
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Micellar nanoparticles with tuneable morphologies through interactions between nucleobase-containing synthetic polymers in aqueous solution†
Zan
Hua
a,
Anaïs
Pitto-Barry
 a,
Yan
Kang
a,
Nigel
Kirby
b,
Thomas R.
Wilks
a and
Rachel K.
O'Reilly
*a
a,
Yan
Kang
a,
Nigel
Kirby
b,
Thomas R.
Wilks
a and
Rachel K.
O'Reilly
*a
aDepartment of Chemistry, University of Warwick, Library Rd, Coventry, CV4 7AL, UK. E-mail: Rachel.OReilly@warwick.ac.uk
bAustralian Synchrotron, 800 Blackburn Road, Clayton, Victoria 3168, Australia
First published on 8th June 2016
Abstract
Herein, we report the preparation of nucleobase-containing synthetic amphiphilic diblock copolymers using RAFT polymerization. Well-defined spherical micelles can be formed in aqueous solutions through the self-assembly of the amphiphilic copolymers, with the nucleobase functionality sequestered in the core of the particles. Following assembly, copolymers with the complementary nucleobase were introduced into the preformed micellar solutions. This addition induced a change in nanostructure size and morphology and this reorganization was fully characterized by DLS, TEM, SLS and SAXS analysis. The insertion of copolymers with the complementary nucleobase into formed micelles was also confirmed by 1H NMR and UV-vis spectroscopy. For micelles consisting of moderately short hydrophobic blocks, upon the addition of complementary nucleobase copolymer a decrease in size was observed but without any accompanying morphological change. For micelles formed from longer hydrophobic blocks, a morphological transition from spheres to cylinders and then to smaller spheres was observed upon increasing the amount of the complementary copolymer. This work highlights how complementary nucleobase interactions can be used to induce nanostructure reorganization and through a simple mixing process provide access to different nanostructure sizes and morphologies.
Introduction
The self-assembly of amphiphilic block copolymer micelles has attracted considerable attention due to the wide range of morphologies that can be formed, and the possibility to incorporate specific functional molecules, potentially leading to a number of novel applications. Different morphologies include spherical micelles,1 cylindrical micelles,2–5 bicontinuous structures,6 lamellae7,8 and vesicles,1,9–12 among others. Such morphologies can be predictably accessed through changes to the hydrophobic/hydrophilic weight fraction, which influences the packing parameter.13,14 There is significant interest in tuning nanostructure size and morphology as these are critical parameters in desired applications. For example, nanostructures with different morphologies can show distinct properties in vivo. Indeed, cylindrical micelles can persist in the circulation up to ten times longer than their spherical counterparts.15 Different morphologies can be accessed through using standard post-polymerization self-assembly methods,16–18 or more recently polymerization-induced self-assembly (PISA).19–22 There is also increasing interest in the utility of responsive polymers to enable reversible morphology transitions in polymeric nanostructures.23–25Nucleobase-containing synthetic polymers inspired by Nature have been widely exploited to mediate polymer tacticity,26 to control polymer composition or sequence27,28 and to template polymerizations.29–31 All these studies have been inspired by the sequence specificity and selective recognition of nucleobase functionalities which can be exploited to create DNA materials32–34 and perform DNA template chemistry.35–37
Nucleobase interactions have also been utilized to drive self-assembly38–43 and for achieving a biomimetic segregation/templating approach to polymer synthesis.44 More recently, our work has shown that distinct self-assembled morphologies were formed with different nucleobase copolymer compositions and different morphological evolutions were observed in CHCl3 and 1,4-dioxane during the RAFT dispersion polymerization of nucleobase-functional monomers.28,45,46 The poor solubility and slow hydrolysis of the monomers in water, however, limit their applications in aqueous solutions. To the best of our knowledge, there is relatively little research into the self-assembly of nucleobase-containing polymers in aqueous solutions.42,47–50 A study by the van Hest group reported the self-assembly behavior of nucleobase-containing amphiphilic copolymers in aqueous solutions and most notably an increase of overall hydrophilicity of the diblock, due to complementary (adenine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :thymine) nucleobase interactions, was observed.42
:thymine) nucleobase interactions, was observed.42
A new family of amphiphilic block copolymers, known as DNA block copolymers (DBCs), have been described, which are composed of a hydrophobic synthetic polymer component attached to a single-stranded (ss) DNA.51–53 The introduction of DBCs into water leads to the formation of micelles with a hydrophobic polymer core and a hydrophilic DNA corona. Precise pairings between complementary functional DNA can be used to modify the micelle at particular locations, from the hydrophobic–hydrophilic interface to the whole corona.54,55 However, the complementary DNA interactions can only be achieved in the corona. Pioneering work reported by the Gianneschi group demonstrated the size and shape of micelles can be controlled through complementary base pairing interactions in the corona.56 But this approach requires expensive DNA building blocks for polymer coupling and this to date has hindered their large-scale and widespread application in self-assembly. Hence, there is significant interest in the exploration of complementary nucleobase interactions embedded within synthetic polymers to allow for access to new classes of responsive and tunable materials and nanostructures.
