
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Activated carbonates: enabling the synthesis of differentiated polycarbonate resins via melt transcarbonation†
J. H.
Kamps
*ab,
T.
Hoeks
a,
E.
Kung‡
a,
J. P.
Lens§
a,
P. J.
McCloskey
c,
B. A. J.
Noordover¶
b and
J. P. A.
Heuts
b
aSABIC, Plasticslaan 1, 4612PX Bergen op Zoom, The Netherlands. E-mail: janhenk.kamps@sabic.com
bDepartment of Chemical Engineering and Chemistry, Eindhoven University of Technology, P.O. Box 513, 5600 MB Eindhoven, The Netherlands
cGE Global Research, 1 Research Circle Niskayuna, NY 12309, USA
First published on 5th August 2016
Abstract
Activated carbonates facilitate the preparation of polycarbonates based on monomers that are unsuitable for traditional melt polymerization at high temperatures. Bis(methyl salicyl) carbonate (BMSC) clearly shows reactivity benefits over diphenyl carbonate in melt polymerization reactions, resulting in shorter reaction times and reduced heat exposure during polymerization. The increased reactivity enables the melt polymerization of a wide range of monomers, as demonstrated by two examples using volatile resorcinol and sterically hindered tert-butyl hydroquinone as monomers in the preparation of (co)polycarbonates.
Introduction
Polycarbonate resins based on bisphenol acetone (BPA) have an extraordinary property profile, combining ductility, strength and durability with high transparency and acceptable temperature stability. Materials based on this resin are applied in compact discs, appliances, helmets, packaging materials, sunglasses, automotive headlamp lenses, etc. Over the past 50 years, the production and processing of polycarbonate has become a multibillion dollar industry,1 serving a 3 million tonnes market in 2009.Generally, two industrial production methods have been demonstrated to produce high-quality polycarbonate resins and remain economically viable. These processes are the two-phase interfacial process (phosgene-based) and the melt transcarbonation process which is based on diphenyl carbonate (DPC).2,3 The traditional melt transcarbonation process, which is known for its solvent free character and yielding neat polymer of high quality, also has some less favorable characteristics, including long residence times at elevated temperatures and potential side reactions which gives rise to color formation and make it less suitable for thermally sensitive monomers. To reduce the required reaction temperature and/or residence time, DPC can be replaced by an activated carbonate source.4 As a common feature, activated carbonate species have an electron-withdrawing functionality substituted on the phenolic leaving group, shown as Chart 1.
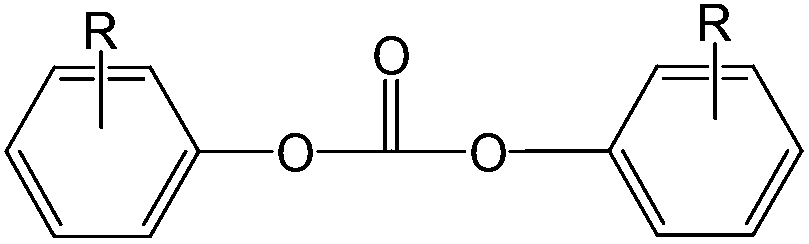 | ||
| Chart 1 General structure of an activated carbonate; R = NO2, Cl, CO2CH3 or other electron withdrawing groups. | ||
With these electron-withdrawing groups, the substituted phenol derivative is a better leaving group than a phenol without the substituents. In principle, the introduction of any known electron-withdrawing group on the ortho or para position would lead to an activated carbonate. One could consider activated carbonates, including phosgene, di-, or triphosgene2 or other examples such as o-nitrophenyl carbonates4 and bis(pentafluorophenyl) carbonate5 to polymerize thermally sensitive monomers. Each of these approaches comes with reactivity benefits, but also with complex design and safety measures (and the corresponding equipment considerations) due to the instability and toxicity of these compounds.
The specific use of such an activated monomer in melt polymerizations would require temperature stability at relatively high temperatures (>200 °C), without unfavorable degradation products (for example halogens) and the absence of color issues (as anticipated for nitrogen containing structures) would be desired. The structure meeting these requirements is bis(methyl salicyl) carbonate (BMSC, Scheme 1, structure I), which has been reported as monomer used in polycondensation reactions by Brunelle.6 Polymerizations with BMSC found some interest recently by industrial research groups,7–9 but only very limited information can be found in scientific publications regarding the advantages and reactivity differences versus the traditional polymerization methods.10 Additionally, activated carbonates can be applied in the broader organic synthetic field; their potential in different synthetic pathways6,11–14 is shown in Scheme 1. All of these variations can be combined in polymerization reactions to yield copolymers of monomers with a wide range of functionalities. In all cases, the by-product of the reaction is methyl salicylate, which can be re-used as a starting material in the synthesis of BMSC.15
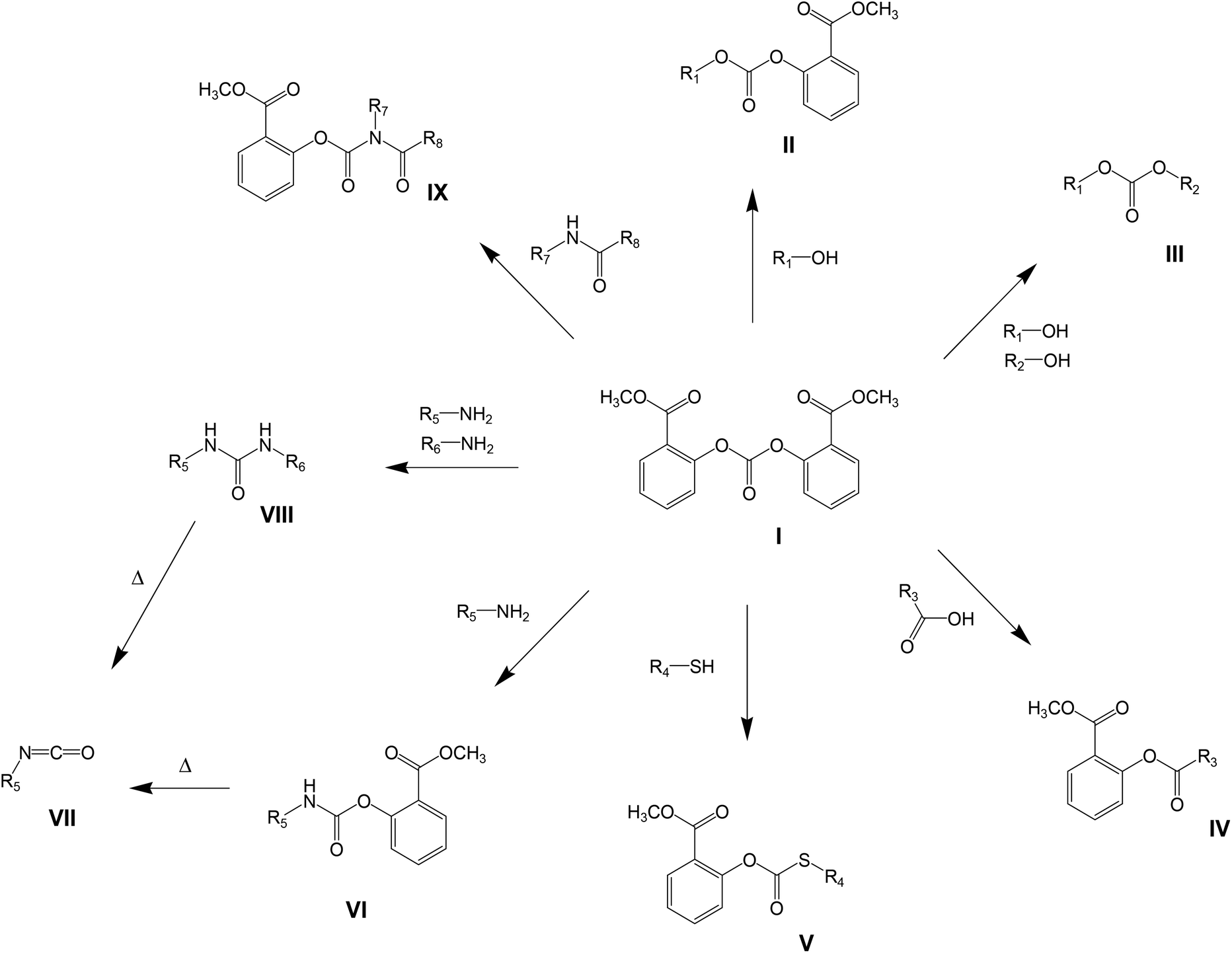 | ||
| Scheme 1 Synthetic routes involving activated carbonates. | ||
This article focusses on this new development in the melt transcarbonation process. First the use of octylphenol as a model system for the kinetic comparison between the reactivity of BMSC and DPC will be discussed. Subsequently, (co)polymerization reactions are described to demonstrate the applicability of the activated carbonates in polymerizations using diols that would be challenging to incorporate in conventional polymerization processes using DPC or phosgene, due to their volatility, limited reactivity, steric hindrance and solubility limitations of the monomer itself or the resulting polymer.
