,N′,N′-tetramethylenethylendiamine (TMEDA), CuBr, and triethylamine (approx. 1 eq. each with regards to azide functionality) and the mixture was purged with argon for 5 min. Rhodamine B propargyl ester (3 eq. with regards to azide functionality) was added, the reaction flask sealed and stirred for 24 h at 50 °C. Modified microspheres were isolated by filtering and washing with DMF, water, ethanol, and acetone (25 mL each) until washings remained colorless, followed by drying in vacuum. Analytical data is presented and discussed in the main text.Characterization
Surface morphology and uniformity of particles were determined by scanning electron microscopy (SEM, LEO1525). Particles were dispersed in ethanol with sonication and spread onto an aluminum specimen stub. After drying, samples were coated with a thin layer of carbon in vacuum. Number-average particle diameters (dn) and diameter dispersities (Đd) were calculated by measuring at least 100 particles using image J software. The coefficient of variation (CV), weight- (dw) and number-average (dn) diameters were calculated using the following equations where ni is the number of particles of diameter di and N is the total number of particles.36,53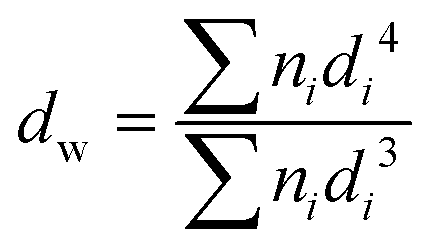

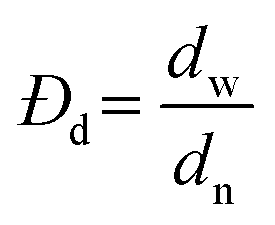

Chemical transformations of microspheres were monitored by FT-IR spectroscopy on an ATR-FT-IR spectrometer Nicolet iS10 (Thermo Fisher Scientific). Spectra were baseline corrected for easier interpretation.
Solution NMR spectra were measured on a Bruker Avance 400 spectrometer using CDCl3 as solvent and the internal solvent signal δ(CHCl3) = 7.26 ppm as reference. Solid state NMR experiments were performed on a Bruker Avance 400 spectrometer, equipped with a 4 mm double resonance probe. 13C cross polarization (CP) magic angle spinning (MAS) spectra were acquired using ramped polarization transfer at an operating frequency of 100.66 MHz. The experimental conditions were: 1H 90° pulse length 4.0 μs, contact time 2 ms, repetition delay 5 s, and spinning rate 14 kHz. Two-phase modulation (TPPM) decoupling was used during the acquisition. All the measurements were carried out at room temperature.
CHN elemental analysis was performed on EuroVector Euro EA and Elementar Vario EL III instruments. Combustion gases were detected by a gas chromatography (Euro EA) or a head conductivity detector (Vario EL III).
Differential scanning calorimetry (DSC), performed on a Mettler Toledo DSC 1 Star System, was used to determine glass transition temperatures (Tg) and the thermal behavior of the polymer samples. The heating rate was 10 °C min−1 and experiments were done under nitrogen atmosphere with a flow rate of 20 mL min−1.
Thermogravimetric analysis (TGA) of samples was performed on a Mettler Toledo TGA 1 Star System thermogravimeter with a heating rate of 10 °C min−1 in a nitrogen atmosphere with a flow rate of 20 mL min−1.
Results and discussion
This study comprised the synthesis of azide functional particles, their partial photo-crosslinking, and the click-modification of residual azide functionality, Scheme 1.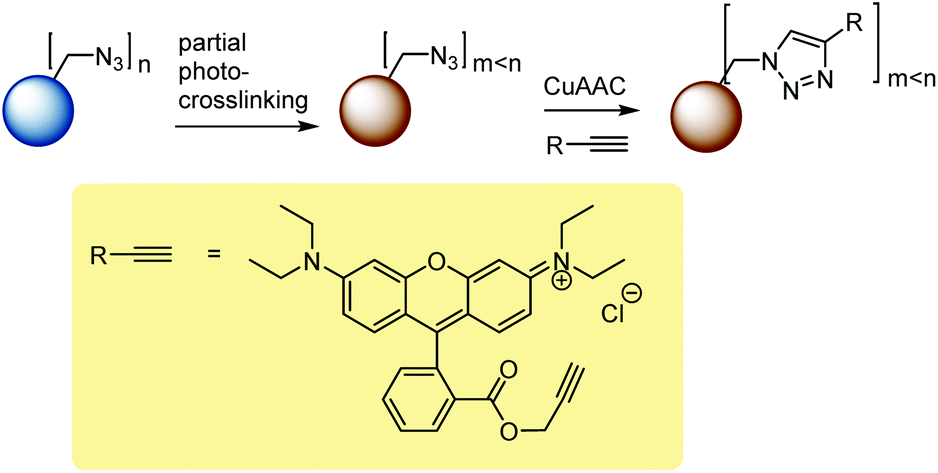 | ||
| Scheme 1 Reaction scheme. | ||
Microsphere synthesis
Styrene-based microspheres were prepared by dispersion polymerization using ethanol as solvent, AIBN as initiator, and PVP-K30 and Aliquat 336 as stabilizers in fixed mass ratios relative to monomers.35 As mentioned above, arguably the most common method to introduce azide functionality into microspheres is the postpolymerization reaction of vinylbenzyl chloride groups with sodium azide. Following this protocol, a series of chloride-functional microspheres was prepared through dispersion copolymerization of styrene (S) and 4-vinylbenzyl chloride (VBC) with S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VBC mass ratios ranging from 99:1 to 0:100. Uniform microspheres of 2–3 μm diameter were obtained as evidenced by SEM imaging (Fig. S1 in the ESI†). However, 1H NMR analysis run on CDCl3 solutions of the copolymers, Fig. S2,† suggested that up to 16% of chloromethyl functionality was lost to nucleophilic substitution with the solvent ethanol, as judged by the appearance of a broad signal at δ = 3.51 ppm attributed to the benzylic ethyl ether (Ar–CH2–OR) (see Scheme 2) and as confirmed through 2D NMR analysis (not shown).
:VBC mass ratios ranging from 99:1 to 0:100. Uniform microspheres of 2–3 μm diameter were obtained as evidenced by SEM imaging (Fig. S1 in the ESI†). However, 1H NMR analysis run on CDCl3 solutions of the copolymers, Fig. S2,† suggested that up to 16% of chloromethyl functionality was lost to nucleophilic substitution with the solvent ethanol, as judged by the appearance of a broad signal at δ = 3.51 ppm attributed to the benzylic ethyl ether (Ar–CH2–OR) (see Scheme 2) and as confirmed through 2D NMR analysis (not shown).
 | ||
| Scheme 2 (A) Side reaction through ethanolysis during the synthesis of chloromethyl-functional microspheres (which can serve as precursors for azide-functional microspheres) and (B) microsphere synthesis through dispersion polymerization of VBA and styrene. | ||
The observed percentage of chloride–ethanol substitution of 12–16 mol% did not depend strongly on the VBC feed content but was reduced to 9 mol% in a trial experiment run without Aliquat 336 (a quaternary ammonium bromide species) as stabilizer, suggesting that the bromide anion may facilitate the substitution reaction through formation of a bromide intermediate, Fig. S3.†
These results encouraged us to investigate the direct copolymerization of 4-vinylbenzyl azide (VBA) and styrene under the same experimental conditions (including the use of Aliquat 336 as stabilizer). Ten microsphere samples with S:VBA comonomer feed ratios between 99:1 and 0:100 wt% were prepared and characterized visually and by 1H solution NMR spectroscopy, scanning electron microscopy (SEM), CHN elemental analysis (EA), FT-IR spectroscopy, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC), see Scheme 2B and Table 1.
