
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ternary organic–inorganic nanostructured hybrid materials by simultaneous twin polymerization†
J.
Weißhuhn
a,
T.
Mark
b,
M.
Martin
a,
P.
Müller
b,
A.
Seifert
a and
S.
Spange
*a
aDepartment of Polymer Chemistry, Institute of Chemistry, Technische Universität Chemnitz, Strasse der Nationen 62, D-09111 Chemnitz, Germany. E-mail: Stefan.spange@chemie.tu-chemnitz.de
bBASF SE, Carl-Bosch Strasse 38, D-67056 Ludwigshafen, Germany
First published on 25th July 2016
Abstract
The acid and base catalyzed simultaneous twin polymerization (STP) of various 2,2′-disubstituted 4H-1,3,2-benzodioxasiline derivatives 2a–d with 2,2′-spirobi[4H-1,3,2-benzodioxasiline] (1) are presented in this paper. The products are nanostructured ternary organic–inorganic hybrid materials consisting of a cross-linked organic polymer, silica and a disubstituted polysiloxane. It can be demonstrated whether and in which extent the copolymerization of the two inorganic fragments of 1 and 2 takes place among the STP and how the molar ratio of the two components determines the structure formation of the resulting hybrid material. Steric and electronic effects of the substituents at the silicon center of 2 on the molecular structure formation and the morphology of the resulting hybrid material were investigated by means of solid state CP MAS 29Si and 13C NMR spectroscopy as well as high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). The mechanical properties (hardness and Young's modulus) of the hybrid materials were analyzed by means of nanoindentation measurements.
Introduction
The fabrication of organic–inorganic hybrid materials has been reported for a variety of material combinations. Hybrid materials can be classified by different categories such as molecular composition, morphology, geometry of internal assembly of the components, external shape, or size.1–3 Therefore, the diversity of possibilities of organic–inorganic hybrid material makes the nomenclature difficult.4 According to Sanchez, they can be classified in terms of the nature of the bonds between the organic and the inorganic phase. Class I hybrid materials have only weak bonds (hydrogen, van der Waals or ionic bonds) between the organic and the inorganic compound, whereas in class II materials both phases are connected by covalent bonds.5The synthesis of organic–inorganic hybrid materials containing two different polymers can be carried out by three different strategies: simultaneous polymerization of two different monomers6–8 (SP), polymerization of heterobifunctional monomers9,12,13 (HP) and twin polymerization10,11 (TP).
The twin polymerization is based on single source monomers which combine an organic moiety directly connected to an inorganic one by covalent bonds. The twin monomer undergoes polymerization to a hybrid material which finally consists of an organic and an inorganic polymer. The two polymers are formed in one mechanistically coupled process either through acid or base catalysis or simply by thermal treatment. If phase separation can be avoided, the resulting polymers form interpenetrating networks. The nature of the catalyst, the molecular structure of the twin monomer used and the turnover of the reaction determine whether a class I or class II hybrid-material is obtained and thus the physical properties thereof.10,11
Hybrid materials derived from TP are not limited to the formation of only two polymers. TP also provides an elegant strategy to generate more than two polymers in one procedure by simple mixing of more than one kind of TM and polymerize them to an organic–inorganic hybrid material using the simultaneous TP (STP).14 If twin monomers with the same organic fragment are combined, two theoretically possible scenarios result according to Fig. 1.
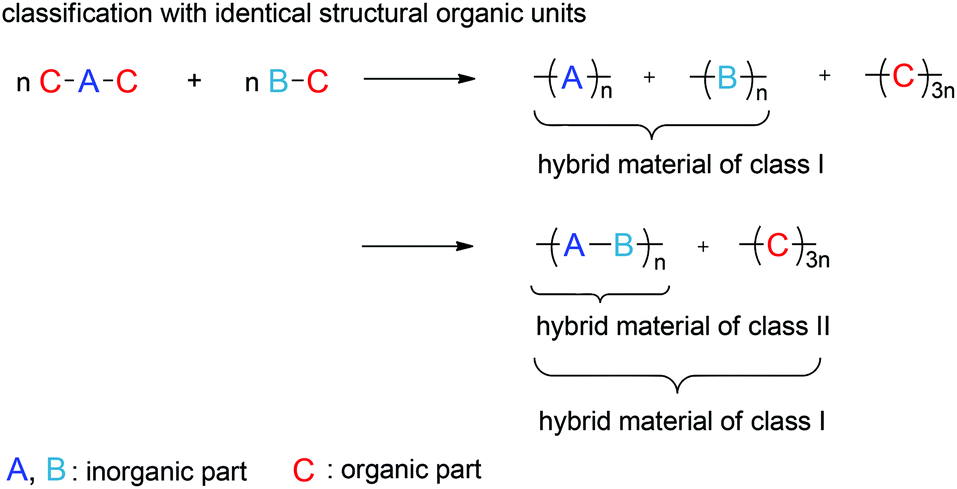 | ||
| Fig. 1 Theoretical scenarios and assumed classification of the composition of ternary organic–inorganic hybrid materials obtained by STP of the monomers C–A–C and B–C. C is the identical structural organic unit in each of the twin monomers.14 | ||
2,2′-Spirobi[4H-1,3,2-benzodioxasiline] 1 bases on salicylic alcohol as organic moiety.10,11,15 It is an ideal twin monomer because polymerization gives phenolic resin and silica without the formation of low molecular weight condensation by-products. The exchange of one unit of salicylic alcohol provides a variety of different monomer structures. The objective of this work is the study of the STP of various 2,2′-disubstituted 4H-1,3,2-benzodioxasiline derivatives 2a–d with 1 (Scheme 1).