The use of resorcinol in the preparation of polycarbonates goes back to the earliest reports on aromatic carbonates.16 At the time, the described resorcinol polycarbonate was mentioned to be a material which melted with decomposition at 190–200 °C. Resorcinol is a meta-substituted dihydroxy benzene compound; the polymers synthesized from this monomer can be expected to have improved flow compared to more linear structures that more easily pack.17
When utilizing the melt polymerization process, in which DPC is used to form the carbonate linkage and the condensation product, phenol, is removed, the use of resorcinol presents several hurdles, mainly related to its volatility. This standard melt polymerization process takes relatively long (up to several hours) and requires high temperatures (up to 300 °C) to ensure that the produced polymer remains liquid and that the formed by-product phenol can be effectively removed from the polymer melt. Inefficient removal of phenol will lead to an equilibrium situation in which the molecular weight build-up is limited by the reverse reaction.18
As is well known for step-growth polymerizations, these processes are very sensitive to stoichiometry fluctuations. Loss of diol monomer during the polymerization results in a reduction of the molecular weight which can be achieved when the loss is not compensated for by adding an excess of the diol monomer at the early stages of the reaction.
Traditionally unhindered, phenolic hydroxyl groups are the preferred building blocks in melt transcarbonation polymerization. The effect of ortho-substitutions in polycondensations is discussed in literature,19 where lower incorporation is found for more hindered alcohols. Studying substituted monomers has drawn attention over the years, several attempts were made to produce polycarbonates with ortho-substitutions, striving for improved properties like hydrolytic stability and enhanced flow behavior,2 but issues with molecular weight build-up are reported by the same authors.
Polymerizations with BMSC are performed in combination with hindered hydroquinones, to demonstrate the use of BMSC when polymerizing hindered phenols, such as tert-butyl hydroquinone. Material properties and a comparison between hydroquinone type monomers are presented in the last part of this article.
Experimental section
Materials
Octylphenol (Sigma Aldrich, 99+%), bis(methyl salicyl) carbonate (SABIC, 99+%), bisphenol A (SABIC, polymerization grade, >99%), di(phenyl) carbonate (SABIC, polymerization grade, >99%), resorcinol (Acros, 99%), tetramethyl ammonium hydroxide (Sachem, aqueous solution, 20%), sodium hydroxide (Acros, NaOH), tetrabutylphosphonium acetate (Sachem, aqueous solution, 40%), tert-butyl hydroquinone (TCI, >98%), di tert-butyl hydroquinone (Acros, 99%), methyl hydroquinone (Acros, 99%), hydroquinone (Acros, 99.5%), acetonitrile (Acros, 99.9%, for HPLC), dichloromethane (Acros, 99.8%, for HPLC), chloroform-d (“100%”, 99.96 atom% D, 0.03% (v/v) TMS).Passivation of glassware
Reactors, flasks etc. used in reactions were passivated by immersion in an aqueous 1 M HCl solution to remove ions from its surface. Before use, the glass surface was rinsed several times with MilliQ water and dried.Kinetic study using octylphenol
A glass round bottom flask, stirred magnetically and heated with an oil bath was charged with equimolar amounts of octylphenol and di(p-cumylphenyl) carbonate (DiPCP) or BMSC to study the conversion of octylphenol to its expected half-product (HP) and bisoctylphenol carbonate. Sodium hydroxide was added as a catalyst (1.5 × 10−6 mol NaOH per mol octylphenol). Conversion of the reactants and formation of the reaction products was followed using HPLC.MATLAB® was used for calculation of the derivatives with central FDM (going fwd/backwards at first and last points) in combination with Adams–Bashfort time integration.20 In the simulation negative reaction constants were artificially set to 0, and concentrations were never allowed to have negative values to conserve mass. In a second step the reaction rate coefficients were manually optimized to fit the experimental data optimally.
Synthesis of (co-)polycarbonate using DPC
A glass reactor equipped with a mechanical stirrer and a vacuum system was charged with BPA, additional diol in case of copolymer preparation, and 25.00 g of DPC, maintaining a molar ratio of [DPC]/[sum of diols] of 1.08. After assembly of the reactor set up, oxygen was removed from the system by repeated vacuum/inert gas cycles. The catalyst was added as 100 μL of an aqueous solution of tetramethyl ammonium hydroxide (TMAH) (2.5 × 10−4 mol per mol diol) and NaOH (1.5 × 10−6 mol per mol diol). The polymerization was carried out in four stages: stage (1) 15 min, 180 °C, atmospheric pressure, stage (2) 60 min, 230 °C, 170 mbar, stage (3) 30 min, 270 °C, 20 mbar, and stage (4) 30 min, 300 °C, 0.5–1.5 mbar. During transitions, care was taken to prevent excessive boiling and loss of monomer to the overhead (condenser, vacuum pump). Subsequently, atmospheric pressure was restored and the polymer was discharged from the reactor and stranded.Synthesis of (co-)polycarbonate using BMSC
A glass reactor equipped with a mechanical stirrer and a vacuum system was charged with BPA, additional diol in case of copolymer preparation, and 20 g of BMSC, maintaining molar ratios of [BMSC]/[sum of diols] between 1 and 1.02. After assembly of the reactor set up, oxygen was removed from the system by repeated vacuum/inert gas cycles. The catalyst was added as 100 μL of an aqueous solution of TBPA (2.5 × 10−4 mol per mol diol) and NaOH (1.5 × 10−6 mol per mol diol). The polymerization was carried out in the following stages: stage (1) 15 min, 180 °C, atmospheric pressure, stage (2) 15 min, 220 °C, 100 mbar, and stage (3) 10 min, 280 °C, 0.5–1.5 mbar. During transitions, care was taken to prevent excessive boiling and loss of monomer to the overhead (condenser, vacuum pump). Subsequently, atmospheric pressure was restored and the polymer was discharged from the reactor and stranded.Characterization
HPLC was used to determine the conversion of the monomers. An Xterra C18 column was used for the analysis on a Perkin Elmer HPLC. The column temperature was maintained at 30 °C. The column was eluted with a ratio of water to acetonitrile of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20. The flow rate of sample in the column was maintained at 1.00 mL min−1 and the amount of sample injected was 5 μL and run time was 23 min.
:20. The flow rate of sample in the column was maintained at 1.00 mL min−1 and the amount of sample injected was 5 μL and run time was 23 min.
SEC molecular weights are reported as number-average (Mn) or weight-average (Mw) molecular weight and were determined by size exclusion chromatography using polymer solutions comprising the product (co)polycarbonates at a concentration of about 1 mg mL−1 in methylene chloride (CH2Cl2) using a cross-linked styrene–divinylbenzene column and related to a polystyrene (PS) reference, calibrated using PS standards (ranging Mw from 1000 to 2000000 g mol−1).