| Code | Feed | SEM analysis | CHN analysis | |||
|---|---|---|---|---|---|---|
| Styrene [wt%] | VBA [wt%] |
d
na [μm] |
Đ
db |
CVc [%] | Azided [mmol g−1] | |
| a Number-average particle diameter. b Dispersity of the particle diameter. c Coefficient of variation of the particle diameter. d Molar azide content calculated from elemental nitrogen analysis. | ||||||
| MS1 | 99 | 1 | 2.77 | 1.002 | 0.12 | |
| MS2 | 98 | 2 | 2.63 | 1.005 | 7.3 | 0.17 |
| MS5 | 95 | 5 | 2.59 | 1.004 | 6.4 | 0.36 |
| MS10 | 90 | 10 | 2.49 | 1.002 | 4.6 | 0.66 |
| MS20 | 80 | 20 | 2.12 | 1.07 | 23.1 | 1.31 |
| MS30 | 70 | 30 | 1.68 | 1.28 | 52.8 | 1.95 |
| MS40 | 60 | 40 | 1.96 | 1.16 | 40.45 | 2.55 |
| MS50 | 50 | 50 | 1.34 | 1.06 | 25.9 | 3.19 |
| MS75 | 25 | 75 | 1.28 | 1.08 | 29.07 | 4.26 |
| MS100 | 0 | 100 | 0.84 | 1.08 | 29.6 | 5.58 |
Dried microspheres presented as powders that were colorless (low VBA content) to yellow (higher VBA content), see Fig. S4.†
1H NMR spectroscopic analysis of microspheres dissolved in CDCl3, Fig. 1, showed the expected signals of the aromatic region (δ = 7.20–6.40 ppm) and backbone (1.90–1.10 ppm), and indicated the successful incorporation of VBA units through the appearance of a signal at 4.22 ppm, attributed to the Ar–CH2N3 resonance. Notably, 1H NMR spectra gave no evidence of side reactions with the solvent and the integration of the benzyl groups and comparison with the aromatic and backbone signals agreed well with the S:VBA comonomer feed ratios.
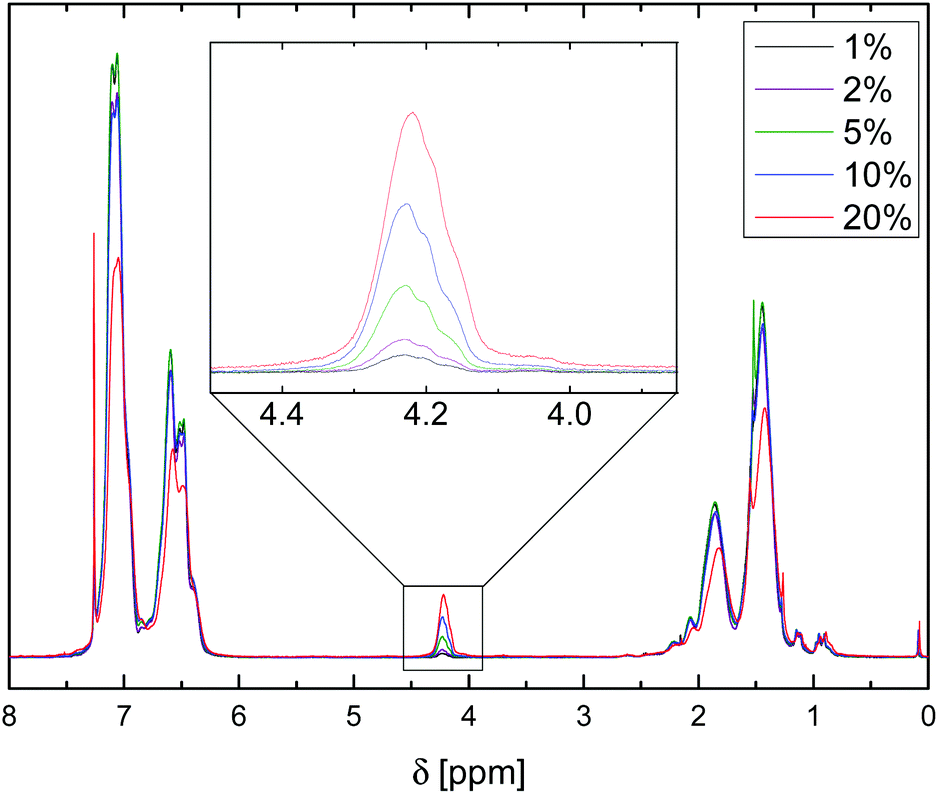 | ||
| Fig. 1 1H NMR spectra of dissolved P(S-co-VBA) microspheres with the signals associated with the azidomethyl groups enlarged for microsphere samples with varying VBA feed contents. | ||
SEM analysis, Fig. 2 and 3, showed spherical particles for all S–VBA compositions and typical diameters of approx. 2.7 μm. For VBA contents below 20 wt%, microspheres were highly regular with monomodal size distributions and size dispersities as low as 1.002 (Table 1). For VBA contents above 20 wt%, samples showed bimodal size distributions featuring large particles of approx. 2.8 μm diameter and a population of smaller spheres with diameters around 1 μm, see Fig. 4 for two representative size histograms. At very high VBA feed ratio, the average microsphere diameter dropped to 0.8 μm with the larger particles in sample MS100 measuring approx. 1.2 μm in diameter. A small degree of particle agglomeration was observed for samples of highest VBA content. Several factors have been shown to influence particle sizes and distributions and include stabiliser molecular weight, stabiliser concentration, and solvent polarity.54 In the present case, the observed sizes and size distributions are likely caused by the polar nature of VBA (compared to S) and a presumed incompatibility of the growing S-co-VBA oligomers with the ethanol–S–VBA solvent mixture which would lead to earlier precipitation and a larger number of smaller particles.27,28,54
 | ||
| Fig. 2 SEM images of microsphere samples (a) MS1; (b) MS5 (two images); and (c) MS10. | ||
 | ||
| Fig. 3 SEM images of microsphere samples (a) MS20; (b) MS40 and (c) MS100. | ||
 | ||
| Fig. 4 Size distribution histograms of (a) MS10 and (b) MS30. | ||
Disc centrifuge measurements, Fig. S5,† expectedly indicated narrow particle size distributions for samples with low to medium VBA contents. For higher VBA contents, however, results were inconclusive, presumably due to a slight crosslinking of polymers through thermal azide degradation and nitrene-based coupling reactions during polymerization.55 Indeed, solubility tests of samples with more than 50 wt% VBA in THF (a good solvent for the base copolymers) resulted in the swelling (rather than dissolution) of particles suggesting crosslinking. With 20–50 wt% VBA, microsphere samples dissolved partially with insoluble components forming a swollen gel. With less than 20 wt% of VBA, on the other hand, microspheres dissolved fully in THF and CDCl3. In order to estimate the amount of azide loss and degree of crosslinking formed during the polymerization with higher certainty than through 1H NMR analysis, elemental analysis was performed of all microsphere samples. Taking into account the molar masses of monomers (and the amount of nitrogen contributed by the cyanopropyl initiating group originating from AIBN), the measured nitrogen content was used to calculate microsphere azide loadings (Table 1) and the ratio of found-to-expected azide content. These values, plotted in Fig. 5, ranged between 0.93 and 1.00 corroborated an, albeit small, loss of azide functionality during polymerization for samples of higher VBA content, in agreement with previous studies on thermal azide degradation in soluble polymer samples.55 With regards to the synthesis of azide-functional microspheres, we recently described the preparation of porous microspheres through a templated copolymerization of S, VBA, and divinylbenzene inside PS seed particles, which lead to found-to-expected azide content ratios as low as 0.37 and below 0.70 for most samples. In spite of some loss (and associated slight crosslinking) observed in the present study, dispersion (co)polymerization of VBA presents a viable avenue toward microspheres with measured azide loadings as high as 5.58 mmol g−1 (see Table 1).
 | ||
| Fig. 5 Ratio of azide content found by elemental analysis of microspheres to expected azide content calculated from the feed ratio in dependence of VBA feed. | ||
As eventual photo-crosslinking of particles was intended, a low degree of crosslinking for the VBA-rich samples was not an impediment. It is, however, for sake of synthetic scope, briefly mentioned that we additionally performed dispersion polymerization syntheses of azide-rich particles (up to 100% VBA) at 30 °C using 2,2′-azobis(4-methoxy-2,4-dimethyl valeronitrile) (V-70) as low-temperature radical initiator under otherwise unchanged reaction conditions.56 All of these microspheres dissolved in THF and chloroform, indicating the successful synthesis of microspheres with very high azide loadings without any observed crosslinking.