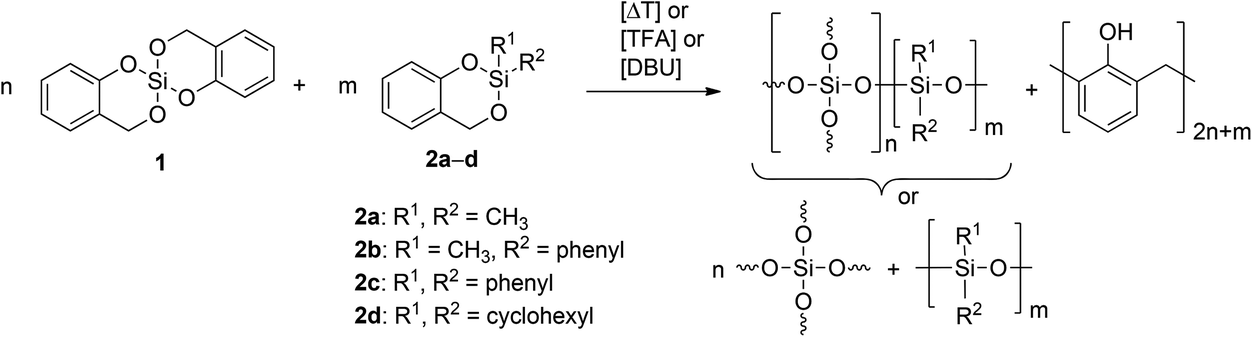 | ||
| Scheme 1 Suggested scenarios of product formation of the STP of 2,2′-spirobi[4H-1,3,2-benzodioxasiline] 1 with various 2,2-disubstituted 4H-1,3,2-benzodioxasilines 2. | ||
The twin polymerization of monomer 1 has been well documented in the literature.16–19 Thermal polymerization, acid or base catalysis is possible to achieve product formation. The STP of 1 and 2 can produce ternary organic–inorganic hybrid materials consisting of a cross-linked organic polymer, silica, and a disubstituted polysiloxane. Silica formed during TP of 1 builds a rigid inorganic network. TP of monomers 2a–d theoretically produces a hybrid material consisting of a polysiloxane as an inorganic polymer and phenolic resin as an organic polymer (Scheme 1). Polysiloxanes as components in hybrid materials have been studied and widely used in industrial fields for many years.20 They are known for their low glass transition temperatures (Tg) due to the long Si–O bond and wide O–Si–O bond angle. The glass transition temperature of polydimethylsiloxane (PDMS) is −123 °C.20 With an increase in the size of the groups R1 and R2 the Tg also increases, as polymethylphenylsiloxane (PMPS) and polydiphenylsiloxane (PDPS) show glass transition temperatures of −28 °C and 40 °C, respectively.20 This low temperature elasticity of the inorganic network and the restriction to the formation of linear polymer chains or rings are the main differences to monomer 1. This needs to be considered when investigating the properties of the resulting polymers from STP of 1 and 2. Phenolic resin as an organic polymer is built from both monomers during STP.
In principle, the simultaneous polymerization of the two monomers 1 and 2 can cause different scenarios depending on the inorganic fragment of the monomers which do or do not react with each other to copolymers. According to Sanchez, the different scenarios would result in different classification of the materials (Fig. 1). Especially the mechanical properties could be influenced by the copolymerization of the inorganic fragments. The polysiloxane of monomer 2 can act as internal plasticizer in the hybrid material due to its low glass transition temperatures (Tg). This covalent linkage is an advantage compared to external plasticizers which can migrate easily and have lower long term stability.
The aim of this study is to analyze whether and in which extent the copolymerization of the two silicon fragments of 1 and 2 takes place among STP and how the molar ratio of the two components affects the overall structure formation and properties of the resulting hybrid material.
Experimental
Materials and methods
All solvents were purified and dried by applying standard techniques. The reactions were carried out with freshly distilled, dried solvents and under inert atmosphere. Salicylic alcohol (99%) was used as purchased from Alfa Aesar. Ethyldichlorosilane (97%) was used as purchased from TCI. Methylphenyldichlorosilane (98%) was used as purchased from Fluka. Diphenyldichlorosilane (99%) and dicyclohexyldichlorosilane (97%) were used as purchased from ABCR. Triethylamine was used as purchased from Roth.1H, 13C{1H} and 29Si{1H} NMR spectra for all compounds were recorded in CDCl3 on a Bruker “Avance DRX 250” (250.1, 63 and 49.7 MHz, respectively) operating at room temperature. Chemical shifts (δ) are reported in parts per million and referenced with the residual undeuterated solvent.
Elemental analyses were determined through a “Vario MICRO” of Elementar Analysensysteme GmbH. Melting point was evaluated using the apparatus “Polytherm A” from WAGNER & MUNZ. Differential scanning calorimetry curves were recorded on a “DSC 30” from Mettler Toledo in nitrogen atmosphere at a volume flow of 50 mL min−1. The measurements were performed at a heating rate of 10 K min−1 from 0 to 300 °C in 40 μl closed aluminium crucibles.
Solid state NMR measurements were performed at 9.4 T on a Bruker Avance 400 spectrometer which were equipped with double-tuned probes capable of MAS (magic angle spinning). The samples were packed in 3.2 mm rotors made of zirconium oxide spinning at 15 (13C) and 12 kHz (29Si). 13C- and 29Si-{1H}-CP-MAS-NMR spectra were acquired using cross polarization (CP) technique with contact time of 3 ms to enhance sensitivity, with a recycle delay of 6 s and 1H decoupling using a TPPM (two puls phase modulation) puls sequence. The spectra are referenced with respect to tetramethyl silane (TMS) using TTSS (tetrakis(trimethylsilyl)silane) as a secondary standard (3.55 ppm for 13C, −9.5 ppm for 29Si). If not stated otherwise, all spectra were acquired at room temperature.
In principle, the cross polarization technique used is not quantitative because the efficiency of polarization transfer from 1H to 29Si is different for the different silicon-species, depending on experimental parameters like the contact time. The intensity of NMR-signals of silicon atoms with hydrogen in close proximity (D-species) is enhanced. As a consequence it is not useful to integrate these spectra (D vs. Q) because it will not give the real concentration. However, all measurements were performed under identical experimental conditions. This allows to compare the spectra of the hybrid materials among each other.