Herein, we report the assembly of nucleobase-containing micelles with tunable morphologies through the introduction of complementary nucleobase interactions within the core of the micelles. The initial micellar constructs in water consist of synthetic nucleic acid cores and hydrophilic coronas (poly(4-acryloylmorpholine)-b-poly(3-(thymin-1-yl)propyl acrylamide) (PNAM-b-PTAm)). Another diblock copolymer with the complementary nucleobase (poly(4-acryloylmorpholine)-b-poly(3-(adenine-9-yl)propyl acrylamide) (PNAM-b-PAAm)) was utilized to induce a morphological change in these micelles. For micelles with TAm blocks of moderate length, a gradual decrease in the size of the spherical micelles was observed with the introduction of increasing amounts of the complementary diblock copolymer, PNAM-b-PAAm. Micelles with a much longer TAm block core demonstrated different behaviors as they showed an initial increase of hydrodynamic diameters through insertion of PNAM-b-PAAm into the micellar core, which was then followed by a morphological change from spheres to cylinders, and finally to smaller spheres. The utilization of unique complementary A–T interactions in the micellar cores enables us to progressively tailor nanostructure sizes and morphologies and provides a new route to access functional nanomaterials.
Experimental section
Materials
2,2′-Azo-bis(isobutyronitrile) (AIBN) was obtained from Molekula and recrystallized from methanol. 2,2′-Azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044, Wako) was used without further purification. Adenine was obtained from Sigma and thymine was bought from Acros. Sodium hydride (60% dispersion mineral oil) was purchased from Aldrich. 4-Acryloylmorpholine (NAM) was bought from Aldrich and was purified by vacuum distillation. DMF, DMSO, triethylamine and other chemicals were obtained from Fisher Chemicals and used without further purification. Dry solvents used in the experiments were obtained by passing over a column of activated alumina using an Innovative Technologies solvent purification system. Dialysis membranes (MWCO = 3.5–5 kDa) were purchased from Spectra/Por. The syntheses of nucleobase-containing monomers 3-(adenine-9-yl)propyl acrylamide (AAm), 3-(thymin-1-yl)propyl acrylamide (TAm) and 2-(((butylthio)carbonothiolyl)thio)propanoic acid are described in the ESI (Fig. S1–S3 and Schemes S1, S2†).Instrumentation
1H NMR and 13C NMR spectra were recorded on a Bruker DPX-300, DPX-400 or HD500 spectrometer with DMSO-d6 or CDCl3 as the solvent. The chemical shifts of protons were relative to tetramethylsilane (TMS) at δ = 0 ppm when using CDCl3 or solvent residues (DMSO 2.50 ppm). UV-vis spectra were recorded on a Perkin-Elmer Lambda 35 UV-vis instrument. Scans from 200 to 700 nm were taken using a quartz cuvette at different times after mixing. High resolution mass spectrometry (HR-MS) was conducted on a Bruker UHR-Q-TOF MaXis with electrospray ionization (ESI). Size exclusion chromatography (SEC) data were obtained in HPLC grade DMF containing 5 mM NH4BF4 at 50 °C, with a flow rate of 1.0 mL min−1, on a set of two PLgel 5 μm Mixed-D columns, and a guard column. SEC data were analyzed with Cirrus SEC software calibrated using polymethyl methacrylate (PMMA) standards.Hydrodynamic diameters (Dh) and size distributions of the self-assemblies were