FT-IR analysis of microspheres, Fig. S6,† showed the expected spectra of the P(S-co-VBA) base material including a strong band at 2095 cm−1 associated with the NNN asymmetric stretching and characteristic bands at ν/cm−1 = 1246, 1050, 878, 816 and 755 that varied with the comonomer feed ratio. The azide band served as an expedient means for the quantification of the azide content (see below).
Given this first dispersion-based synthesis of microspheres with very high azide contents, their potential in the materials science field, and the known thermal lability of the azide functionality, the thermal properties of all samples were characterized by DSC and TGA. DSC was used to determine glass transition temperatures, Tg, and the energy, E, released as exothermic heat flow during the thermal decomposition of the azide groups of all samples. A representative DSC curve, measured of sample MS50, is given in Fig. 6 (bottom curve) and values of Tg and E are plotted vs. VBA feed ratio in Fig. 7. The glass transition temperature showed a linear dependence on the S–VBA composition, decreasing from 102 °C found for sample MS1 (close to the literature value of 100 °C for homo-PS)57 to 66 °C measured for sample MS100, presumably due to increased sterical hindrance associated with the azidomethyl groups and less compact packing of polymer chains. The measured heat flow during azide decomposition varied in a linear fashion with azide content, increasing from 20 J g−1 for sample MS1 to 1299 J g−1 for sample MS100. Importantly, this data was used as a calibration and allowed for the estimation of azide contents in microspheres after partial crosslinking (see below). Above 350 °C, the onset of an endothermic peak indicated the decomposition of the polymer.
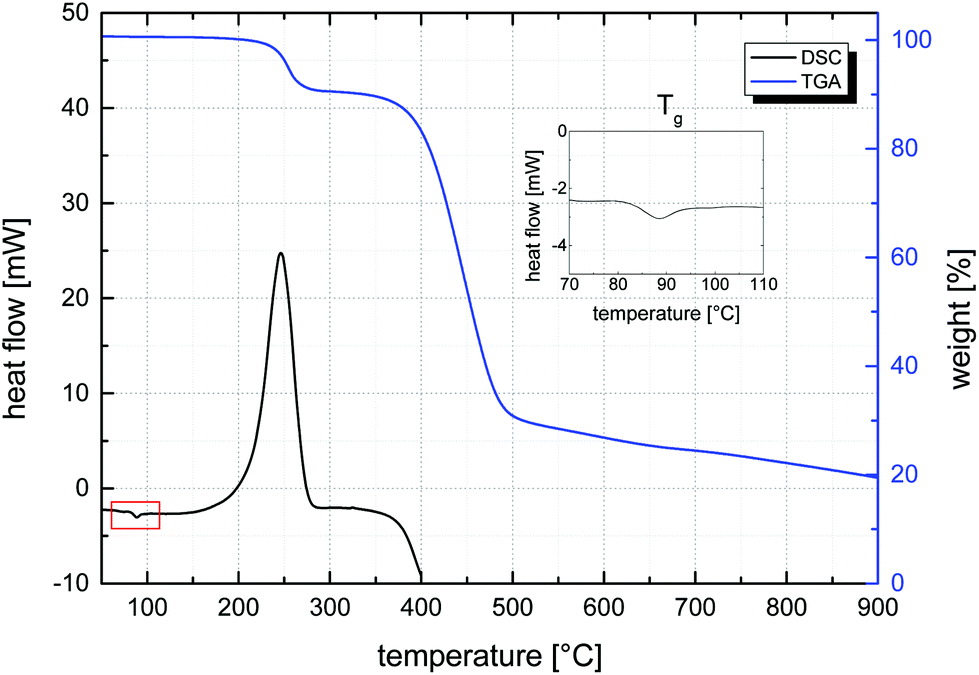 | ||
| Fig. 6 Representative DSC (bottom curve, left axis) and TGA (top curve, right axis) data of MS50 showing the glass transition temperature (DSC, inset), the exothermic azide decomposition around 250 °C, the polymer decomposition at 380–480 °C, and a residue of approx. 20 wt% at 900 °C (TGA). | ||
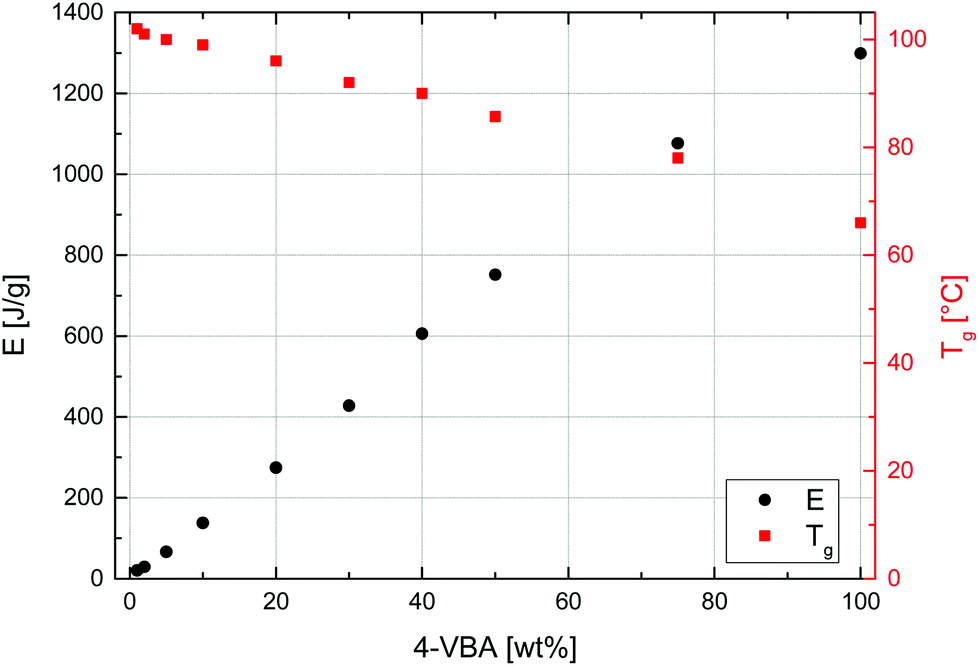 | ||
| Fig. 7 Glass transition temperatures (Tg, squares, right axis) and exothermic heat flow (E, circles, left axis) during azide decomposition measured by differential scanning calorimetry vs. VBA feed. | ||
A representative decomposition curve obtained by TGA is shown in Fig. 6 (top curve) and curves of all samples are shown in Fig. S7.† In agreement with DSC, the loss of the azide group was observed between 200–280 °C through a mass loss corresponding to approximately two thirds of the nitrogen content, in agreement with the loss of one N2 molecule per –N3 group. With higher azide contents, the temperature of highest azide decomposition rate decreased from around 270 °C to 240 °C (see ESI†), making high-concentration azide microspheres somewhat more susceptible to thermal crosslinking at temperatures above 200 °C. This high temperature range is in agreement with the only small degree of azide decomposition found during polymerization at 70 °C. A massive mass loss above 350 °C indicated the decomposition of the polymer material under nitrogen atmosphere. This polymer decomposition temperature increased with initial content of azide groups (which, at temperatures above 350 °C, were expected to have undergone crosslinking reactions). Interestingly and although the material was fully organic, a considerable percentage of mass that increased with azide content to over 25 wt%, remained at 900 °C (Fig. S7†), suggesting considerable thermal stability conferred by the nitrene-based crosslinking.