Ultra-thin samples for TEM were prepared by Ultramicrotomy. The polymer was therefore embedded in Epofix resin (Struers; Denmark) and cut at room temperature. HAADF-STEM images were recorded on a Tecnai G2-F20ST machine (FEI Company, USA) operated at 200 keV. Images were evaluated using the Olympus (Japan) iTEM 5.2 (Build 3554) and FEI TIA 4.1.202 software packages. In the HAADF-STEM images presented in this work, bright contrast corresponds to heavier elements like Si or denser sample regions.
The nanoindentations were conducted using a Nanoindenter UNAT with a Berkovich diamond indenter. The hardness and Young's Modulus were measured using the following experimental parameters: maximum load of 5 mN, the holding time was 180 s, 18–28 experiments of indentation were performed for each sample at 18–28 different locations, the average of all measurement for each sample was used. The experiments were carried out at room temperature. The Oliver and Pharr method was employed to extract the mechanical properties using a Poisson's ratio of 0.4.
Synthetic procedures
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) (cm−1) = 3023, 2961, 1607, 1485, 1275, 1125, 1030, 912, 830; EA: Calc. for C14H14O2Si (242.35): C 69.38, H 5.82; Found: C 69.20, H 5.93.
(cm−1) = 3034, 2967, 1607, 1485, 1252, 1117, 1022, 910, 695; EA: Calc. for C19H16O2Si (304.42): C 74.96, H 5.30; Found: C 74.97, H 5.33.
(cm−1) = 3024, 2921, 2666, 1607, 1485, 1275, 1105, 1032, 909, 754; EA: Calc. for C19H28O2Si (316.51): C 72.10, H 8.92; Found: C 71.59, H 8.99.
(cm−1) = 3023, 2961, 1607, 1485, 1275, 1125, 1030, 912, 830; EA: Calc. for C14H14O2Si (242.35): C 69.38, H 5.82; Found: C 69.20, H 5.93.
(cm−1) = 3034, 2967, 1607, 1485, 1252, 1117, 1022, 910, 695; EA: Calc. for C19H16O2Si (304.42): C 74.96, H 5.30; Found: C 74.97, H 5.33.
(cm−1) = 3024, 2921, 2666, 1607, 1485, 1275, 1105, 1032, 909, 754; EA: Calc. for C19H28O2Si (316.51): C 72.10, H 8.92; Found: C 71.59, H 8.99.
Polymerization
The combination of the desired monomers 1 and 2a–d was mixed and heated at 80 °C to give a clear melt. After cooling the mixture at room temperature for 5 minutes the catalyst was added. Coloured monolithic polymers are obtained due to the phenolic resin compound. The polymers cooled to room temperature and dried in a desiccator over calcium chloride. In case of the trifluoromethanesulfonic acid (TFA) the temperature was 85 °C with a monomer/catalyst ratio of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Base catalysis with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) used temperatures between 100 to 140 °C and a monomer/catalyst ratio of 50:1 or 100:1.
:1. Base catalysis with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) used temperatures between 100 to 140 °C and a monomer/catalyst ratio of 50:1 or 100:1.
Extraction
About 1 g of the resulting powdered polymer material was placed in an extraction thimble (19 × 90 mm) of Fischerbrand and soxhlet-extracted with 200 mL dichloromethane for 30 h. The residue was dried at 40 °C and 50 mbar until the weight was constant. Excess of dichloromethane was removed under reduced pressure to give the extract.Results and discussion
Simultaneous twin polymerization of two monomers – ternary organic–inorganic hybrid materials
The synthesis of the twin monomers 2a–d is shown in Scheme 2. Homo twin polymerization of 2a–d was also performed. The base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, pKaqa = 13.4, pKMeCNa = 24.34)21,22 and the acid trifluoroacetic acid (TFA, pKaqa = 0.0)23 were used as catalysts. The homo twin polymerization of monomer 2 shows high amounts of extractable content, especially concerning the polydialkyl(aryl) siloxane component. Detailed results and discussions can be found in the ESI.† The solubility of the inorganic component of 2 can be prevented by copolymerization with the silicon network derived from 1. Hence, the acid and base catalyzed STP of 1 and 2 was investigated. To adjust the polymerization temperatures for the STP, the mixtures of monomer 1 and monomer 2 were analysed by DSC measurements. Exothermic peaks in the DSC curves can indicate a polymerization of the monomer due to the heat of polymerization. The results of the DSC measurements of the monomer mixtures as function of type of catalysis are presented in Table 1. The corresponding DSC curves are shown in the ESI (Fig. S2, S3, S6 and S7†). The onset temperatures for the thermally induced twin polymerization were monitored by exothermic peaks varying between 184 and 219 °C. There is no clear observable trend regarding the molecular structure of monomer 2. A slightly reduced trigger temperature, compared to pure 1, was found only for the mixture of 1 and 2c.
| Monomer mixture (1:1 w eq.) |
T onset, exo [°C] | ||
|---|---|---|---|
| Thermally induced | DBU | TFA | |
| 1 | 197 | 130 | 25 |
| 1 + 2a | 219 | 106 | 30 |
| 1 + 2b | 210 | 108 | 72 |
| 1 + 2c | 184 | 107 | 25 |
| 1 + 2d | 205 | 107 | 80 |
Base catalyzed twin polymerization is readily suitable for all mixtures of monomers 1 and 2, with polymerization temperatures about 107 °C. Herein the combination of both monomers (except 2d) reduces the trigger temperatures, compared to the single monomers 2, when catalyzed with DBU (Fig. S3†).
In comparison to the single monomers 2 with TFA (Fig. S2†), the polymerization trigger temperatures of the monomer mixtures with TFA are reduced below 85 °C.
According to the results of the DSC measurement, the polymerization experiments were performed in monomer melt with TFA and DBU at temperatures of 85 °C and 110 °C, respectively.