determined by dynamic light scattering (DLS). The DLS instrumentation consisted of a Malvern Zetasizer NanoS instrument operating at 25 °C with a 4 mW He–Ne 633 nm laser module. Measurements were made at a detection angle of 173° (back scattering), and Malvern DTS 7.03 software was used to analyze the data. Dh was calculated by fitting the apparent diffusion coefficient in the Stokes–Einstein equation Dh = kT/(3πηDapp), where k, T and η are the Boltzmann constant, the temperature and the viscosity of the solvent, respectively. When only the measured sample is a solution of monodispersed spherical micelles, Dh coincides to the real hydrodynamic diameter as Dapp is equal to the translational diffusion coefficient (Dt). For anisotropic nanoparticles such as cylinders, the non-negligible rotational diffusion also contributes to the Dapp. Therefore, the measured Dh for these samples only represents a relative value and provides polydispersity information to detect multiple populations. Static light scattering (SLS) measurements were conducted with an ALV CGS3 (λ = 632 nm) at 20 °C. The data were collected from 50° to 150° with an interval of 10° against a toluene standard. The self-assembled solutions were filtered through 0.45 μm nylon filters prior to analysis. In SLS analysis, the angular dependence of the absolute excess time-average scattering intensity, the Rayleigh ratio Rθ, can lead to the weight-average molar mass Mw, the radius of gyration Rg and the second virial coefficient A2 by:
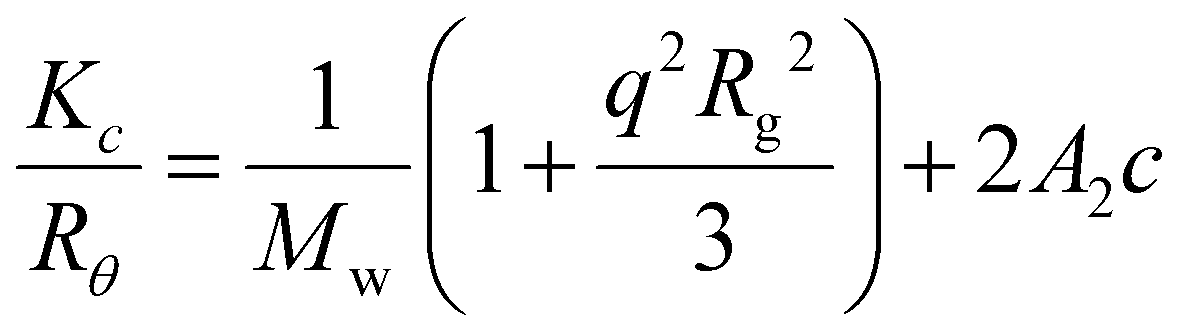 | (1) |
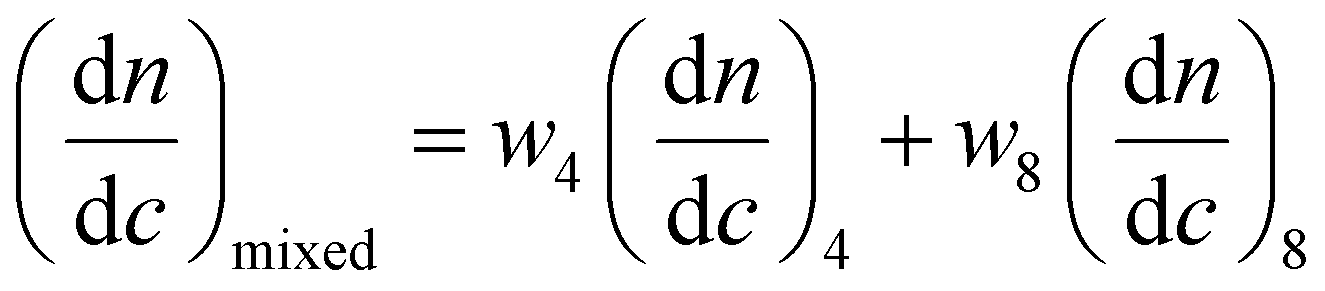 | (2) |
TEM observations were performed on a JEOL 2000FX electron microscope at an acceleration voltage of 200 kV. All TEM samples were prepared on graphene-oxide (GO)-coated lacey carbon grids (400 Mesh, Cu, Agar Scientific), to enable high contrast TEM images without any staining.57 Generally, a drop of sample (10 μL) was pipetted on a grid and left for several minutes, then blotted away. TEM images were analyzed using the ImageJ software, and over 100 particles were counted for each sample to obtain number-average diameter Dn (for spherical micelles) or number-average width Wn (for cylindrical micelles).
Small-angle X-ray scattering (SAXS) experiments were performed on the SAXS/WAXS beamline at the Australian Synchrotron facility at a photon energy of 12 keV. The samples in solutions were run using 1.5 mm diameter quartz capillaries. The measurements were collected at 25 °C with a sample-to-detector distance of 3.252 or 7.160 m to give q ranges of 0.005 to 0.22 and 0.002 to 0.10 Å−1, respectively, where q is the scattering vector and is related to the scattering angle (2θ) and the photon wavelength (λ) by the following equation:
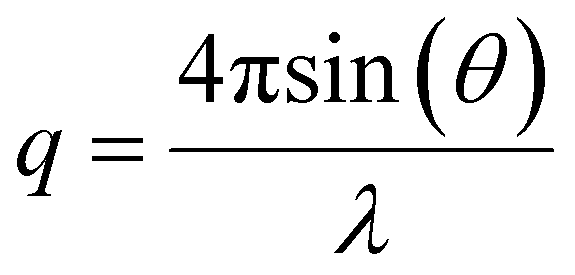 | (3) |
All patterns were normalized to a fixed transmitted flux using a quantitative beam stop detector. The two-dimensional SAXS images were converted into one-dimensional SAXS profile (I(q) versus q) by circular averaging, where I(q) is the scattering intensity. ScatterBrain, Primus and NCNR Data Analysis IGOR PRO software were used to plot and analyze SAXS data.58,59 The scattering length density of the solvents and monomers were calculated using the “Scattering Length Density Calculator” provided by the NIST center for Neutron Research.