Controlled photo-crosslinking
With a set of well-defined azide-functional microspheres in hand, their crosslinking through controlled UV irradiation was studied next. Upon irradiation, the azide group sheds a molecule of nitrogen and forms a highly reactive nitrene group which can undergo several addition and insertion reactions with nearby molecules forming covalent crosslinks between different polymer chains. Main expected reaction pathways for the crosslinking of azide-functional microspheres are shown in Scheme 3.51 Notably, most of these reactions do not rely on dimerization (as do other common photo-crosslinkers) but offer universal attachment to nearby groups.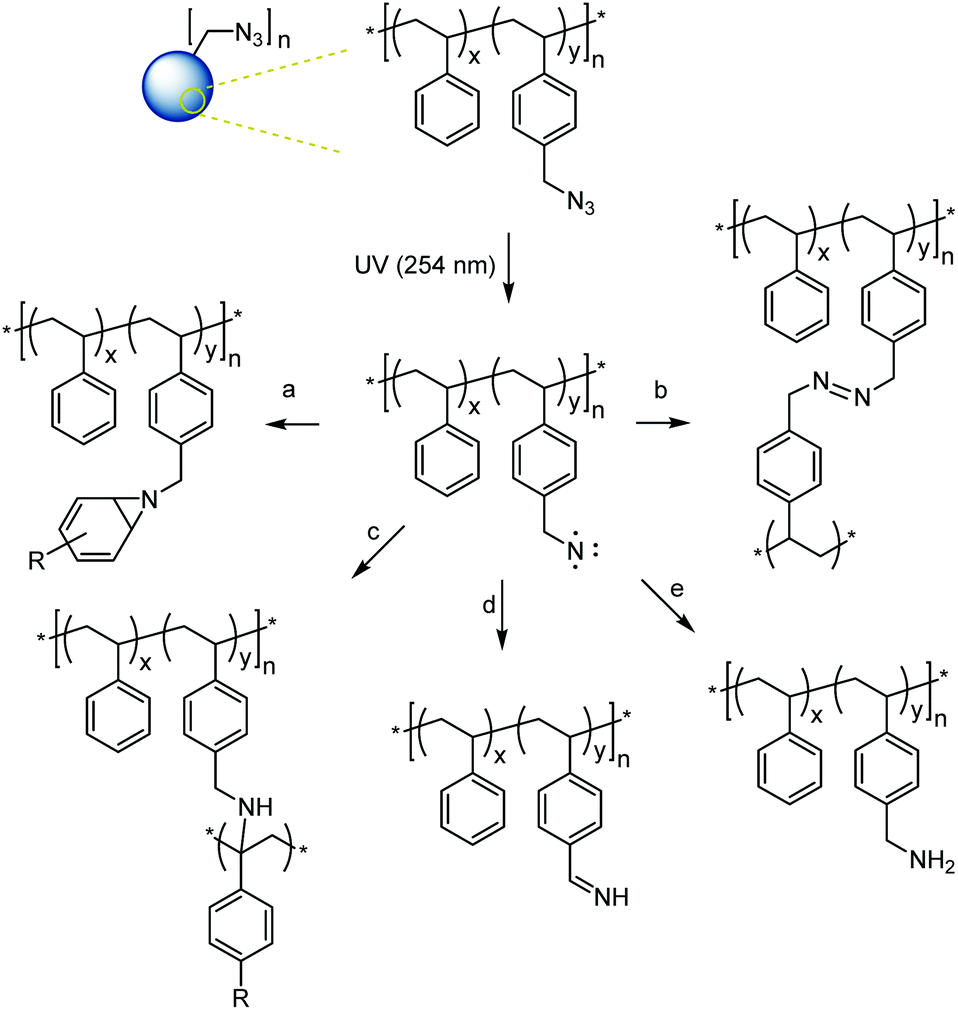 | ||
| Scheme 3 Possible reactions of the singlet nitrene formed after azide irradiation:51 (a) addition to double bond; (b) nitrene–nitrene coupling; and (c) C–H bond insertion (shown at benzylic position) leading to crosslinking and (d), (e) H abstractions not leading to crosslinking. | ||
A UV lamp emitting a wavelength of 254 nm, ideal for the formation of nitrenes from azides50 and designed for irradiation of liquid samples, was dipped directly into a stirred suspension of particles in water which was kept at room temperature through a cooling tube dipped into the solution. Irradiation times were varied between 30 s and 1 h and the conversion based on the amount of residual azide functionality was determined by three methods: (i) FT-IR spectroscopy, through integration of the azide vibrational band at 2095 cm−1 and calibrating the absorbance of samples before irradiation to 100% (Fig. S6†); (ii) elemental analysis by determining the nitrogen content and taking into account the original nitrogen content before irradiation and the expected remainder of a third of this mass after quantitative reaction (Scheme 3); (iii) DSC by measuring the exothermic heat flow during the thermal degradation of residual azide groups using the above data (Fig. 7) for calibration. As a representative example, the azide conversion for the irradiation of sample MS50 as determined by IR, EA, and DSC is shown in Fig. 8. Under the reactions conditions, a conversion of 50% of azide groups was achieved in 3 minutes, a conversion of approx. 95% was found after 20 min of irradiation, and essentially complete reaction was confirmed after 60 min. Importantly, the data from all three analytical methods was in excellent agreement, confirming the reliability of each technique, which was also apparent through repeated experiments. Additionally, this agreement also indicated that the penetration depth of the UV light during irradiation was sufficient to perform reactions inside the microspheres.