Acid catalyzed STP
The polymerization experiments for the TFA catalysis are summarized in Table S3† and Fig. 2. The monomer ratio 1:2 n% was varied. Optically transparent and homogenous hybrid materials were obtained for the monomers 2a, 2b and 2c with 1 (Table S3†). The transparency of the hybrid material decreases with increasing amount of monomer 2d. Fig. 2 shows the hybrid materials derived from STP of 1 + 2b as an example in comparison to the materials obtained from STP of 1 + 2d.
 | ||
| Fig. 2 Mass loss by extraction as a function of the monomer composition 1:2a–d in the hybrid material obtained from STP using TFA as catalyst and pictures of the monoliths of 1 + 2b and 1 + 2d with different monomer ratios derived from TFA catalyzed STP. | ||
The effect of silicon alkyl/aryl substituents and monomer ratios of 1 and 2 on the molecular structure of the hybrid materials were studied by solid state NMR spectroscopy. The 13C-{1H}-CP-MAS-NMR spectra show typical signals for phenolic resin and additional signals for the dialkyl(aryl)siloxane units, which can be observed at 28 ppm for the dicyclohexylgroups of monomer 2d, at 115 to 135 ppm for the diphenylgroups of monomer 2c, at 115 to 135 ppm and 0.5 ppm for the phenyl and methyl groups of monomer 2b and at 0.5 ppm for the dimethyl group of monomer 2a.
Methylene groups of phenolic resin give a signal at 31–36 ppm. The substitution pattern of the phenolic resin can be estimated from the chemical shifts of the aromatic carbons and of the methylene groups. Non-substituted aromatic carbon atoms in the ortho position to the –OR′-group show signals at 115–119 ppm (o,p′-substitution, see signal 4 in Fig. 3A) and in para position at 120–124 ppm (o,o′-substitution, see signal 3 in Fig. 3A). meta Position linkages can be monitored at 127–135 ppm, but they overlap with residual signals from ortho- and para-substituted phenolic carbon atoms (signals 2 in Fig. 3A). The phenoxy carbon atoms appear at 149–156 ppm for both, Caryl–OH– and Caryl–O–Si-units, and can therefore not be separated.26 Although 13C-{1H}-CP-MAS-NMR spectra are not really quantitative, there is a high amount of o,p′-substitution in the hybrid material as signal 4 is more intense than signal 3 (Fig. 3A), assuming the polarization transfer for all aromatic carbons is in the same order of magnitude.
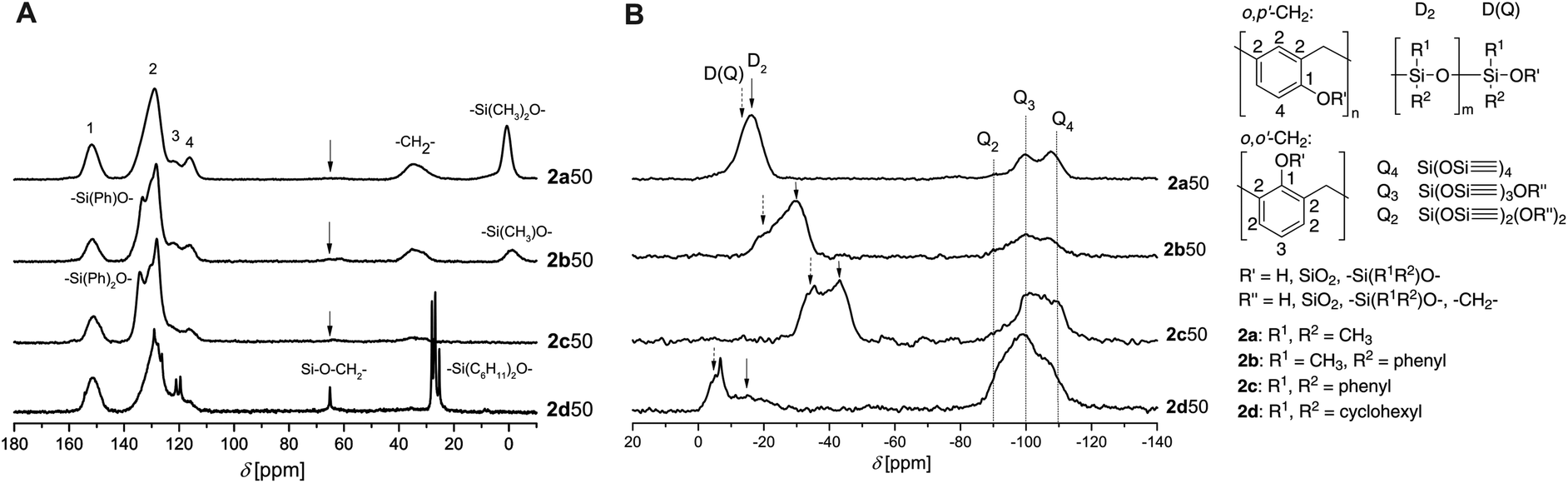 | ||
| Fig. 3
A
13C-{1H}-CP-MAS-NMR spectra and B29Si-{1H}-CP-MAS-NMR spectra of hybrid materials produced from different monomers 2 at a monomer ratio 1:2 of 50:50 n% (before extraction). | ||
Additional signals at 58–73 ppm (Fig. 3A) indicate methylene carbon atoms27 due to still existing Si–O–CH2 groups of the monomers 2a–d. These units are reactive which is shown in DSC measurements of the hybrid materials derived from monomers 2a–c. An exothermic peak due to thermal post-curing is detected (Fig. S9†). In contrast, no exothermic peaks in DSC curves are observed for hybrid materials based on 2d although there are many sharp signals in the 13C-{1H}-CP-MAS-NMR spectra denoting high mobility in the solid state. These signals originate from unconverted residuals of monomer 2d (signal at 65 ppm) which is also verified through extraction experiments with DCM (Table S3†). Extraction experiments give the possibility to determine the amount of conversion and to study the structure of side reactions and oligomeric products which are removable from the hybrid material. The mass loss after extraction of all samples (STP of 1 and 2) varies significantly from 6–100%, whereas a clear dependence of the extractable fraction on the monomer ratio can be identified (Fig. 2). The mass loss increases with increasing amount of monomer 2 in the hybrid material which relates to the not crosslinked inorganic compound derived from this monomer.