1 H-NMR spectra were measured on a Bruker Avance 400 MHz spectrometer at a temperature of 44 °C. For each analysis, a solution of 5% wt/v in CDCl3 99.8% D with 0.03 wt% TMS was prepared. All spectra were obtained with a spin-rate of 20 Hz. The 1H-NMR measurements were performed at 400 MHz with a 5 mm diameter quadruple nucleus probe, using a sweep width of 6410 Hz (13 to −3 ppm chemical shift range) with a 30° flip angle, 10 s relaxation time, 32k data points and 256 scans. Processing included apodization with 0.3 Hz for 1H NMR.
13
C-NMR spectra were measured on either a GE NMR Instruments Omega 500 or Bruker Avance 500 spectrometer operating at 125.75 MHz for 13C. Approximately 200 mg of the polymer sample was dissolved in 3.5 mL of chloroform-d. The central peak of the chloroform-d resonance was used as an internal chemical shift standard (= 77.0 ppm). Acquisition parameters included a 30.3 kHz spectral width and 32k data points resulting in a 1.08 s acquisition time. A pulse delay of 1 s and flip angle of 45° were used. Broadband proton decoupling was carried out using the Waltz-16 pulse sequence. Between 1200 and 16000 (overnight) scans were acquired in blocks of 600. When high resolution spectra were needed, the data was processed without any exponential apodization. A 1 Hz exponential apodization was applied for standard 13C spectra. In addition, a polynomial baseline correction was applied.
31 P-NMR data were acquired at 202.5 MHz on a Bruker Avance 500 MHz NMR spectrometer. Acquisition parameters included a 30° flip angle, 32k data points with a sweep width of 14.1 kHz (110–180 ppm chemical shift range), and 4 dummy scans. The pulse delay was chosen so that the total recycle time was 3 s. The total number of scans varied depending upon the concentration of end groups in the polymer.
DSC glass transition temperatures (Tg) were determined by using a Perkin Elmer DSC7. The Tg was calculated, based on the “one half Cp” method (heat capacity at constant pressure). Melting points were derived from the second heating curve. The following profile was used; step 1 (first heating): heat from 30 °C to 360 °C at 20 °C min−1, step 2: cool from 360 °C to 170 °C at 20 °C min−1, step 3: hold at 170 °C for 30 min, step 4 (second heating step): heat from 170 °C to 360 °C at 20 °C min−1, step 5: cool from 360 °C to 30 °C at 20 °C min−1.
MVR was determined at 300 °C and 1.2 kg according to standard ISO 1133 using a Zwick B4106.200 set up.
Results and discussion
The effect of BMSC on the polymerization conditions for BPA polycarbonate is evident from a simple comparison with DPC targeting comparable molecular weights as illustrated in Table 1.| 1 | 2 | |
|---|---|---|
| a Relative to PS standards. | ||
| Carbonate source | DPC | BMSC |
| Step 1 (t, T, p) | 15 min, 180 °C, atm | 15 min, 180 °C, atm |
| Step 2 (t, T, p) | 60 min, 230 °C, 170 mbar | 15 min, 220 °C, 100 mbar |
| Step 3 (t, T, p) | 30 min, 270 °C, 20 mbar | 10 min, 280 °C, 0.5–1.5 mbar |
| Step 4 (t, T, p) | 30 min, 300 °C, 0.5–1.5 mbar | — |
| Overall reaction time | 135 min | 40 min |
| M w (dalton)a | 38.5 × 103 | 40.0 × 103 |
| M n (dalton)a | 17.4 × 103 | 17.7 × 103 |
| T g (°C) | 144.1 | 144.4 |
As shown in Table 1, the differences between the polymerization profiles using DPC (entry 1) and BMSC (entry 2) indicate increased reaction rates when BMSC is used. Using BMSC requires less than one third of the total reaction time and also uses lower temperatures during the polymerization stages to achieve a similar molecular weight.
To investigate this behavior further a kinetic study was done, followed by the investigation on the use of BMSC in the preparation of (co)polycarbonates.
Kinetic study
Kinetic studies regarding the formation of (poly)carbonates are studied and published by various groups. Gross et al. published equilibrium constants for the reaction between DPC and BPA18 (Scheme 2) and Brunelle reported reactivity studies of (ortho nitro-substituted) activated carbonates.4
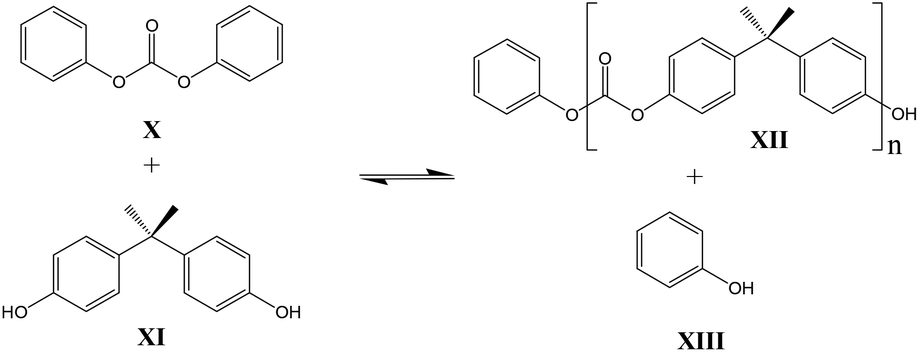 | ||
| Scheme 2 Transcarbonation of DPC with BPA. | ||
In contrast to these earlier studies, in the current study octylphenol was selected as a model compound rather than BPA, to prevent oligomerization. To overcome further potential issues when studying the DPC reactions (Scheme 2, structure X), caused by the relatively low boiling point of phenol, DiPCP carbonate (Scheme 3, structure XIV) was used as a model for DPC to ensure that the condensation product remains in the reaction mixture. The para-cumyl phenol (PCP) (Scheme 3, structure XVIII) is believed to have the same characteristics in the transcarbonation reaction in terms of reactivity.
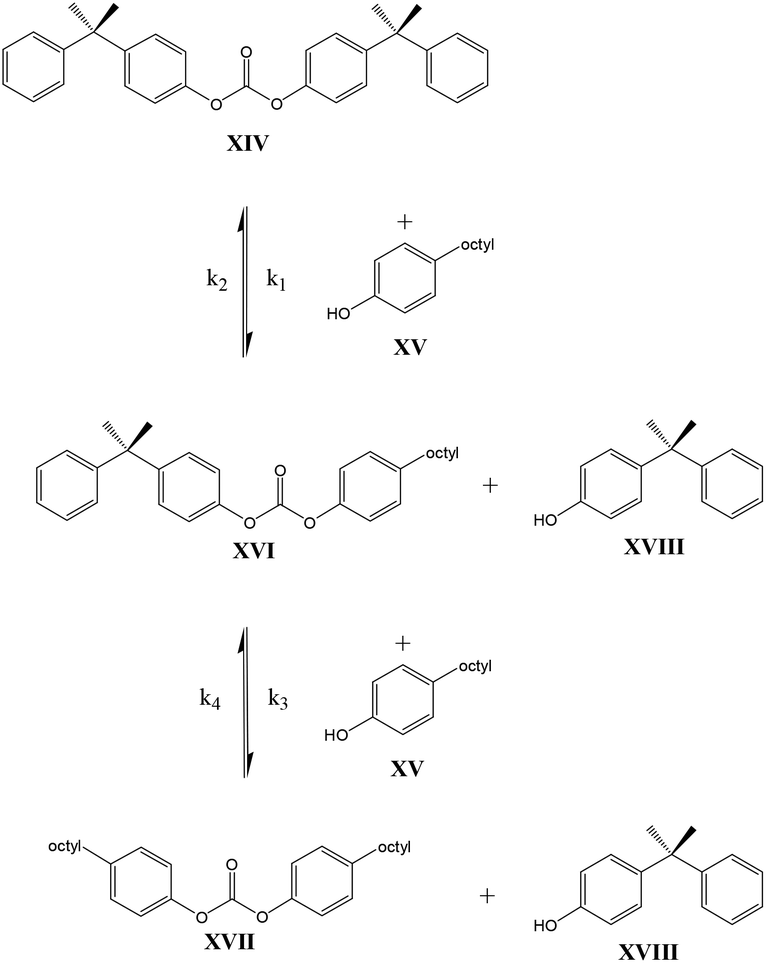 | ||
| Scheme 3 DPC model compound reaction. | ||
As shown in Schemes 3 and 4, the reaction with octylphenol involves 2 steps, replacing one or two arylates on the carbonate, forming the octylphenol carbonate (half product – HP) or the dioctyl carbonate full product (full product – FP). The individual components were analyzed using HPLC of the mixtures during the equilibration, following the conversion of reactants and formation of the half and full product versus time (25 minutes), as plotted in Fig. 1 and 2. (The complete dataset is available in the ESI, Tables S1 and S2†).