Synthesis of poly(4-acryloylmorpholine) (PNAM) macro-CTA via RAFT polymerization
A 10 mL ampoule was charged with NAM (126 μL, 1.0 mmol), 2-(((butylthio)carbonothiolyl)thio)propanoic acid (2.4 mg, 0.01 mmol), VA-044 (0.32 mg, 0.001 mmol) and a mixture of 1,4-dioxane and water (0.5 mL, v:v 1:4).60 The mixture was thoroughly degassed via 4 freeze–pump–thaw cycles, filled with nitrogen and then immersed in an oil bath at 70 °C for 2 h. The polymerization solution was precipitated three times from cold CH3OH. The light yellow polymer was dried in a vacuum oven overnight at room temperature and analyzed by 1H NMR spectroscopy and DMF SEC (Fig. S4 and S5†). The degree of polymerization (DP) of this PNAM macro-CTA, 1, was calculated to be 96 using 1H NMR spectroscopy by comparing the integrated signals corresponding to the backbone signals (δ = 1.62 ppm) with those of the methyl group from the CTA (δ = 0.87 ppm).
Synthesis of PNAM96-b-PTAmn and PNAM96-b-PAAmm diblock copolymers
The typical procedures are as follows. For PNAM96-b-PTAm114, PNAM96 (35 mg, 0.0025 mmol), TAm (71 mg, 0.30 mmol), and AIBN (0.04 mg, 0.00025 mmol) were dissolved in 0.5 mL DMSO. The mixture was thoroughly degassed via 4 freeze–pump–thaw cycles, filled with nitrogen and then immersed in an oil bath at 70 °C overnight. An aliquot of the crude product was taken and analyzed by 1H NMR spectroscopy to calculate the conversion. The residual solution was precipitated three times from cold CH3OH. The light yellow polymer was dried in a vacuum oven overnight at room temperature and analyzed by 1H NMR spectroscopy and DMF SEC (Fig. S4 and S5†). See Table 1 for NMR and SEC characterization of polymers 2–8.| Polymer |
M
n,NMRa (kDa) |
M
n,SECb (kDa) |
Đ
Mb |
|---|---|---|---|
| a Measured by 1H NMR spectroscopy (400 MHz) in deuterated DMSO. b Measured by DMF SEC. | |||
| PNAM961 | 13.8 | 13.9 | 1.06 |
| PNAM96-b-PTAm172 | 17.8 | 19.1 | 1.09 |
| PNAM96-b-PTAm343 | 21.8 | 22.9 | 1.10 |
| PNAM96-b-PTAm1144 | 40.8 | 33.7 | 1.24 |
| PNAM96-b-PTAm3015 | 85.1 | 68.4 | 1.29 |
| PNAM96-b-PTAm636 | 28.7 | 26.0 | 1.18 |
| PNAM96-b-PTAm1607 | 51.7 | 40.6 | 1.28 |
| PNAM96-b-PAAm198 | 18.5 | 19.9 | 1.08 |
Self-assembly of PNAM-b-PTAm diblock copolymer in water
For the copolymer with moderate hydrophobic TAm blocks (4, 6 and 7), a vial was charged with 10 mg copolymer, 1 mL water and a stirrer bar. The vial was sealed and left at 70 °C overnight with stirring and then cooled to room temperature to afford well-defined nanostructures. The micelle solutions were then diluted to 0.5 mg mL−1 with 18.2 MΩ cm water at room temperature for TEM and DLS analyses. For the copolymer PNAM96-b-PTAm301 (5) with a longer hydrophobic block, a solvent switch method was used to afford self-assembly. Specifically, the copolymer was dissolved in DMF (at 8 mg mL−1) and stirred for 2 h. Then an excess of 18.2 MΩ cm water was added via a syringe pump at a rate of 1 mL h−1. The final volume ratio between water and organic solvent was 8:1. The solution was then dialyzed against 18.2 MΩ cm water, incorporating at least 6 water changes, to afford self-assemblies at a concentration of ca. 1 mg mL−1.