The inorganic moieties of the hybrid material derived from 1 and 2 can either build two homo polymers of SiO2 and polysiloxane or react with each other to build a copolymer. In case of monomer 2a the formation of copolymer was proven by solid state NMR spectroscopy and extraction experiments of the hybrid material with acetone and DCM.14Fig. 3B shows 29Si-{1H}-CP-MAS-NMR spectra of hybrid materials obtained from TFA catalyzed STP of 1 and 2a–d with signals arising from silicon dioxide and polydialkyl(aryl)siloxane units (–SiR1R2O–). Silicon dioxide is characterized through Q2 (−90 ppm), Q3 (−100 ppm) and Q4 (−110 ppm) signals.28,29 Whereas Q4 indicates the formation of a complete condensed SiO2 species with four siloxane bridges connecting to the inorganic network, Q2 and Q3 display partly unconverted Si–O–C or Si–OH units (see inset in Fig. 3).30 D-Structures in the 29Si NMR chemical shift range of 0 ppm to −45 ppm result from the inorganic moiety of monomer 2 and give information about the degree of condensation and the neighbors of the dialkyl(aryl)subtituted Si-species. The D-signals can be separated in D2 and D(Q) signals (see inset in Fig. 3). The products obtained from monomers 2b and 2c with 1 show both signals. This proves the formation of a copolymer with the SiO2 network because the D(Q) type of signal occurs if D-type silicon atoms are bound covalently to silica (Q-species).
Extraction of the hybrid materials with DCM reduces the intensity of the D2 signal compared to the D(Q) signal (Fig. S8†). This is due to the solubility of the polysiloxane homopolymer in the extraction solvent DCM, whereas the polysiloxane cannot be extracted if covalently bound to the silica (= copolymer).
The hybrid material based on monomer 2d also shows additional sharp signals in the 29Si solid state NMR which relates to the 13C solid state NMR spectrum. After extraction with DCM, only weak D2 and D(Q) signals remain in the 29Si solid state NMR spectrum indicating low tendency of copolymerization of polydicyclohexylsiloxane (PDHS) with SiO2 and a higher part of oligomers of PDHS (Fig. 4A). No homopolymerization of 2d to large cycles or high molecular weight PDHS can be noticed which correspondes well with the results of DSC measurements. This confirms the decreasing reactivity of 2 in STP in accordiance to the steric demand of the monomers because monomer 2d has, compared to monomer 2a, very sterically demanding substituents.
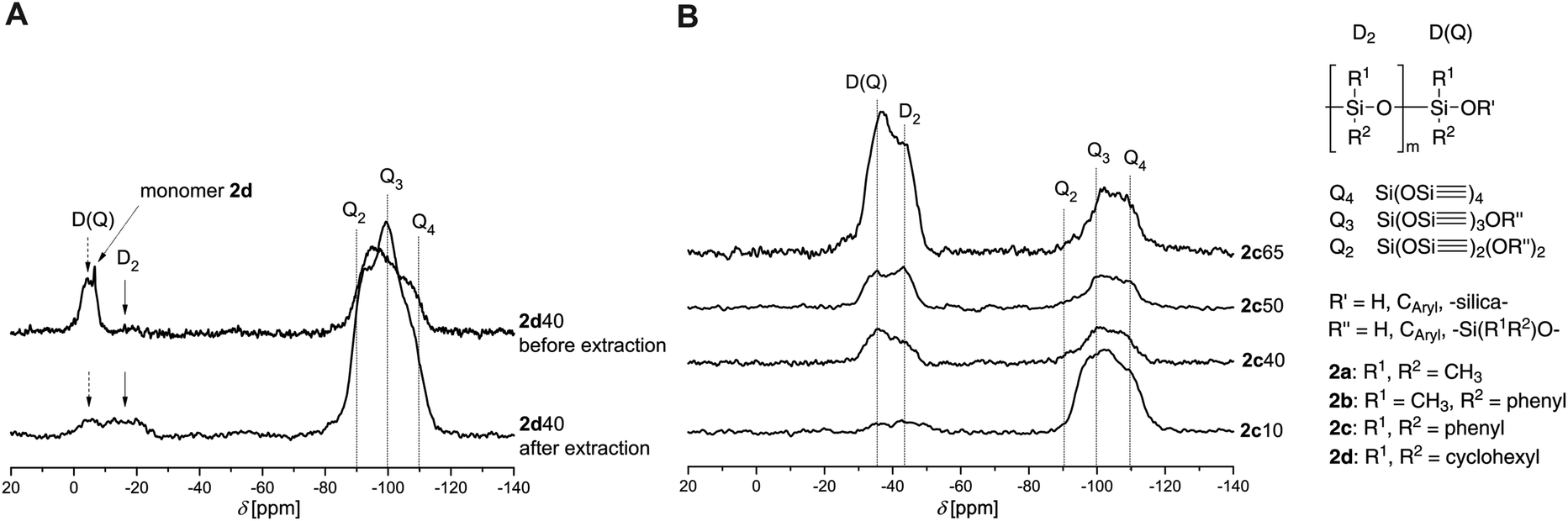 | ||
| Fig. 4
A
29Si-{1H}-CP-MAS-NMR spectra of the hybrid material 2d40 before and after extraction. B29Si-{1H}-CP-MAS-NMR spectra of hybrid materials containing monomer 2c at different monomer ratios (n%) and a concentration of TFA of 20:1 nM:nTFA (before extraction). | ||
Moreover, hybrid materials derived from 2a–c show similar ratios of Q2, Q3 and Q4. In case of 2d a higher amount of Q2 is detected. Monomer 2d seems to act as a solvent, to dilute the polymerization mixture and to disrupt the TP of 1 due to its very low tendency to polymerize.
Different monomer ratios used for the synthesis of the hybrid materials are, of course, also reflected in the corresponding intensities of D- and Q-signals in the 29Si solid state NMR spectra (Fig. 4B). The intensity of the D signals increases with increasing amount of monomer 2 in the polymerization mixture which is shown for monomer 2c as an example. It must be noted that the used cross polarization (CP) technique is not quantitative. However, all measurements were performed under identical experimental conditions and this allows to compare the spectra of the hybrid materials among each other.