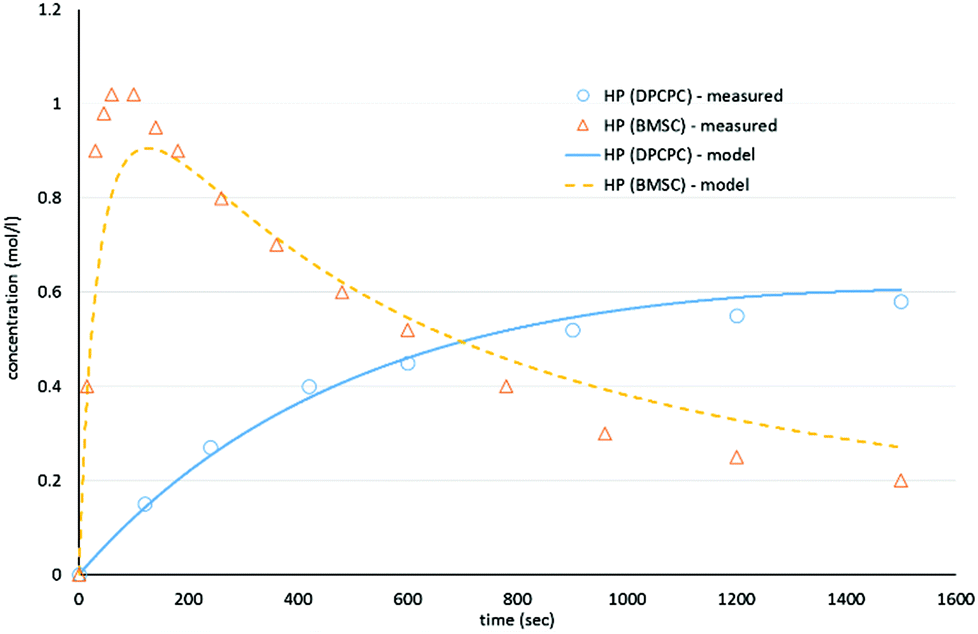 | ||
| Fig. 1 Model compound equilibration for the formation of the half product from DiPCP carbonate (XIV) and BMSC (XIX) at 200 °C. | ||
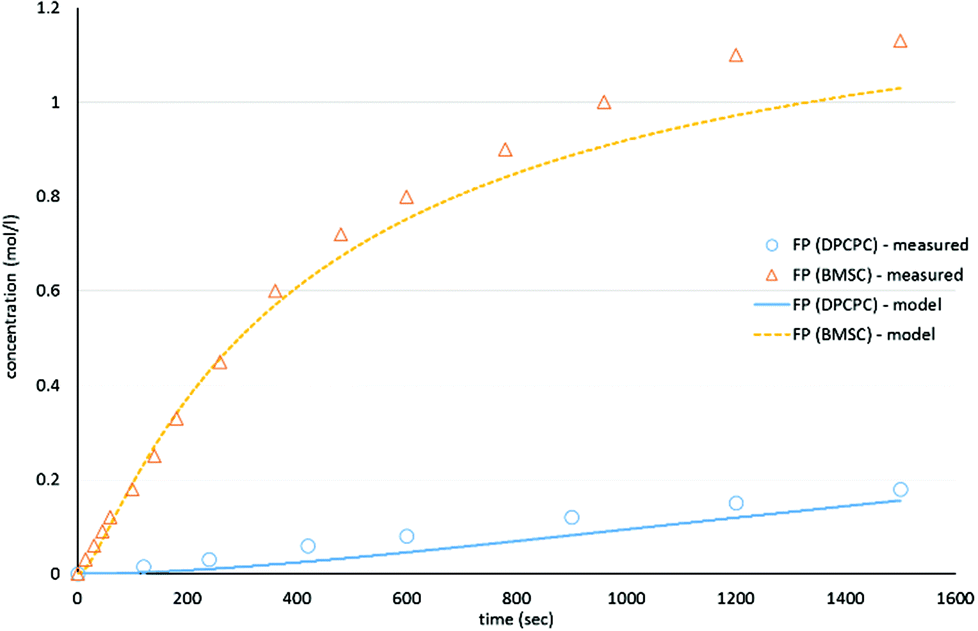 | ||
| Fig. 2 Model compound equilibration for the formation of the full product from DiPCP carbonate (XIV) and BMSC (XIX) at 200 °C. | ||
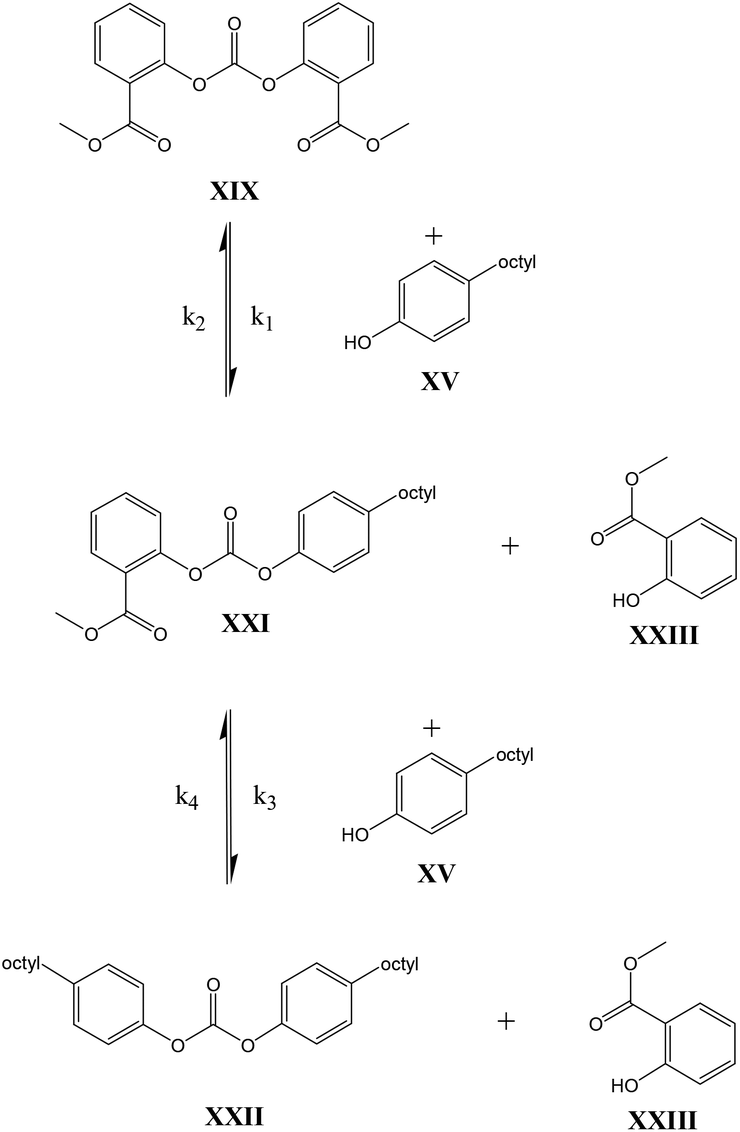 | ||
| Scheme 4 BMSC model compound reaction. | ||
MATLAB® was used to calculate the reaction rate coefficients of the different steps occurring (as shown in Schemes 3 and 4), using the expressions displayed in Scheme 5 (based on pseudo second order kinetics). The derived reaction rate coefficients were used to model the reactions, the results of which are displayed in Fig. 2 (for the formation of the half product) and Fig. 3 (for the formation of the full product) plotted together with the actual measured values for comparison. A simple approach was used to demonstrate the reactivity advantage of the activated carbonate, effects of catalyst etc. are not part of the model and taken into account in the reaction rate coefficients, giving a realistic reflection of the reaction rate differences.