Addition of complementary copolymer PNAM-b-PAAm into PNAM-b-PTAm micellar solution
The diblock copolymer PNAM96-b-PAAm19 (8) was dissolved in H2O at 10 mg mL−1. This was then added to the micellar solution of PNAM96-b-PTAmn (0.5 mg mL−1) dropwise with stirring. The molar ratios were calculated according to the Mn determined from 1H NMR spectroscopic analyses and their mass concentration (see ESI†). The mixture was then sealed and allowed to stir at room temperature overnight. The solutions were then analyzed by DLS, TEM, SLS and SAXS analysis.Results and discussion
Synthesis and characterization of nucleobase-containing synthetic diblock copolymers
A PNAM macro-CTA was synthesized by RAFT polymerization in a mixture of water and 1,4-dioxane (Scheme 1).60 This PNAM96 macro-CTA (1) was then chain-extended with various amounts of TAm or AAm in DMSO to yield a series of PNAM96-b-PTAm/PAAm diblock copolymers (2–8). Each TAm/AAm polymerization was carried out for 24 h at 70 °C prior to analysis by 1H NMR spectroscopy and SEC with DMF as eluent.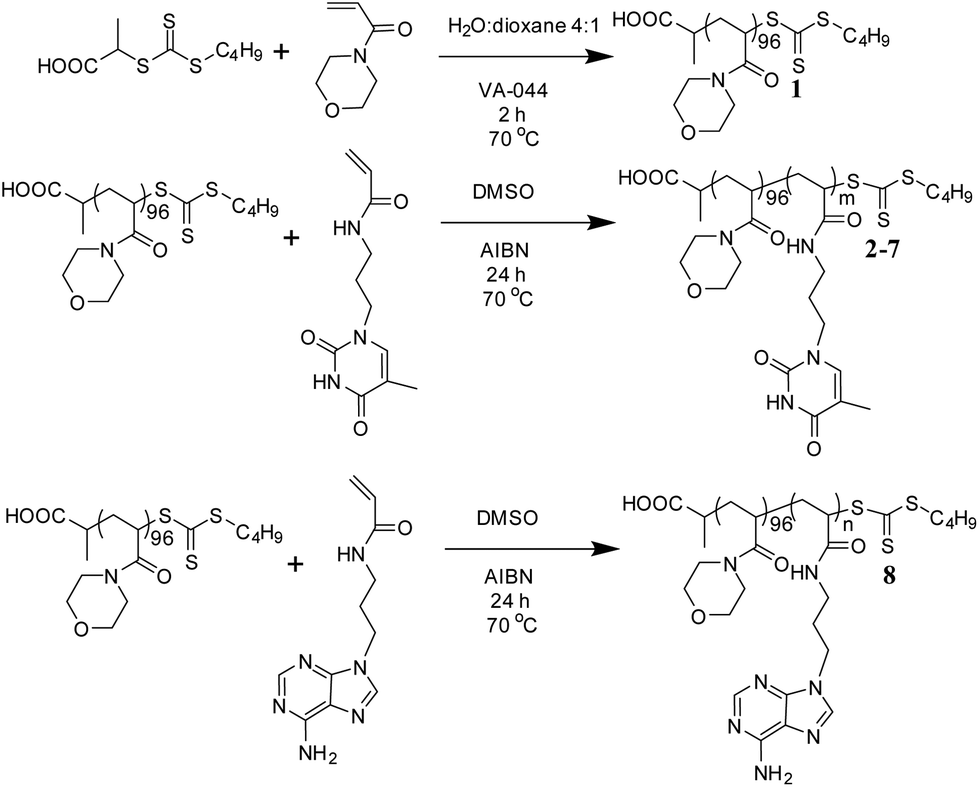 | ||
| Scheme 1 Synthetic route for the preparation of PNAM96 and the nucleobase-containing block copolymers, 2–8. | ||
The characterization data for the polymers are summarized in Table 1. 1H NMR spectroscopy was used to calculate the DP by comparing the integrated signals corresponding to characteristic signals from the nucleobases (δ = 11.23 ppm) with those assigned to the end group of the CTA (δ = 0.84 ppm) (Fig. S4†). SEC traces revealed unimodal molecular weight distributions (ĐM < 1.30) and minimal contamination of unreacted PNAM96 macro-CTA (Fig. S5†). These results indicated diblock nucleobase copolymers were obtained with relatively high blocking efficiencies through RAFT polymerization.