Base catalyzed STP
The polymerization experiments of STP catalyzed with DBU are summarized in Table 2. The following experimental parameters were modified: concentration of catalyst, polymerization time and polymerization temperature. The monomer ratio 1:2 was 50:50 n% for all experiments.
:1 = 50:50 [n%]. The naming of the samples is based on the experimental parameters used. The first part of the name represents the molar ratio of monomers to catalyst (M:I) like 50:1 n% is abbreviated as 50. The next part indicates the specific monomer 2 and the molar ratio of that monomer used in the mixture
| Experiment | Concentration of catalyst M:I [n%] |
Time | Temperature | Mass loss by extraction [%] |
|---|---|---|---|---|
| 50/2a50 | 50:1 |
4 h | 110 °C | 2 |
| 100/2a50 | 100:1 |
8 h | 110 °C | — |
| 300/2a50 | 300:1 |
4 h | 130 °C | — |
| 50/2b50 | 50:1 |
3 h | 110 °C | 14 |
| 100/2b50 | 100:1 |
3 h | 110 °C | 25 |
| 50/2c50 | 50:1 |
6 h | 140 °C | 23 |
| 100/2c50 | 100:1 |
6 h | 140 °C | 28 |
| 50/2d50 | 50:1 |
8 h | 110 °C | 62 |
Compared to TFA catalysis, the polymerizations were performed with reduced catalyst concentrations because otherwise optically inhomogeneous products are formed.31 This is due to a mixing problem when adding the catalyst to the monomer mixtures resulting in very high local concentrations of active species at high concentrations of DBU. The concentration of catalyst strongly influences the necessary polymerization time and temperature required to form a hybrid material. And therefore, in comparison to the acid catalyzed STP, the base catalyzed STP demands longer polymerization times and higher polymerization temperatures to form solid hybrid materials. Although the temperature of 110 °C was found to be a suitable polymerization temperature for all mixtures according to previous DSC measurements (Table 1), the temperature was increased up to 140 °C for three experiments to ensure solidification of the hybrid materials. The polymerization time differs from 3 to 8 hours. The products are optically transparent and homogenous hybrid materials (Table S4†). Only the material obtained from monomer 2d is non transparent and flexible due to 2d showing low tendency to perform TP as shown for TFA catalyzed STP.
The 13C-{1H}-CP-MAS-NMR spectra of the hybrid materials show the typical signals for phenolic resin and dialkyl(aryl)siloxane units, indicating that the molecular structure is similar to the acid catalyzed polymerization. Fig. 5 shows the CP-MAS-NMR spectra of the products obtained from 2b with different concentration of catalyst as an example. It can be noticed that the ratio of signal 3 and 4 indicating o,p′- and o,o′-substitution is different from the acid catalyzed polymerization. In case of the base catalyzed polymerization o,o′-substitution is preferred due to higher polymerization temperatures. In case of thermal TP of 1, o,o′-substitution is clearly obtained in majority. The effect of rearrangement of o,p′-substitution to o,o′-substitution at higher temperatures has been described in literature.32
 | ||
| Fig. 5 A 13C-{1H}-CP-MAS-NMR spectra and B29Si-{1H}-CP-MAS-NMR spectra of hybrid materials produced from monomer 2b as an example with different concentration of DBU (both before extraction). | ||
The 29Si-{1H}-CP-MAS-NMR spectra of the hybrid materials show D signals for the polydialkyl(aryl)siloxane units and Q signals for the SiO2 network. The D signals consist of a D2 and a D(Q) signal which proves copolymerization of the polydialkyl(aryl)siloxane and the silica. Compared to Q2 and Q3 signals, the silica network shows large amounts of Q4 signals. This indicates an early formation of the SiO2 network at an already low conversion of monomers which can be traced back to the high polymerization temperatures. Nevertheless, many reactive SiOR intermediates remain which can be removed for example by extraction of the hybrid materials (Table S4†). The extracts also consist of oligomers of the phenolic resin built during STP. However, a higher concentration of catalyst decreases the mass loss by extraction. Thus, conversion of monomers is enhanced in case of M:I of 50:1 n% in comparison to 100:1 n%. For both concentrations of DBU the resulting hybrid materials can be post cured as monitored by DSC measurements which is shown for monomer 2b (Fig. S11†). Higher concentrations of DBU (50:1 n%) give an exothermic signal starting at 71 °C during heat treatment. This temperature is typical for the base catalyzed STP of 2b and 1 (Table 1). Thus, small amounts of catalytic active species remain in the hybrid material. In this case, increasing the polymerization time can increase the conversion of the monomers. If a smaller amount of catalyst is applied, the post curing of the material occurs at 168 °C. This temperature is close to the thermal induced polymerization of 2b and 1 which takes place at 200 °C.
Morphology and mechanical properties
HAADF-STEM analysis of selected samples show fine and dense silicon rich structures with nanoclusters of 1–2 nm in size. The size of nanostructures can be controlled by the ratio of the two monomers 1 and 2, as shown in Fig. 6 for monomer 2a. An increasing amount of monomer 1 in the reactant mixture produces smaller nano domains of the final product. | ||
| Fig. 6 HAADF-STEM images of hybrid materials derived from 1 and 2a initiated with TFA containing 75%, 50% and 25% of 2a. | ||
All hybrid materials derived from TFA catalyzed STP of 1 and 2 show similar nanostructures without any clear trend regarding the chemical structure of different monomers 2a–d (Fig. 7). Electron micrographs of the hybrid material obtained with 1 and 2d in equimolar ratio (2d50) show a special feature within the samples: a very porous structure with holes in the size of several hundred nanometers is visible at lower magnification. The pores result from unconverted monomer 2d which is dissolved during sample preparation for STEM measurements. The remaining material consists mainly of polymer derived from 1 with only small amount of 2d and shows the nanostructured domains of the phenolic resin and the silica/oligodicyclohexanesiloxane. The high amount of unconverted monomer 2d and hence the phase separation between monomer and polymer in the nm scale is the reason for the macroscopic non-transparent monoliths which is also confirmed by the STEM analysis. DBU catalysis of monomers 1 and 2 in equimolar ratio results in hybrid materials which typically show nanostructure domains of 1–2 nm that are similar to acid catalysis.