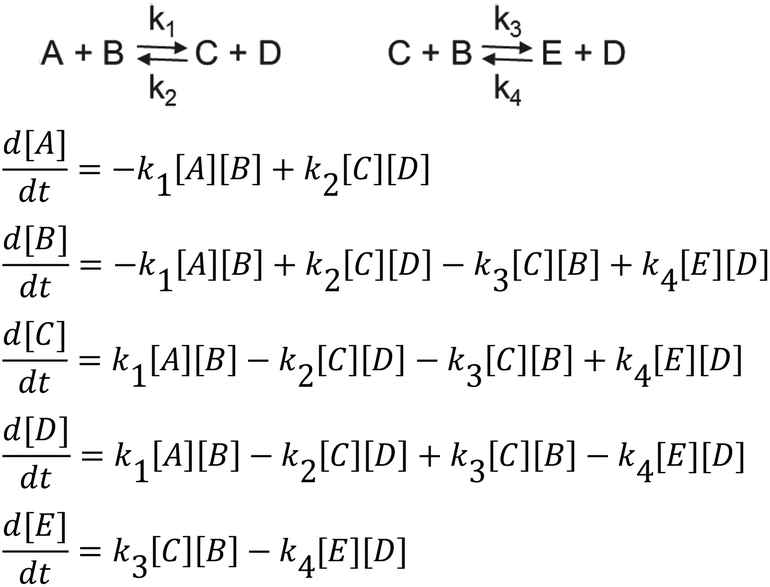 | ||
| Scheme 5 Kinetic model. | ||
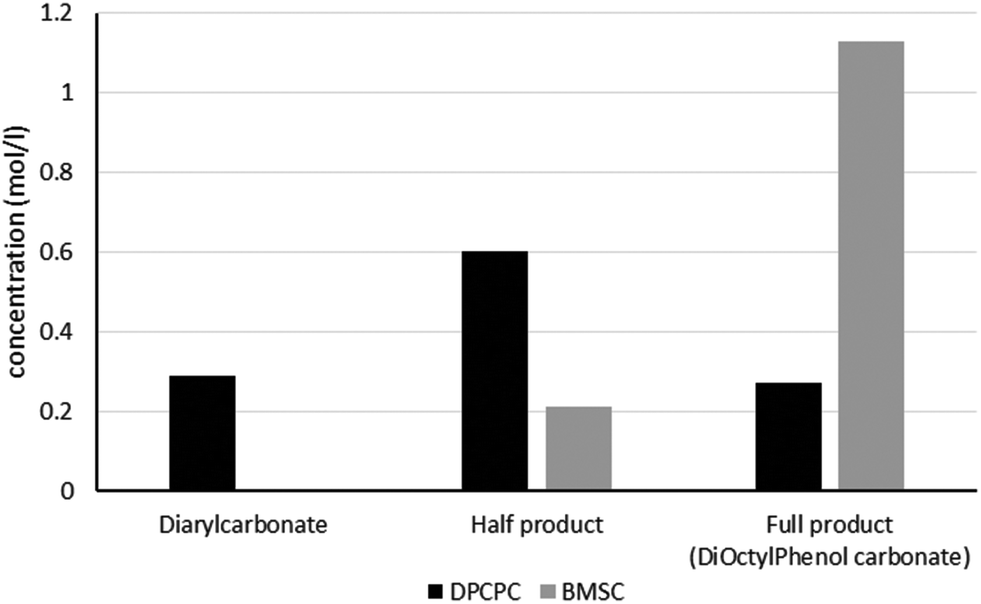
As can be seen in Fig. 1, the formation of half product in the reaction with BMSC quickly reaches a maximum, after which the consumption of half product (which reacts with another molecule of octylphenol to form the full product) takes over and reduces the total amount of half product again over time. For the reaction with the DPC analogue the reaction reaches equilibrium at a conversion of around 50% (Fig. 2).
The calculated values for the reaction rate coefficients are presented in Table 2. For the initial forward step, i.e. formation of half product, the reaction rate coefficient k1 is approximately 30 times higher for the BMSC reaction, compared to the DPCPC reaction. Also note that the reaction rate coefficient for the formation of the full product (k3) is about 9 times higher for the activated carbonate. The reverse reactions to form the initial carbonate from the half product is considerably higher in the case of the DPCPC reactions, where k2 is about half of the k1. In the experiments with BMSC the k2 is around three orders of magnitude lower than k1.
| DPCPC | BMSC | |
|---|---|---|
| k 1 (l mol−1 s−1) | 5.30 × 10−4 | 1.60 × 10−2 |
| k 2 (l mol−1 s−1) | 2.60 × 10−4 | 1.67 × 10−5 |
| k 3 (l mol−1 s−1) | 2.00 × 10−4 | 1.80 × 10−3 |
| k 4 (l mol−1 s−1) | 4.56 × 10−4 | 3.70 × 10−5 |
| K eq. HP | 2.0 | 1.0 × 103 |
| K eq. FP | 4.4 × 10−1 | 4.9 × 101 |
The equilibrium concentrations of the reaction mixtures are represented in Fig. 3, where it becomes apparent that, even at equilibrium, considerable amounts of DPCPC remain present in the reaction mixture. For both reaction steps the equilibrium constant K (Keq.) can be calculated using eqn (1), where the leaving group is either structure XVIII (Scheme 3) or XXIII (Scheme 4) and carbonate is either structure XIV (Scheme 3) or XIX (Scheme 4).
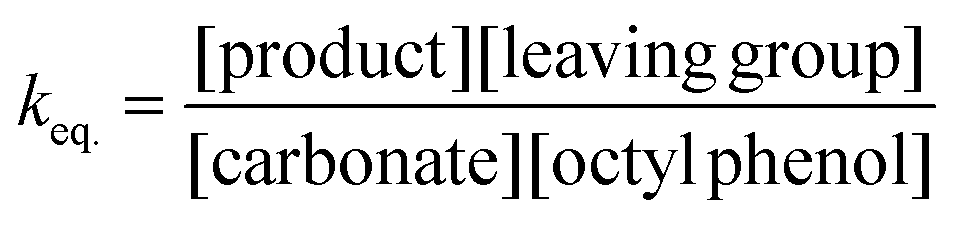 | (1) |
As represented in Fig. 3, and as also indicated by the values of Keq., it can be observed that the experiments with BMSC drive toward the formation of full product, while main reaction product in the experiments with DPC(PC) is the half product.
Besides the faster conversion, the methyl salicylate produced as condensation product during the polymerization has favorable properties compared to the unsubstituted phenolic leaving groups (as produced in the traditional melt transcarbonation process). Its lower solubility in the polymer product mixture and an internal hydrogen bond that stabilizes the structure (Chart 2) makes it less likely to react with the carbonate linkages of the formed polymer chains.
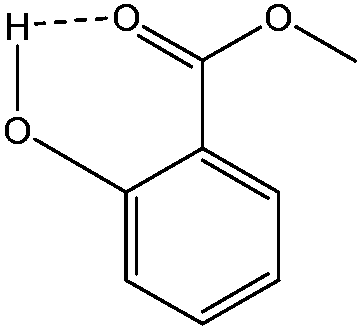 | ||
| Chart 2 Methyl salicylate, showing internal hydrogen bond. | ||
The reactivity benefit of BMSC, as shown by reaction rate coefficients and the equilibrium constant, can lead to various advantages in the polymerization process, enabling reduced reaction times and possibly lower polymerization temperatures, making the polymerization of thermally unstable and/or volatile monomers possible.