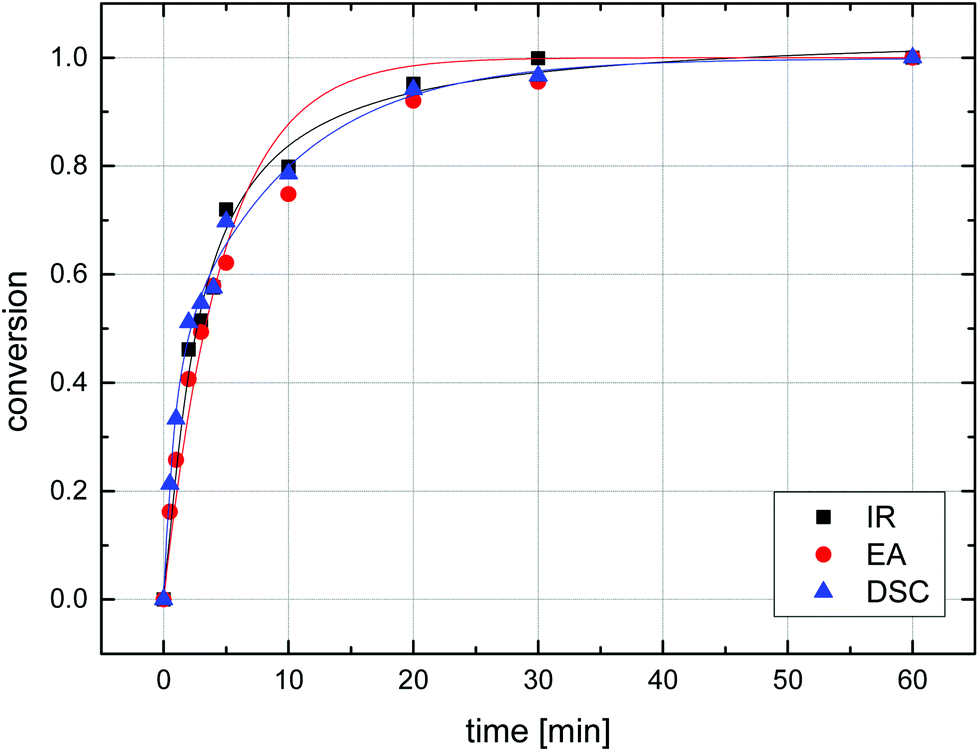 | ||
| Fig. 8 Azide decomposition (conversion = 1 − residual azide) vs. time during the irradiation of P(S-co-VBA) microsphere sample MS50 determined by FT-IR spectroscopy (squares), elemental analysis (circles), and differential scanning calorimetry (triangles). The curves are exponential fits of the data to guide the eye. | ||
For microsphere samples of varying azide loadings, the observed reaction rate increased with decreasing azide content, Fig. 9. For sample MS10, for example, (near-) complete reaction was found after 5 minutes of irradiation, while sample MS100 required 30 min to reach approx. 98% conversion. Exponential fitting of data suggested first order kinetics for the azide decomposition of sample MS100, though notably, other samples deviated from first order kinetics with observed reaction rates generally lower than predicted by first-order theory, Fig. S8.† The azide decomposition rate may be influenced by several factors including UV absorbance by the azide itself and the polystyrene matrix leading to lower flux of light inside particles, and a matrix absorptivity that depends on the crosslink density.
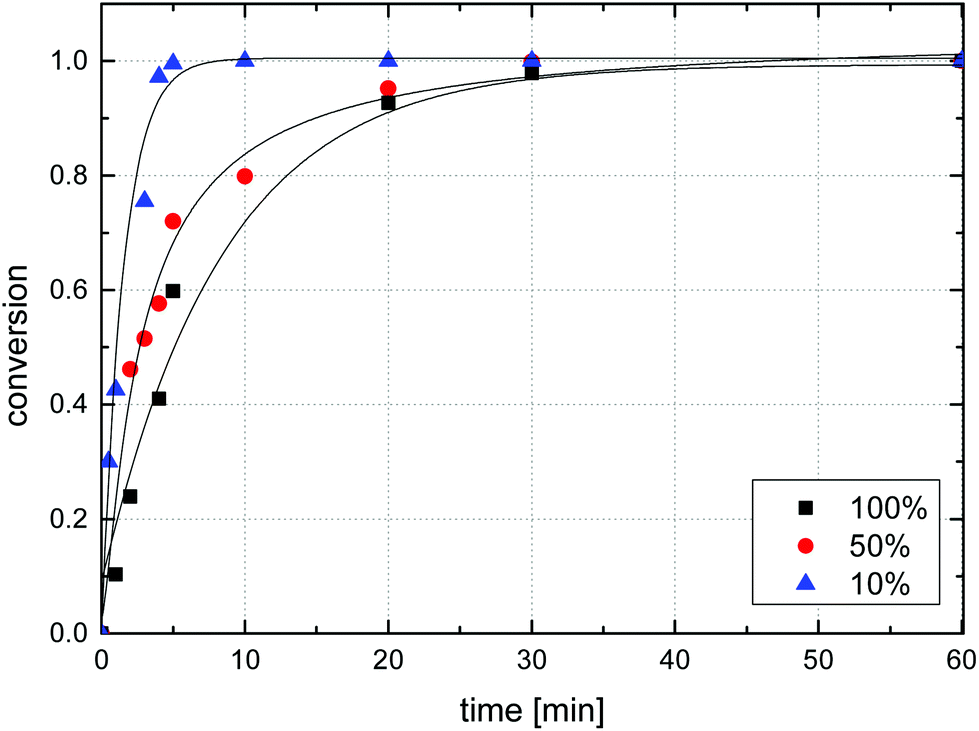 | ||
| Fig. 9 Comparison of azide conversion during the irradiation of microsphere samples MS10, MS50, and MS100 determined by IR spectroscopy. | ||
The local concentration of azides has also been shown to influence the preferred reaction pathway of the nitrene intermediates (Scheme 3) with crosslinking through nitrene–nitrene coupling typically only found for high azide concentrations and pathways (a) and (c) (Scheme 3) being the most common crosslinking reactions at low or intermediate azide concentration.51 Notably, some possible reaction pathways involving H abstraction from a C–H bond without combination with the C-centered radical do not lead to crosslinking reactions. FT-IR measurements and 13C solid state NMR spectroscopy were run on samples MS10, MS50, and MS100 before and after exhaustive crosslinking. Both measurements confirmed the disappearance of the azide group (disappearance of the NNN vibration at 2095 cm−1 in the IR spectrum and of the 13C NMR resonance assigned to the –CH2N3 group at 54.6 ppm, Fig. S9†). However, the results were inconclusive with regards to identifying preferred nitrene reaction pathways as shown in Scheme 3. While reaction pathway (a) (leading to crosslinking through CC addition) was expected to prevail, it was not possible to determine the exact crosslinking density from the azide conversion.
Notably, most published examples on azide-functional microspheres, films, or other architectures describe exhaustive irradiation and crosslinking. Significantly, the combination of a reproducible experimental setup, and use of either IR, EA, or DSC analysis described herein enabled the partial crosslinking of microspheres by sparing a defined and predetermined amount of azide functionality which can then be available for further modification. This protocol thus allows for tuning of the initial azide content (Table 1) and the proportion of azide functionality used for crosslinking. Taking advantage of this, microspheres with different extents of crosslinking and residual azide functionality were prepared and characterized by SEM imaging and swelling studies, Table 2.
| Microsphere sample | SEM analysis before crosslinking | Crosslinking extenta | Residual azide loadingb | SEM analysis after crosslinking | Solvent behaviorc | |||||
|---|---|---|---|---|---|---|---|---|---|---|
|
d
nd [μm] |
Đ
de |
CVf [%] | [%] | [mmol g−1] |
d
na [μm] |
Đ
db |
CVc [%] | Appearance | ||
| a Percentage of azide functionality used during irradiation determined through FT-IR analysis of the residual azide band at 2095 cm−1. b Estimated from initial azide loading, azide decomposition and average molar mass. c Crosslinked microspheres were kept in THF for 24 h. d Number-average particle diameter. e Dispersity of the particle diameter. f Coefficient of variation of the particle diameter. | ||||||||||
| MS10 | 2.49 | 1.002 | 4.6 | 50 | 0.33 | 2.57 | 1.001 | 2.5 | Individual particles | Swelling and dissolution of particles |
| 100 | 0 | 2.55 | 1.001 | 2.9 | Individual particles | swelling and light agglomeration | ||||
| MS50 | 1.34 | 1.06 | 25.9 | 50 | 1.68 | 1.34 | 1.06 | 23.8 | Individual particles | Swelling, with no gelation or disintegration |
| 100 | 0 | 1.26 | 1.05 | 22.8 | Some particles glued together | n.d. | ||||
| MS100 | 0.84 | 1.08 | 29.6 | 50 | 0 | 0.94 | 1.07 | 27.2 | Individual particles | Swelling, with no gelation or disintegration |
| 100 | 3.06 | 0.89 | 1.07 | 24.5 | Some particles glued together | n.d. | ||||
SEM analysis of irradiated microspheres showed that the spherical shapes, particle diameters, and particle size distributions were not affected by the photo-crosslinking. In fact, for most samples, SEM images showed no differences to the images recorded before crosslinking, see Fig. 10 and average particle diameters and dispersities given in Table 2. For two samples, MS50 and MS100, both with high azide loadings, however, exhaustive crosslinking for 60 minutes was found to result in particles being ‘glued’ together as observed by SEM, Fig. 11. Presumably, microspheres aggregated in their aqueous suspension during the irradiation and nitrene reactions formed covalent links between particles.