Self-assembly of PNAM-b-PTAm diblock copolymers
The self-assembly of the diblock copolymers was achieved by direct dissolution of the polymers in water at a concentration of 10 mg mL−1 at 70 °C, with overnight stirring. DLS analysis of the solution showed a diameter of 58 nm for copolymer 4 (Fig. 1a). Notably, no self-assembly was observed for the diblock copolymers 2 and 3, most likely as a consequence of the short TAm block providing insufficient hydrophobic interactions to form a stable hydrophobic core (Fig. S6†).61 TEM images confirmed the spherical nature of the micelles for copolymer 4 (Fig. 1b). The image analysis revealed that the diameter of the spherical micelles was narrowly dispersed (Fig. S7†). The diameter from TEM analysis is slightly smaller than that from DLS analyses partly due to the insensitivity of the former technique to the hydrophilic corona domain.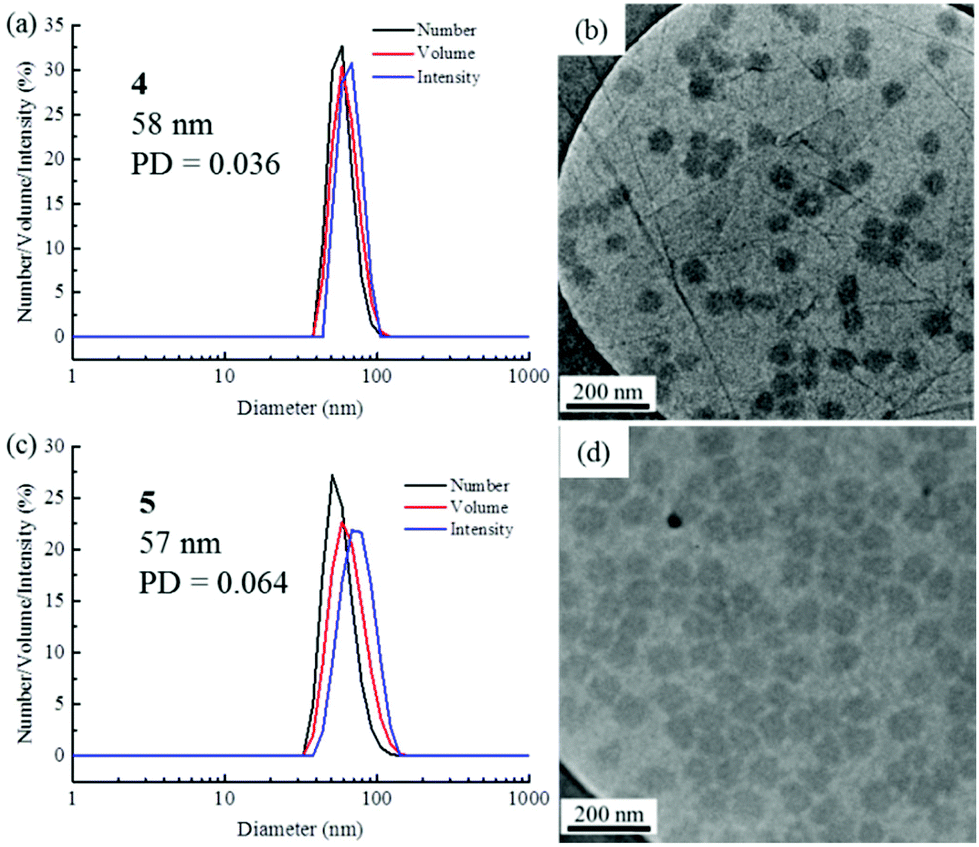 | ||
| Fig. 1 DLS analyses and TEM images of self-assembled micelles. (a and b) PNAM96-b-PTAm1144; (c and d) PNAM96-b-PTAm3015. | ||
For polymer 5, which has a much longer hydrophobic block, a solvent switch method was used as using a direct dissolution method formed ill-defined nanostructures. DLS analysis showed a bimodal distribution and TEM images also indicated that the sample contained both worms and spheres (Fig. S8†). No change was observed even after heating at 70 °C for several days. The long hydrophobic block could result in a large energy barrier for unimer exchange, leading to the formation of ‘frozen’ micelles.62 Compared with the direct dissolution method, it was found that well-defined micelles could be easily formed through a solvent switch method, especially for the crew-cut aggregates. Specifically, the copolymer 5 was dissolved in DMF at 8 mg mL−1 and water was added at a very slow rate (1 mL h−1) to make a final solution of ca. 1 mg mL−1. The DMF was then removed by dialysis against water incorporating at least 6 water changes. DLS and TEM analyses indicated well-defined nanostructures were formed (Fig. 1c and d). The low concentration of the self-assembled solution is expected to limit the further fusion of the spherical micelles into more complex structures.63 It is noteworthy that micelles of both 4 and 5 had a similar Dh and Nagg, however, a much larger hydrophobic core was formed for 5 as the hydrophobic block was much longer (Fig. S9, S10 and Table S1†).