 | ||
| Fig. 7 HAADF-STEM images of hybrid materials derived from STP 1 and 2 using TFA and DBU as catalyst. | ||
The Young's modulus and hardness of the surface of the hybrid materials obtained from the acid catalyzed STP of 1 and 2 were measured by nanoindentation analysis (Table S5†). The mechanical attributes as a function of the monomer ratio were measured using monomer 2b as selected example. Fig. 8 illustrates a linear trend of Young's modulus and hardness as function of the 1:2b ratio. Both, Young's modulus and hardness, increase with an increasing amount of monomer 1 in the hybrid material. This trend was also observed for the combination of monomer 1 and 2a in the same order of magnitude.15 For this reason, the experimental values were hereafter compared to values of pure PDMS found in the literature.
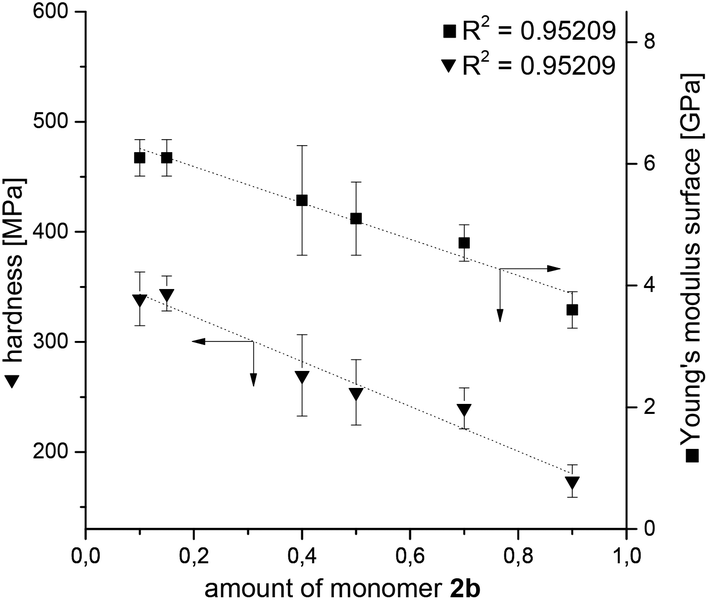 | ||
| Fig. 8 Hardness and elastic modulus as function of monomer composition of 2b:1 using TFA as catalyst (n = 17–28, error bars cover 3σ). | ||
Whereas pure PDMS samples have a hardness of 28 MPa33 and a Young's modulus of about 3.2 GPa32,33 the nanocomposites with SiO2, PMPS and phenolic resin exceed this value and reach values of 344.0 MPa and 6.1 GPa, respectively. This observation can be explained by an increasing SiO2 content and therefore the total inorganic amount increases, hence stronger formation of Si–O–Si bonds occurs. This results in an increasing resistance to deformation.
For nanometer particle-reinforced polymer composites a Young's modulus of 4.2 GPa and a hardness of 0.25 × 103 MPa was observed through nanoindentation.34 These hybrid materials were prepared by ultraviolet curable technique based on epoxy acrylate as continuous organic phase and silica nanoparticles (5%) as discontinuous inorganic phase In this case, nanoindentation hardness also increased with the content of nanosilica.
Both, Young's modulus and hardness, decrease with an increasing steric demand of the substituents of 2 for a 1:2 ratio of 50:50 (Table S5, Fig. S12†). No values were measured for the material TFA/2d50/20 due to its softness. It is assumed that the mechanical properties of the resulting hybrid materials are also influenced by the glass transition temperatures of the polysiloxanes originating from monomer 2. The Tg increases from PDMS and PMPS to PDPS with increasing size of the substituents at the silicon atom.20 However, it could be shown that the values for hardness and Young's modulus decrease from PDMS to PDPS. It is important to note, that the nanoindentation was measured at the not extracted hybrid materials. These show a certain amount of monomer or oligomers resulting from an incomplete turnover which are removable and make the material soft. This amount increases from PDMS to PDPS although the materials were treated at the same reaction parameters (Fig. 2). The effect of different organofunctional groups on the hydrolysis reaction at the silicon of silane esters for the acid and base catalysed reaction was studied in the literature.35 Herein the reaction rate decreased from methyl- to phenyl- to cyclohexyl-substitution. This is in correlation with the different reactivity of the monomer 2a–2d and hence less turnover during polymerization reaction resulting in less hard hybrid materials.
Conclusion
Ternary organic hybrid materials consisting of phenolic resin, SiO2 and polysiloxane were synthesized by acid or base catalyzed simultaneous twin polymerization of two twin monomers (1 and 2a–2d). Phenolic resin as organic polymer is formed by both of the monomers, but the different monomers 1 and 2 deliver different inorganic polymers. In case of monomer 2a, 2b and 2c the inorganic fragments react with each other to copolymers. 2d shows only low tendency to form copolymers with SiO2. The corresponding covalent linkage were monitored by 29Si-{1H}-CP-MAS-NMR spectroscopy showing D(Q) signals. Hence, STP of 1 and 2 proceeds effective according to the reactivity of the monomers. Increasing steric demand of the substituent in the 2,2′-disubstituted 4H-1,3,2-benzodioxasiline lead to low and incomplete conversion. The nature of the catalyst influences the course of reaction and the molecular structure of the products.According to Sanchez and Mackenzie, the products represent a new class of hybrid materials having one organic polymer mixed on the nanoscale with two inorganic polymers linked through covalent bond (Co-STP). HAADF-STEM analysis verified the dense nanostructure with domains of 1–2 nm in size. The Young's modulus and hardness at the surface of the hybrid materials can be adjusted by the monomer ratio 1:2.