Resorcinol copolycarbonate
To demonstrate the benefits of activated carbonates, the copolymerization of resorcinol (RS) with BPA is studied. Resorcinol is a monomer expected to have improved flow and therefor a structure of interest for the preparation of polycarbonates (Chart 3). We were not able to find any literature references for the glass transition temperature of the resorcinol homopolycarbonate, but based on extrapolation using the Fox–Flory equation on known resorcinol copolymers,17 we expect this Tg to be ∼85 °C. This value is less interesting for commercial applications and as such, resorcinol is used as comonomer in combination with BPA. | ||
| Chart 3 Resorcinol carbonate. | ||
Due to its relatively low boiling point (277 °C), loss of monomer during the polymerization is a potential issue and a limiting factor in the conventional melt process. Experiments were carried out, comparing the two types of diaryl carbonates (DAC) and their effect on the polymerization of BPA with resorcinol as a comonomer (Table 3). For both systems the optimal conditions were selected, varying temperature and pressure profile and also ratio of reactants, targeting Mn > 15 × 103 dalton. The polymerization using BMSC is shorter than that using DPC, requiring 40 min total reaction time versus 135 min for the polymerization with DPC. Also, the required temperature in the last step of the polymerization is lower, 280 °C with BMSC vs. 300 °C required for polymerizations with DPC. Even with this milder profile, the molecular weight build-up is comparable. In these experiments the benefit of the higher reactivity of BMSC manifests itself in the higher percentage incorporation of resorcinol, where entry 1 (DPC) shows only 65.4% incorporation of the total charged amount of resorcinol. The resorcinol which is not accounted for in the polymer is lost to the overhead along with the phenol stream. Entry 2 (BMSC) has 94% incorporation of resorcinol into the polymer, and only a limited amount of comonomer is lost during the polymerization.
| 1 | 2 | |
|---|---|---|
| a Diaryl carbonate (DAC). b Dihydroxy aromatic compound (DHA). c Relative to PS standards. | ||
| Mol ratio BPA:RS charged |
80:20 |
80:20 |
| DACa |
DPC | BMSC |
| DAC/DHAb |
1:08 |
1:03 |
| M w (dalton)c | 36.5 × 103 | 37.2 × 103 |
| M n (dalton)c | 15.9 × 103 | 16.2 × 103 |
| BPA:Res (%) measured |
87:13 |
81:19 |
| % incorporation of resorcinol | 65.4 | 94.0 |
| T g (°C) | 134 | 128 |
| Theoretical Mn (at p = 1 and no loss) (dalton) | ∼3 × 103 | ∼15 × 103 |
The incorporation of resorcinol was characterized using 13C-NMR; Fig. 4 shows the NMR spectrum of a copolymer consisting of resorcinol and BPA. From these results, it is concluded that the incorporation of resorcinol is much more efficient when using BMSC compared to the case of DPC.
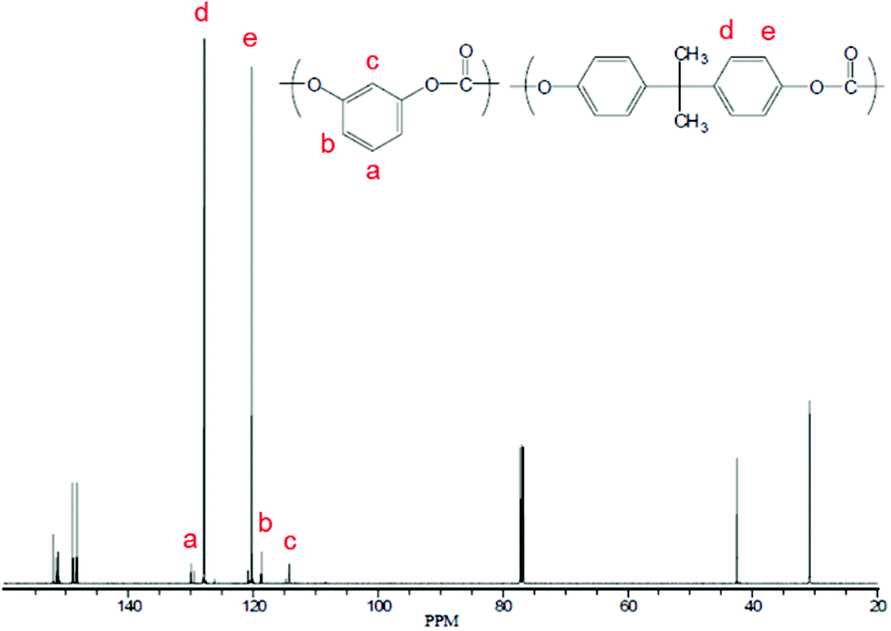 | ||
| Fig. 4 13C spectrum of 80/20 BPA/RS copolycarbonate prepared via the DPC process. | ||
The difference in DAC/DHA ratio between entries 1 and 2 (Table 3) is reflecting the reaction rate coefficient differences for the polymerization with DPC and BMSC, where BMSC is much more effective in reaching full conversion, as indicated by the kinetic studies. Using the Carothers equation (extent of reaction p = 1 and the starting stoichiometric ratio), the theoretical Mn was calculated (included in Table 3). The BMSC-based polymerization closely follows the theoretical predictions, while the DPC-based polymerization follows a different profile. The experiment carried out at a DAC/DHA ratio of 1.08 results in a much higher Mn value than theoretically expected, indicating that the molecular weight built is not only controlled by the formulated monomers.
Next to this Mn result, also the end-group analysis of the materials shows the same pattern. The polymers produced with BMSC have complete capping with methyl salicylate groups, where the DPC based polymer has 73 mol% phenol capped (as determined by 31P-NMR characterization of the OH end-groups) and 27 mol% available OH groups, although the charged ratio of monomers contained a considerable excess of DPC. This can be explained by the fact that DPC is lost from the reactor during polymerization because it forms an azeotrope with phenol as described by Woo and coworkers.21
Both the end-group analysis and the comparison to the theoretical Mn indicates that the molecular weight built with BMSC is mainly determined by the stoichiometry of the reactants. In the case of DPC, the process parameters controlling the removal of condensate and balancing loss of monomers versus temperature (viscosity) are the determining factors for polymerization.
The difference between the incorporated percentages of resorcinol for entries 1 and 2 in Table 3 is reflected in the measured Tg for these materials. With the higher amount of resorcinol, the BMSC-based copolymer has a lower Tg than the DPC-based copolymer, showing a reduction by approx. 17 °C for the 19 mol% copolymer compared to BPA homopolymer (Tg = 145 °C for comparable Mn).
Sterically hindered diols
Another diol, tert-butyl hydroquinone (tBHQ) was studied in its polymerization with BMSC to form polycarbonate in a melt polymerization (Chart 4), investigating the effect of the steric hindrance caused by the tert-butyl group.19 When tBHQ is used in the interfacial process with phosgene, cyclization is observed, which limits the molecular weight build-up.22 Melt polymerizations with DPC were also attempted, resulting in a brown colored material and low Mn.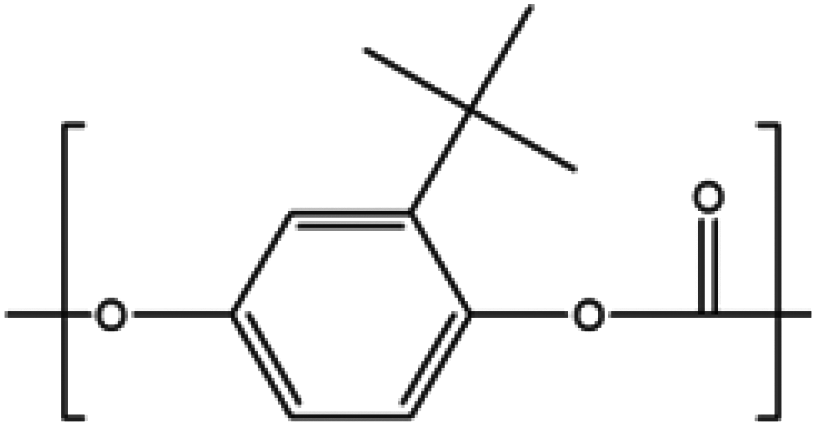 | ||
| Chart 4 tert-Butyl hydroquinone polycarbonate. | ||
In the current study we clearly demonstrate that, although being sterically hindered, the production of the homopolymer of tert-butyl hydroquinone is possible in a robust and scalable manner. A range of molecular weights was produced and their flow properties compared to those of BPA polycarbonate.