Tuning spherical micelle sizes using complementary nucleobase interactions
The formed self-assembled micelles contained TAm cores, which can interact with AAm through hydrogen-bonding. To explore this, copolymer 8 containing the complementary nucleobase adenine was added to the micellar solutions described above with the aim of inducing a response as a result of the formation of complementary hydrogen bonds within the confined core domain. It is notable that copolymer 8 was observed to form small aggregates in water with Nagg of ca. 13 as measured by SLS and SAXS analysis (Fig. S11†). It is expected that copolymers with longer AAm blocks could form larger and more stable aggregates, which may limit their ability to induce such complementary interactions upon addition to the parent micelles. Hence, we explored the addition of copolymer 8 to micelle solutions of 4 and 5.To explore this reorganization, different molar ratios of 8 were added to micelle solutions of 4 and left to stir overnight at room temperature. The solutions were then analyzed by DLS, which indicated a significant decrease in micelle sizes with an increased quantity of complementary copolymers (Fig. 2b). The pristine micelles had a diameter of ca. 58 nm, which decreased to just 38 nm after the addition of 1 molar eq. of 8. Further, smaller micelles with diameters of ca. 34 nm and 29 nm were obtained upon addition of 3 molar eq. and 5 molar eq. of 8, respectively (Fig. 2b). DLS data of the newly-formed micelles also indicated the formation of monodispersed nanostructures induced by the complementary copolymer interactions (Fig. S12†). Further TEM images confirmed smaller spherical micelles were produced by mixing initial micelles composed of 4 and complementary copolymer 8 (Fig. 2c, d and S12†). Meanwhile, there was no obvious changes observed when mixing initial micelles of 4 and non-complementary compolymer 2 (Fig. S13†). For self-assembled spherical micelles composed of 6 and 7 with a slightly shorter or longer hydrophobic block respectively, a similar change of micelle sizes was observed (Fig. S13–S15†).
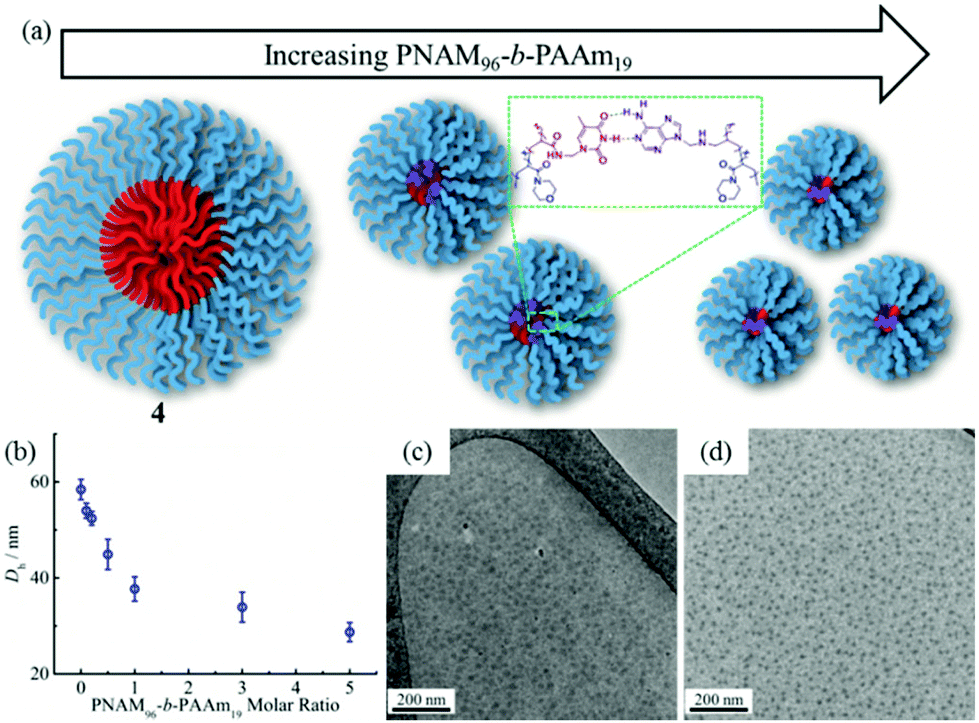 | ||
| Fig. 2 Interactions between 8 and micelles of 4 result in a change in nanoparticle sizes. (a) Schematic of the micellar size decrease through hydrogen-bonding interactions between 8 and micelles of 4; (b) DLS hydrodynamic diameters (Dh) of micellar solutions of 4 with increasing molar ratios of 8; (c and d) TEM images of micellar solutions of 4 upon addition of 1 molar eq. and 5 molar eq. of 8. | ||
In order to further corroborate the interactions between 4 and 8, 1H NMR and UV-vis spectroscopy were used to characterize the micellar mixtures. The 1H NMR spectrum of 8 in D2O showed the protons of adenine at 7.62 ppm (Fig. 3a). Conversely, only the peaks of the hydrophilic NAM block in micelles of 4 were observed by 1H NMR analysis, which indicated the thymine block formed a hydrophobic core in D2O. If the copolymer 8 could interact with the hydrophobic thymine core of 4, the AAm block was confined into the insoluble hydrophobic core and the relaxation time increased significantly, both of which decreased the peak intensity of the protons attributable to the adenine functionality. Indeed, an obvious decrease or disappearance of the adenine peaks was observed in the 1H NMR spectra of the mixtures of 4 and 8 (Fig. 3a and S16†). Thus, it was proposed that copolymer 8 could insert into the thymine core of micelles composed of 4.