 | ||
| Fig. 10 SEM images of photo-crosslinked microspheres after irradiation (left images) and after stirring in THF for 24 h showing swelling (right images) of samples (a) MS10 after 100% azide conversion; (b) MS50 with 50% of azide functionality used for crosslinking and (c) MS100 with 50% of azide functionality used for crosslinking. Photo-crosslinking was not found to affect the size dispersity of microspheres while swelling of particles with a low degree of crosslinking was observed in THF. | ||
 | ||
| Fig. 11 SEM images of microspheres (a) MS50 and (b) MS100 each after UV irradiation for 60 min leading to some extent of particle agglomeration. | ||
Successful crosslinking was demonstrated by adding irradiated microspheres into THF (a good solvent for the polystyrene-based copolymers that dissolves non-crosslinked particles). The sample with the lowest crosslinking density, MS10 after using 100% of azide functionality for crosslinking swelled considerably with particles sticking together as seen by SEM analysis, Fig. 10 (top right). For all other samples, particles did not disintegrate in THF indicating sufficient crosslinking to prevent dissolution. These observations suggested that more than 10 wt% VBA feed were necessary to ensure adequate crosslinking and that for samples containing 50 wt% or more of VBA, irradiation times should be kept below 60 min under these reaction conditions to prevent particle agglomeration.
Click modification of partially crosslinked microspheres
As mentioned above, the chemical modification of microspheres is crucial for many applications and the Cu-catalyzed azide–alkyne cycloaddition (CuAAC) reaction has been documented to be amenable to the modification of azide (and alkyne)43-functional microspheres. Herein, after demonstrating the preparation of microspheres with a tunable degree of crosslinking and a tunable amount of residual azide functionality through partial photo-crosslinking, the chemical accessibility of the residual azide groups was demonstrated through CuAAC-modification with an alkyne-functional fluorescent dye, Rhodamine B propargyl ester, Scheme 1. This alkyne was chosen because of the obvious visual confirmation of successful modification, the importance of fluorescent microspheres as probes for optical measurements, but also because, as a larger, sterically hindered alkyne, it represents a suitable way of probing the accessibility of the azide groups with challenging alkyne reagents. As substrate, a sample of MS50, photo-crosslinked for 3 min and with 48% of residual azide functionality (as determined by IR-spectroscopy), was chosen. Rhodamine B propargyl ester was provided in 3-fold excess to azide groups and CuBr, N,N,N′,N′-tetramethylethylendiamine, and triethylamine were used as the catalytic system in DMF as solvent at room temperature.35,43 After intensive washing of the dye-modified microspheres until washings were colorless, the product was obtained as a deeply purple powder, Fig. 12. IR spectroscopy, Fig. 13, indicated a remainder of approx. 9% of the initial azide content. As CuAAC represents the only viable reaction that diminishes the azide concentration, this data suggested that after 52% of azide groups had been used for crosslinking, another 39% of initial azide groups were modified through CuAAC, or that 81% of the azide groups spared during photo-crosslinking were successfully conjugated to the fluorescent dye—a fairly high reaction efficiency when comparing to the literature and considering the bulky nature of the dye alkyne.35,43 The IR spectrum run on the dye-modified microsphere sample additionally showed characteristic vibrations of the Rhodamine dye, qualitatively confirming its presence in the modified particles. | ||
| Fig. 12 Photograph of microspheres MS50 as synthesized, featuring azide-functionality and presenting as an off-white powder (left); after partial photo-crosslinking, presenting as slightly more yellow powder (middle); and after modification of the residual azide groups with Rhodamine B propargyl ester showing bright purple color (right). | ||
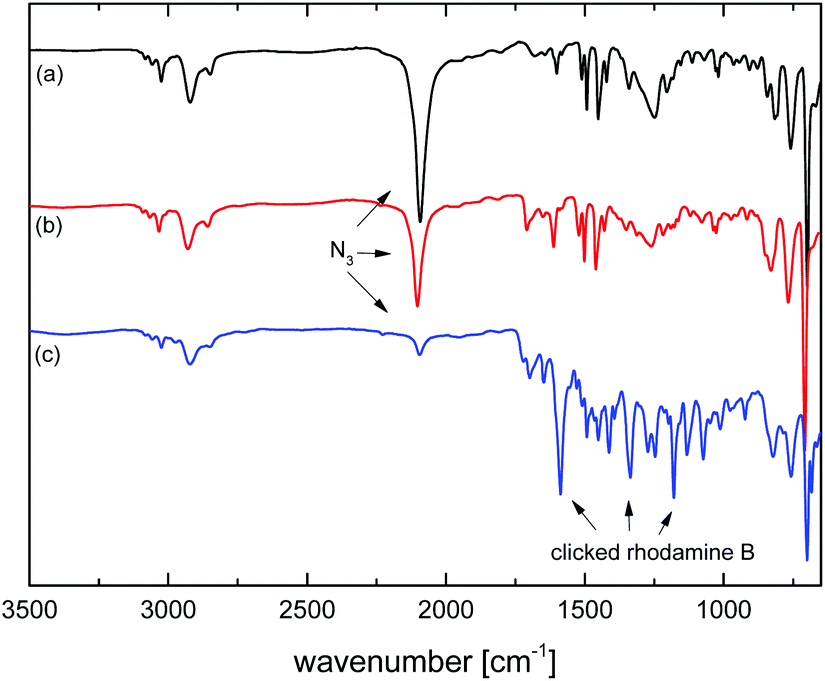 | ||
| Fig. 13 FT-IR spectra of microspheres MS50 (a) as synthesized, (b) after partial photo-crosslinking for 3 min sparing 48% of azide functionality, and (c) after CuAAC modification with Rhodamine B propargyl ester after which 9% of initial azide functionality remained. The NNN asymmetric stretch of the azide group and characteristic vibrations of the dye are marked. | ||
Conclusions
This study exploited several key characteristics of the azide functionality for the preparation of crosslinked and functionalized polystyrene-based microspheres: (i) while the benzyl chloride functional group was found to undergo nucleophilic substitution with the alcoholic solvent during polymerization, the benzyl azide functional group was found to be stable under the same reaction conditions. This allowed for a first description of dispersion (co-)polymerizations based on vinylbenzyl azide to give well-defined microspheres with tunable azide loadings. (ii) Upon irradiation at 254 nm, the azide group forms a highly reactive nitrene intermediate that can undergo a combination of reactions with nearby C–H and CC bonds. This was exploited to crosslink the microspheres while preserving their sizes and size distributions. (iii) As documented in the literature for the modification of microspheres, the azide group is amenable to highly efficient copper-catalyzed cycloaddition reactions with alkynes, which was used herein to conjugate residual azide functionality to a fluorescent dye. The pivotal point of this work is that the photo-crosslinking can be done under controlled conditions that allow for the conversion of a predetermined amount of azide functionality and that this conversion can be easily monitored by FT-IR spectroscopy, elemental analysis, and differential scanning calorimetry. As a result, the degree of crosslinking and the amount of unreacted azide functionality can be tuned and the azide group, as a single functionality, can be exploited for both photo-crosslinking and click-modification. Given the inherent difficulties in producing crosslinked and functional microspheres associated with many common synthetic strategies, the presented methodology offers a simple, robust, and versatile avenue toward well-defined microsphere materials that can be customized depending on application requirements.