Acknowledgements
The authors gratefully acknowledge financial support from the DFG Forschergruppe 1497 and DFG Sp 392/34-2. Thanks are also given to BASF SE for financial support and TEM measurements. Furthermore the authors thank Alexandra Schuberth and Natalia Nier for nanoindentation measurements.Notes and references
- G. Kickelbick, Prog. Polym. Sci., 2003, 28, 83–114 CrossRef CAS.
- C. Sanchez, B. Julian, P. Bellville and M. Popall, J. Mater. Chem., 2005, 15, 3559–3592 RSC.
- V. A. Gerasin, E. M. Antipov, V. V. Karbushev, V. G. Kulichikhin, G. P. Karpacheva, R. V. Talroze and Y. V. Kudryavtsev, Russ. Chem. Rev., 2013, 82, 303–332 CrossRef.
- J. D. Mackenzie, J. Sol–Gel Sci. Technol., 1994, 2, 81–86 CrossRef CAS.
- C. Sanchez and F. Ribot, New J. Chem., 1994, 18, 1007–1047 CAS.
- H. Müller, P. Rehak, C. Jäger, J. Hartmann, N. Meyer and S. Spange, Adv. Mater., 2000, 12, 1671–1675 CrossRef.
- S. Grund, A. Seifert, G. Baumann, W. Baumann, G. Marx, M. Kehr and S. Spange, Microporous Mesoporous Mater., 2006, 95, 206–212 CrossRef CAS.
- C.-L. Chiang, C.-C. Ma, D.-L. Wu and H.-C. Kuan, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 905–913 CrossRef CAS.
- B. M. Novak, Adv. Mater., 1993, 6, 422–433 CrossRef.
- S. Spange and S. Grund, Adv. Mater., 2009, 21, 2111–2116 CrossRef CAS.
- S. Spange, P. Kempe, A. Seifert, A. A. Auer, P. Ecorchard, H. Lang, M. Falke, A. Pohlers, M. Hietschold, W. Hoyer, G. Cox, E. Kockrick and S. Kaskel, Angew. Chem., Int. Ed., 2009, 48(44), 8254–8258 CrossRef CAS PubMed.
- L. Ni, N. Moreau, A. Chemtob and C. Croutxé-Barghorn, J. Sol–Gel Sci. Technol., 2012, 64, 500–509 CrossRef CAS.
- A. Chemtob, C. Belon, C. Croutxé-Barghorn, S. Rigolet, L. Vidal, J. Brendle, J. Mandel and N. Blanchard, Polym. Eng. Sci., 2011, 51, 1466–1475 CAS.
- T. Löschner, A. Mehner, S. Grund, A. Seifert, A. Pohlers, A. Lange, G. Cox, H. J. Hähnle and S. Spange, Angew. Chem., Int. Ed., 2012, 51, 3258 CrossRef PubMed.
- T. Ebert, A. Seifert and S. Spange, Macromol. Rapid Commun., 2015, 36, 1623–1163 CrossRef CAS PubMed.
- P. Kempe, T. Löschner, A. A. Auer, A. Seifert, G. Cox and S. Spange, Chem. – Eur. J., 2014, 20, 8040–8053 CrossRef CAS PubMed.
- P. Kempe, T. Löschner, D. Adner and S. Spange, New J. Chem., 2011, 35, 2735–2739 RSC.
- T. Ebert, G. Cox, E. Sheremet, O. Gordan, D. R. T. Zahn, F. Simon and S. Spange, Chem. Commun., 2012, 48, 9867–9869 RSC.
- I. Tschernook, J. Prehl and J. Friedrich, Polymer, 2015, 60, 241–251 CrossRef.
- R. G. Jones, W. Ando and J. Chownjowski, Silicon containing polymers, Springer, 2000, p. 185 Search PubMed.
- M. S. Miran, H. Kinoshita, T. Yasuda, M. A. B. H. Susan and M. Wantanabe, Phys. Chem. Chem. Phys., 2012, 14, 5178–5186 RSC.
- I. Kaljurand, A. Kütt, L. Sooväli, T. Rodima, V. Mäemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS PubMed.
- D. MacFarlane, J. Pringle, K. Johansson, S. Forsyth and M. Forsyth, Chem. Commun., 2006, 1905–1917 RSC.
- R. Cragg and M. Nazery, J. Organomet. Chem., 1974, 71, 225–230 CrossRef CAS.
- R. H. Cragg and R. D. Lane, J. Organomet. Chem., 1978, 154, C6–C8 CrossRef CAS.
- M. Stevens, Polymer Chemistry: An Introduction, Oxford University Press, New York, Oxford, 1999 Search PubMed.
- R. Rego, P. Adriaensens, R. Carleer and J. Gelan, Polymer, 2004, 45, 33–38 CrossRef CAS.
- E. Lippmaa, M. Mägi, A. Samoson, G. Engelhardt and A.-R. Grimmer, J. Am. Chem. Soc., 1980, 102, 4889–4893 CrossRef CAS.
- G. Maciel and D. Sindorf, J. Am. Chem. Soc., 1980, 102, 7607–7608 CrossRef.
- K. Haraguchi, Y. Usami and Y. Ono, J. Mater. Sci., 1998, 33, 3337–3334 CrossRef CAS.
- T. Löschner, Ph. D. Thesis, TU Chemnitz, 2013 Search PubMed.
- M. Rahman, R. Dammel and D. Durham, Proc. SPIE - Int. Soc. Opt. Eng., 1994, 2195, 685–695 CrossRef CAS.
- C. A. Charitidis and E. P. Koumoulos, Plast., Rubber Compos., 2012, 41, 88–93 CrossRef CAS.
- G. C. Xu, A. Y. Li, L. D. Zhang, X. Y. Yu, T. Xie and G. S. Wu, J. Reinf. Plast. Compos., 2004, 23, 1365–1372 CrossRef CAS.
- F. D. Osterholtz and E. R. Pohl, J. Adhes. Sci. Technol., 1992, 6, 127–149 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py00903d |
| This journal is © The Royal Society of Chemistry 2016 |