NMR characterization of this material (Fig. 5) showed that the incorporation of the monomer happens in a random fashion.
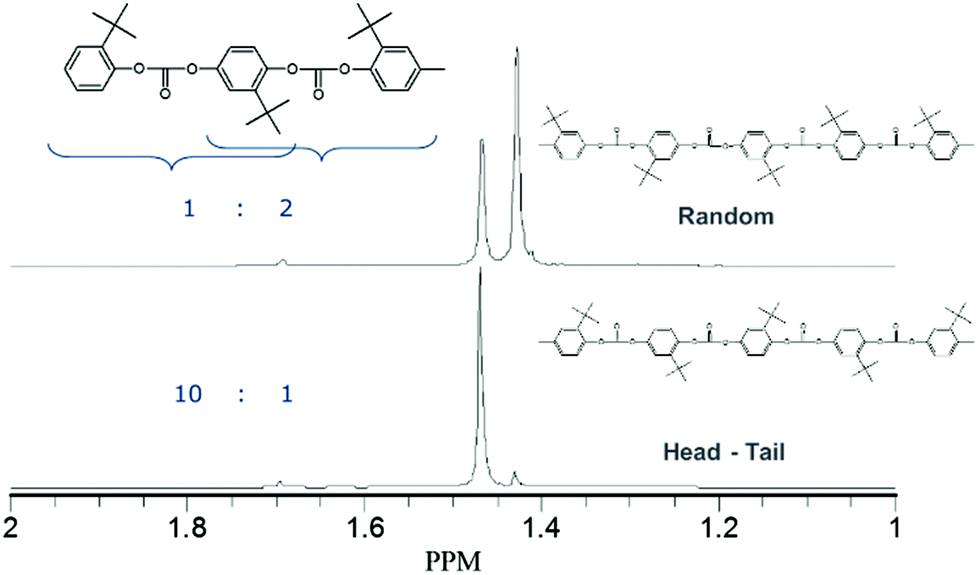 | ||
| Fig. 5 13C spectrum of the tBHQ homopolymer showing random (top) and head-tail (bottom) architectures. | ||
A comparative sample was produced using phosgenation,23 which resulted in a “head to tail” architecture (also shown in Fig. 5), where the distribution of the peaks show a 10:1 ratio in occurrence, clearly much less random than the 1:2 ratio found for the polymer produced in melt polymerization. It cannot be excluded that interface effects during phosgenation play a role in this observation. The random incorporation of tBHQ suggests that the tert-butyl group is not a hindrance for the polymerization when using BMSC as a carbonyl source, but the effect of transcarbonation between polymer chains in the melt, which would lead to the same finding, cannot be excluded from these results.
As indicated by Freitag et al.,2 the substitution on an aromatic ring can have an influence on the flow behavior of the corresponding polymer. Using BMSC in polymerization with tBHQ, hydroquinone (HQ, Chart 5a) and BPA enabled the production of a series of polymers which were compared in terms of melt volume-flow rate (MVR) versus their measured molecular weights (Fig. 6). As can be seen from the graph, a higher flow at similar Mw is measured for the 50% copolymer of tBHQ and BPA vs. the copolymer of HQ and BPA. Clearly, the tBHQ homopolymer showed the highest improvement in melt volume-flow rate over the BPA homopolymer.
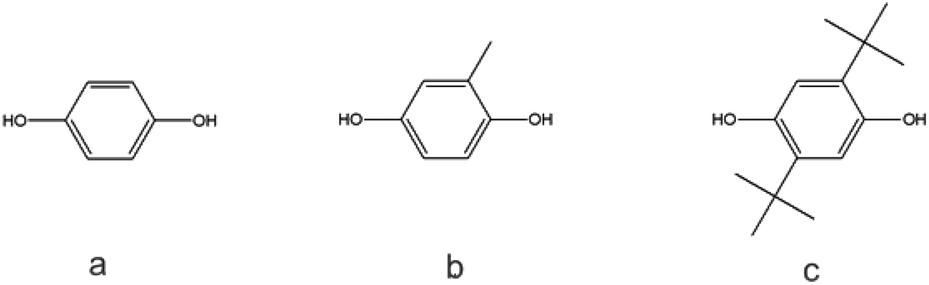 | ||
| Chart 5 (a) Hydroquinone, (b) methyl hydroquinone, (c) di-tert-butyl hydroquinone. | ||
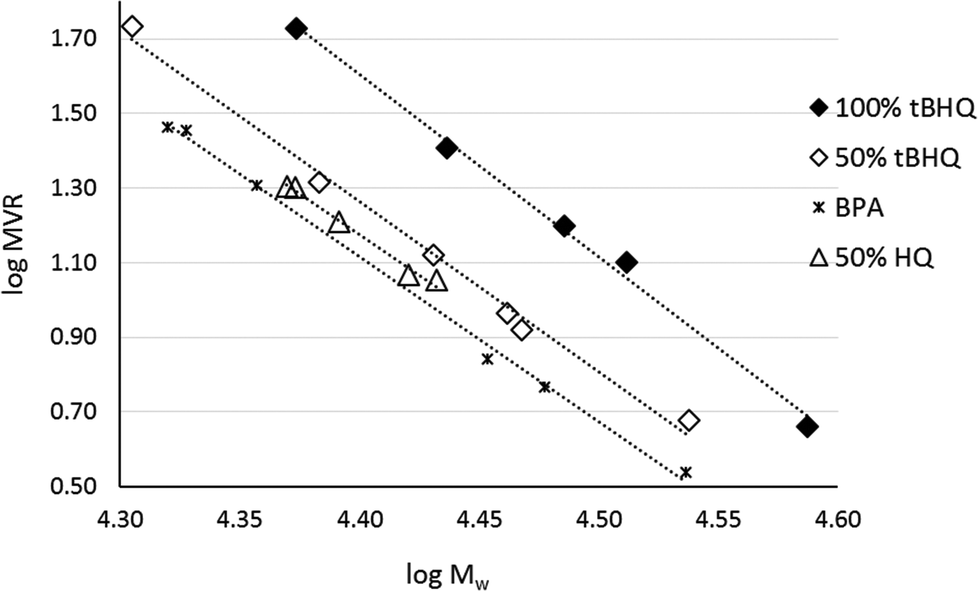 | ||
| Fig. 6 Molecular weight dependence of the melt volume-flow rate for a range of copolycarbonates. | ||
During experimentation, it was found that copolymers containing a higher hydroquinone content showed crystallization. BMSC polymerizations were used to produce a range of hydroquinone-type polycarbonates and to study this effect further. In addition to HQ and tBHQ, also the methyl substituted (MeHQ) and the di tert-butyl substituted monomers (dtBHQ) were used (Chart 5a–c).
The use of BMSC in these polymerizations enabled high conversions and acceptable molecular weight build-up.
Interestingly, the tBHQ resulted in amorphous polymers up to homopolymer, which showed a Tg of close to 130 °C, where the hydroquinone type monomers used for comparison (Chart 5a–c) resulted in polymers showing crystalline behavior when the hydroquinone type monomer was used in high mol%. At lower levels also these polymers showed amorphous behavior.