Acknowledgements
The authors thank Renate Walter (Zoological Institute, University of Hamburg) for SEM measurements. Dr Young Joo Lee at the NMR center of the Chemistry Department, University of Hamburg is acknowledged for solid state NMR measurements. M. A., W. P., and H.-U. M. acknowledge the German Federal Ministry of Education and Research (BMBF) for financial support (13N11312).References
- J. Wang, P. A. G. Cormack, D. C. Sherrington and E. Khoshdel, Monodisperse, Molecularly Imprinted Polymer Microspheres Prepared by Precipitation Polymerization for Affinity Separation Applications, Angew. Chem., Int. Ed., 2003, 42(43), 5336–5338 CrossRef CAS PubMed.
- M. R. Buchmeiser, New synthetic ways for the preparation of high-performance liquid chromatography supports, J. Chromatogr., A, 2001, 918(2), 233–266 CrossRef CAS PubMed.
- E. Unsal, S. T. Camli, S. Senel and A. Tuncel, Chromatographic performance of monodisperse–macroporous particles produced by “modified seeded polymerization.” I: Effect of monomer/seed latex ratio, J. Appl. Polym. Sci., 2004, 92(1), 607–618 CrossRef CAS.
- K. Saralidze, L. H. Koole and M. L. W. Knetsch, Polymeric Microspheres for Medical Applications, Materials, 2010, 3(6), 3537–3564 CrossRef CAS.
- A. Yethiraj, Tunable colloids: control of colloidal phase transitions with tunable interactions, Soft Matter, 2007, 3(9), 1099–1115 RSC.
- B. Peng, E. van der Wee, A. Imhof and A. van Blaaderen, Synthesis of Monodisperse, Highly Cross-Linked, Fluorescent PMMA Particles by Dispersion Polymerization, Langmuir, 2012, 28(17), 6776–6785 CrossRef CAS PubMed.
- D. Zanaga, F. Bleichrodt, T. Altantzis, N. Winckelmans, W. J. Palenstijn, J. Sijbers, B. de Nijs, M. A. van Huis, A. Sanchez-Iglesias, L. M. Liz-Marzan, A. van Blaaderen, K. Joost Batenburg, S. Bals and G. Van Tendeloo, Quantitative 3D analysis of huge nanoparticle assemblies, Nanoscale, 2016, 8(1), 292–299 RSC.
- H. Kawaguchi, Functional polymer microspheres, Prog. Polym. Sci., 2000, 25(8), 1171–1210 CrossRef CAS.
- N. Fontanals, P. Manesiotis, D. C. Sherrington and P. A. G. Cormack, Synthesis of Spherical Ultra-High-Surface-Area Monodisperse Amphipathic Polymer Sponges in the Low-Micrometer Size Range, Adv. Mater., 2008, 20(7), 1298–1302 CrossRef CAS.
- J. Ugelstad, L. Soderberg, A. Berge and J. Bergstrom, Monodisperse polymer particles — a step forward for chromatography, Nature, 1983, 303(5912), 95–96 CrossRef.
- R. P. A. Dullens, E. M. Claesson and W. K. Kegel, Preparation and Properties of Cross-Linked Fluorescent Poly(methyl methacrylate) Latex Colloids, Langmuir, 2004, 20(3), 658–664 CrossRef CAS PubMed.
- A. Tuncel, A. Denizli, M. Abdelaziz, H. Ayhan and E. Piskin, Biologically Modified Polymeric Biomaterial SurfacesAlbumin adsorption on to large-size monodisperse polystyrene latices having functional groups on their surfaces, Clin. Mater., 1992, 11(1), 139–144 CrossRef.
- R. Arshady, Suspension, emulsion, and dispersion polymerization: A methodological survey, Colloid Polym. Sci., 1992, 270(8), 717–732 CAS.
- C. M. Cheng, F. J. Micale, J. W. Vanderhoff and M. S. El-Aasser, Synthesis and characterization of monodisperse porous polymer particles, J. Polym. Sci., Part A: Polym. Chem., 1992, 30(2), 235–244 CrossRef CAS.
- M. T. Gokmen and F. E. Du Prez, Porous polymer particles—A comprehensive guide to synthesis, characterization, functionalization and applications, Prog. Polym. Sci., 2012, 37(3), 365–405 CrossRef CAS.
- M. J. Derry, L. A. Fielding and S. P. Armes, Polymerization-induced self-assembly of block copolymer nanoparticles via RAFT non-aqueous dispersion polymerization, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- J. Rieger, Guidelines for the Synthesis of Block Copolymer Particles of Various Morphologies by RAFT Dispersion Polymerization, Macromol. Rapid Commun., 2015, 36(16), 1458–1471 CrossRef CAS PubMed.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Recent advances in RAFT dispersion polymerization for preparation of block copolymer aggregates, Polym. Chem., 2013, 4(4), 873–881 RSC.
- J. Ugelstad, A. Berge, T. Ellingsen, O. Aune, L. Kilaas, T. N. Nilsen, R. Schmid, P. Stenstad, S. Funderud, G. Kvalheim, K. Nustad, T. Lea, F. Vartdal and H. Danielsen, Monosized magnetic particles and their use in selective cell separation, Makromolekulare Chemie, Macromol. Symp., 1988, 17(1), 177–211 CrossRef CAS.
- J. Ugelstad, K. H. Kaggerud, F. K. Hansen and A. Berge, Absorption of low molecular weight compounds in aqueous dispersions of polymer-oligomer particles, 2. A two step swelling process of polymer particles giving an enormous increase in absorption capacity, Die Makromol. Chem., 1979, 180(3), 737–744 CrossRef CAS.
- J. Ugelstad, H. R. Mfutakamba, P. C. Mørk, T. Ellingsen, A. Berge, R. Schmid, L. Holm, A. Jørgedal, F. K. Hansen and K. Nustad, Preparation and application of monodisperse polymer particles, J. Polym. Sci., Polym. Symp., 1985, 72(1), 225–240 CrossRef CAS.
- J. Ugelstad, P. C. Mórk, K. H. Kaggerud, T. Ellingsen and A. Berge, Swelling of oligomer-polymer particles. New methods of preparation, Adv. Colloid Interface Sci., 1980, 13(1–2), 101–140 CrossRef CAS.
- H. Bamnolker and S. Margel, Dispersion polymerization of styrene in polar solvents: Effect of reaction parameters on microsphere surface composition and surface properties, size and size distribution, and molecular weight, J. Polym. Sci., Part A: Polym. Chem., 1996, 34(10), 1857–1871 CrossRef CAS.
- C.-W. Chen and C.-Y. Chen, Preparation of monodisperse polystyrene microspheres: effect of reaction parameters on particle formation, and optical performances of its diffusive agent application, Colloid Polym. Sci., 2009, 287(12), 1377–1389 CAS.
- S. T. Ha, O. O. Park and S. H. Im, Size control of highly monodisperse polystyrene particles by modified dispersion polymerization, Macromol. Res., 2010, 18(10), 935–943 CrossRef CAS.
- S. Kawaguchi and K. Ito, Dispersion Polymerization, Adv. Polym. Sci., 2005, 175, 299–328 CrossRef CAS.
- S. M. Klein, V. N. Manoharan, D. J. Pine and F. F. Lange, Preparation of monodisperse PMMA microspheres in nonpolar solvents by dispersion polymerization with a macromonomeric stabilizer, Colloid Polym. Sci., 2003, 282(1), 7–13 CAS.
- K. P. Lok and C. K. Ober, Particle size control in dispersion polymerization of polystyrene, Can. J. Chem., 1985, 63(1), 209–216 CrossRef CAS.