A range of polymers were prepared with varying percentage of monomers, resulting in an overview of morphology (amorphous or crystalline, Fig. 7), only showing the results for actual measured samples.
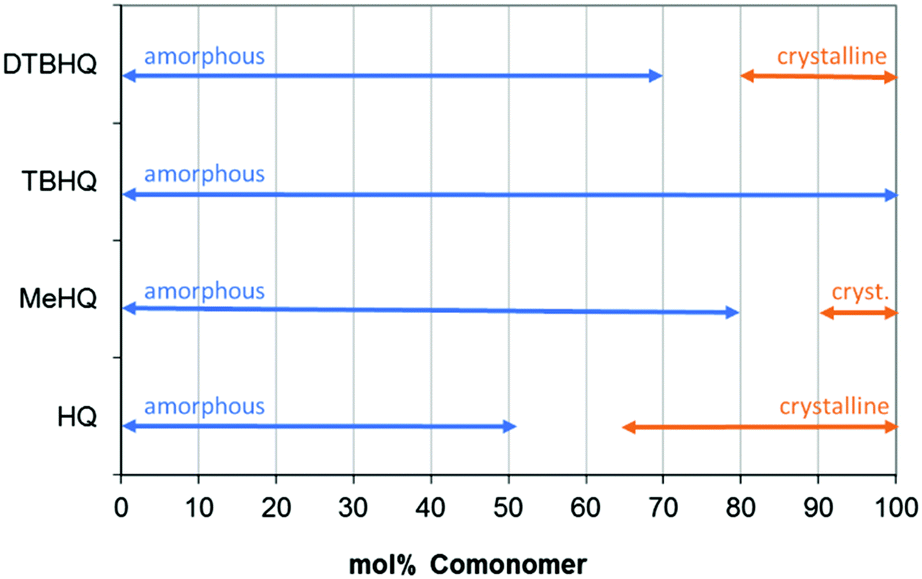 | ||
| Fig. 7 Effect of comonomer content on polymer properties, evaluated using DSC measurements (2nd heating curve). | ||
The crystallization is already observed during the polymerization process (by loss of transparency, turning into powder form and showing crystalline behavior24) and confirmed by DSC afterwards, showing a melting peak in the first heating curve as well as a melting peak in the second heating. Melting temperatures of the materials, measured in the second heating step in DSC, are listed in Table 4. Samples with formulations close to the transition from amorphous materials to crystalline material were selected.
As shown for resorcinol, also the polymerization of tert-butyl hydroquinone benefitted from the use of activated carbonate, with great flexibility in the percentage of comonomer incorporated, noted robustness towards monomer purity and good molecular weight build-up.
Conclusions
The use of bis(methylsalicyl) carbonate (BMSC) as an activated carbonate is a promising approach, enabling the production of polymers containing monomers that are unsuitable for the traditional processes used for polycarbonate synthesis. BMSC clearly shows reactivity benefits in melt polymerization over diphenyl carbonate routes, which was demonstrated by kinetic studies. The advantage in reactivity and ease of process enables the production of copolymers which was demonstrated by examples using resorcinol hydroquinone type of monomers, which showed unique properties.Acknowledgements
The work described in this document is the result of a joint effort from the authors, the SABIC LEXAN™ Technology department, the SABIC Global Analytical Technology team, former colleagues at General Electric (GE) Corporate Research and Development department and many other contributors at SABIC and GE. Special acknowledgement goes to the contributions of Hans Peter Brack, Lina Prada, Pete Davis, Alberto Nisoli and Pieter Janssen.Notes and references
- P. Dutia, Chem. Wkly., 2012, 188–193 Search PubMed.
- D. Freitag, G. Fengler and L. Morbitzer, Angew. Chem., Int. Ed. Engl., 1991, 30, 1598–1610 CrossRef.
- D. G. LeGrand and J. T. Bendler, Handbook of polycarbonate science and technology, Marcel Dekker, 2000 Search PubMed.
- D. J. Brunelle, Macromol. Rep., 1991, 28(Suppl 2), 95–102 Search PubMed.
- K. Mikami, J. Am. Chem. Soc., 2013, 135, 6826–6829 CrossRef CAS PubMed.
- D. J. Brunelle, (Ortho-alkoxycarbonyaryl)-carbonate transesterification, US4323668, 1982.
- H. P. Brack, M. A. J. Campman, J. Garcia Agudo, J. H. Kamps, H. Looij, F. Olmedo Fernancez and D. J. P. M. Willemse, Aliphatic polycarbonates, methods of making, and articles formed therefrom, US7671164, 2010 Search PubMed.
- J. A. Cella, J. H. Kamps, J. P. Lens, K. L. Longley, P. J. McCloskey, N. Ramesh, W. W. Reilly, P. M. Smigelski Jr. and M. B. Wisnudel, Method of polycarbonate preparation, US7115700, 2006 Search PubMed.
- B. Jansen, J. H. Kamps, E. Kung, P. J. McCloskey and P. M. Smigelski Jr., Method of preparing polycarbonate, US7498400, 2009 Search PubMed.
- M. Colonna, C. Berti, E. Binassi, M. Fiorini, S. Sullalti, F. Acquasanta, S. Karanam and D. J. Brunelle, React. Funct. Polym., 2011, 71, 1001–1007 CrossRef CAS.
- H. A. Belfadhel, H. P. Brack, R. Godoy-Lopez and D. J. P. M. Willemse, Method for making carbonates and esters, US7977447, 2011 Search PubMed.
- C. Berti, A. Celli and E. Marianucci, Eur. Polym. J., 2002, 38, 1281–1288 CrossRef CAS.
- H. A. Belfadhel, H. P. Brack, R. Godoy-Lopez and D. J. P. M. Willemse, Method for making Carbamates, Ureas and Isocyanate, US8058469, 2008 Search PubMed.
- J. H. Kamps, E. Kung and J. P. Lens, Poly(carbonate-co-urea) copolymers and melt transesterification method of preparing these copolymers, US7666977, 2010 Search PubMed.
- P. J. McCloskey, T. B. Burnell, D. J. Brunelle, E. W. Shanklin, P. M. Smigelski Jr. and G. Kailasam, Interfacial method of preparing ester-substituted diaryl carbonates, US6420588, 2002 Search PubMed.
- A. Einhorn, Annahlen, 1898, 300, 135 CrossRef CAS.
- R. B. Durairaj, Resorcinol: Chemistry, Technology and Applications, Springer-Verlag, Berlin Heidelberg, 2005, DOI:10.1007/b982897.
- S. M. Gross, W. C. Bunyard, K. Erford, G. W. Roberts, D. J. Kiserow and J. M. DeSimone, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 171–178 CrossRef CAS.
- W. Deits and O. Vogl, J. Macromol. Sci., 1981, 1145–1159 CrossRef CAS.
- R. Butt, Introduction to Numerical Analysis Using MATLAB, Jones and Bartlett Publishers, 2010 Search PubMed.
- B. G. Woo, K. Y. Choi, K. H. Song and S. H. Lee, J. Appl. Polym. Sci., 2001, 80, 1253–1266 CrossRef CAS.
- D. J. Brunelle, Solvent-resistant Polycarbonates, Trends Polym. Sci., 1995, 3, 154–158 CAS.
- J. H. Kamps, E. Kung and B. Mullen, tert-Butylhydroquinone polycarbonates, US7629432, 2009 Search PubMed.
- S. Shafer and J. Lee, Liquid crystal polycarbonates and methods of preparing same, US7211641, 2007 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py00925e |
| ‡ Current address: SABIC, 475 Creamery Way, Exton, PA 19341, USA. |
| § Current address: FRX Polymers, Inc., 200 Turnpike Road, Chelmsford, MA 01824, USA. |
| ¶ Current address: Nuplex Resins, Synthesebaan 1, 4612 RB Bergen op Zoom, the Netherlands. |
| This journal is © The Royal Society of Chemistry 2016 |