- H. Uyama and S. Kobayashi, Dispersion polymerization of styrene in aqueous alcohol solution: Effects of reaction parameters on the polymer particle formation, Polym. Int., 1994, 34(3), 339–344 CrossRef CAS.
- J. Choi, S. Y. Kwak, S. Kang, S. S. Lee, M. Park, S. Lim, J. Kim, C. R. Choe and S. I. Hong, Synthesis of highly crosslinked monodisperse polymer particles: Effect of reaction parameters on the size and size distribution, J. Polym. Sci., Part A: Polym. Chem., 2002, 40(23), 4368–4377 CrossRef CAS.
- J.-S. Song, F. Tronc and M. A. Winnik, Two-Stage Dispersion Polymerization toward Monodisperse, Controlled Micrometer-Sized Copolymer Particles, J. Am. Chem. Soc., 2004, 126(21), 6562–6563 CrossRef CAS PubMed.
- J.-S. Song and M. A. Winnik, Cross-Linked, Monodisperse, Micron-Sized Polystyrene Particles by Two-Stage Dispersion Polymerization, Macromolecules, 2005, 38(20), 8300–8307 CrossRef CAS.
- C. M. Tseng, Y. Y. Lu, M. S. El-Aasser and J. W. Vanderhoff, Uniform polymer particles by dispersion polymerization in alcohol, J. Polym. Sci., Part A: Polym. Chem., 1986, 24(11), 2995–3007 CrossRef CAS.
- J. Tan, X. Rao, X. Wu, H. Deng, J. Yang and Z. Zeng, Photoinitiated RAFT Dispersion Polymerization: A Straightforward Approach toward Highly Monodisperse Functional Microspheres, Macromolecules, 2012, 45(21), 8790–8795 CrossRef CAS.
- M. Albuszis, P. J. Roth, F. Exnowitz, D. L. Wong, W. Pauer and H.-U. Moritz, Synthesis and in-depth characterization of reactive, uniform, crosslinked microparticles based on free radical copolymerization of 4-vinylbenzyl azide, Polym. Chem., 2016, 7(5), 1168–1180 RSC.
- T. Bahar and A. Tuncel, Monodisperse poly(p-chloromethylstyrene) microbeads by dispersion polymerization, Polym. Eng. Sci., 1999, 39(10), 1849–1855 CAS.
- S. Margel, E. Nov and I. Fisher, Polychloromethylstyrene microspheres: Synthesis and characterization, J. Polym. Sci., Part A: Polym. Chem., 1991, 29(3), 347–355 CrossRef CAS.
- J.-P. Montheard, M. Chatzopoulos and M. Camps, Chemical Transformations of Chloromethylated Polystyrene, J. Macromol. Sci., Part C: Polym. Rev., 1988, 28(3–4), 503–592 CrossRef.
- I. Dumistracel, G. Ponchel, G. Danila, D. Duchene and A. Carpov, Poly(vinylbenzyl chloride) microsphere synthesis and their chemical modifications, J. Microencapsulation, 2000, 17(1), 45–55 CrossRef CAS PubMed.
- B. Elmas, S. T. Camli, M. Tuncel, S. Senel and A. Tuncel, DNA-responsive uniform latex particles based on p-chloromethylstyrene, J. Biomater. Sci., Polym. Ed., 2012, 12(3), 283–296 CrossRef.
- Ç. Kip, B. Maraş, O. Evirgen and A. Tuncel, A new type of monodisperse porous, hydrophilic microspheres with reactive chloroalkyl functionality: synthesis and derivatization properties, Colloid Polym. Sci., 2013, 292(1), 219–228 Search PubMed.
- A. Zillessen and E. Bartsch, Synthesis of Photo-Cross-Linkable Microgel Colloids for Cluster Formation Studies, Langmuir, 2010, 26(1), 89–96 CrossRef CAS PubMed.
- M. Albuszis, P. J. Roth, W. Pauer and H.-U. Moritz, Macroporous uniform azide- and alkyne-functional polymer microspheres with tuneable surface area: synthesis, in-depth characterization and click-modification, Polym. Chem., 2014, 5(19), 5689–5699 RSC.
- A. Bayraktar, B. Saracoglu, C. Golgelioglu and A. Tuncel, Click-chemistry for surface modification of monodisperse-macroporous particles, J. Colloid Interface Sci., 2012, 365(1), 63–71 CrossRef CAS PubMed.
- D. R. Breed, R. Thibault, F. Xie, Q. Wang, C. J. Hawker and D. J. Pine, Functionalization of polymer microspheres using click chemistry, Langmuir, 2009, 25(8), 4370–4376 CrossRef CAS PubMed.
- K. Ouadahi, E. Allard, B. Oberleitner and C. Larpent, Synthesis of azide-functionalized nanoparticles by microemulsion polymerization and surface modification by click chemistry in aqueous medium, J. Polym. Sci., Part A: Polym. Chem., 2012, 50(2), 314–328 CrossRef CAS.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes, Angew. Chem., Int. Ed., 2002, 41(14), 2596–2599 CrossRef CAS.
- V. Rodionov, H. Gao, S. Scroggins, D. A. Unruh, A.-J. Avestro and J. M. J. Fréchet, Easy Access to a Family of Polymer Catalysts from Modular Star Polymers, J. Am. Chem. Soc., 2010, 132(8), 2570–2572 CrossRef CAS PubMed.
- M. K. Klein, A. Zumbusch and P. Pfleiderer, Photo-crosslinkable, deformable PMMA colloids, J. Mater. Chem. C, 2013, 1(43), 7228–7236 RSC.
- L. Xu, J. Farrell, R. G. Karunakaran, A. Honglawan and S. Yang, Synthesis of dual-functional copolymer with orthogonally photosensitive groups, J. Polym. Sci., Part A: Polym. Chem., 2013, 51(5), 1215–1222 CrossRef CAS.
- S. Al Akhrass, R.-V. Ostaci, Y. Grohens, E. Drockenmuller and G. Reiter, Influence of Progressive Cross-Linking on Dewetting of Polystyrene Thin Films, Langmuir, 2008, 24(5), 1884–1890 CrossRef CAS PubMed.
- H. Morita, S. Mori, N. Uchino and S. Yokoyama, Magnetic Field and Polymer Matrix Effects on Photocrosslinking Reaction in Solid Polymer Matrices, in Zeitschrift für Physikalische Chemie, 1993, vol. 182, p. 209 Search PubMed.
- Q. Zhang, Y. Han, W. Wang, T. Song and J. Chang, A theoretical and experimental investigation of the size distribution of polystyrene microspheres by seeded polymerization, J. Colloid Interface Sci., 2010, 342(1), 62–67 CrossRef CAS PubMed.
- D. Horák and P. Shapoval, Reactive poly(glycidyl methacrylate) microspheres prepared by dispersion polymerization, J. Polym. Sci., Part A: Polym. Chem., 2000, 38(21), 3855–3863 CrossRef.
- V. Ladmiral, T. M. Legge, Y. Zhao and S. Perrier, “Click” Chemistry and Radical Polymerization: Potential Loss of Orthogonality, Macromolecules, 2008, 41(18), 6728–6732 CrossRef CAS.
- Y. Li, J. Yang and B. C. Benicewicz, Well-controlled polymerization of 2-azidoethyl methacrylate at near room temperature and click functionalization, J. Polym. Sci., Part A: Polym. Chem., 2007, 45(18), 4300–4308 CrossRef CAS.
- R. J. Andrews and E. A. Grulke, Glass Transition Temperatures of Polymers, in Polymer Handbook ed. J. Brandrup, E. H. Immergut and E. A. Grulke, John Wiley & Sohns, fourth edition, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py00937a |
| This journal is © The Royal Society of Chemistry 2016 |