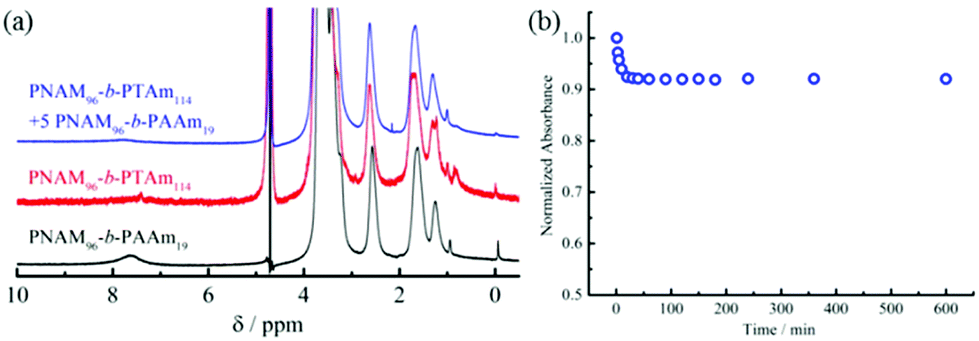 | ||
| Fig. 3 Interactions between 8 and micelles of 4 studied by 1H NMR and UV-vis spectroscopy. (a) 1H NMR spectra in D2O of a micellar solution of 4, a solution of 8, and a micellar solution of 4 containing 5 molar eq. 8; (b) the peak absorbance change of the mixture of 5 molar eq. 8 with a micellar solution of 4 as observed by UV-vis spectroscopy. | ||
The hydrogen-bonding interactions between the adenine and thymine containing amphiphiles were also examined by UV-vis spectroscopy (Fig. 3b and S17†). To explore this, 5 molar eq. of 8 in water was added into a micellar solution of 4 and UV-vis measurements were performed at different times after mixing. The maximum absorption peak was at 262 nm just after mixing. No shift of peak absorbance was detected after 10 h. However, a greater than 9% decrease in peak intensity was observed after 2 h, with no further decrease after this time. The absorption decrease suggested that interactions between the complementary nucleobases adenine and thymine were taking place. This decrease of absorbance is analogous to the hypochromicity for two complementary DNA.64 Thus, we propose that the nucleobase-containing synthetic diblock copolymers 4 and 8 with complementary adenine and thymine residues interacted through hydrogen bonding. Their interactions decreased the interfacial tension of the hydrophobic block with the solvent due to the increasing volume ratio of the hydrophilic block. The energy barrier for chain exchange was thus significantly lowered, leading to reorganization to generate smaller spherical micelles. Another factor, as described previously is that the interactions between complementary nucleobase copolymers can increase the hydrophilicity of the core–corona interface.42 Thus, the interfacial tension also decreased. Moreover, the insertion of the complementary copolymers could lead to the formation of a denser hydrophilic corona, increasing the core-chain stretching and the corona-chain repulsion simultaneously. In order to reduce the total free energy of the system, spherical micelles underwent reorganization into spheres with smaller diameters, whereby the corona-chain repulsion and core-chain stretching were reduced. The micellar reorganization was further facilitated by the low interfacial tension of the polymers with moderately short hydrophobic blocks.
SAXS was utilized to further characterize the change in micelle size upon addition of the complementary diblock copolymer. Scattering curves expressed as double-logarithmic plots of I(q) against q were shown in Fig. 4a. The Guinier fit was performed to determine the Rg while the anisotropy was determined by the Guinier–Porod fit. A significant decrease in diameters of gyration Dg (Dg = 2Rg) was observed, which was consistent with decreasing Dh values from DLS (Fig. 2b and S18†) upon increasing addition of polymer 8. Also, the ratios of Rg/Rh were close to 0.77 for all cases, indicating all of the micelles were spherical in nature (Table S2†).65 Meanwhile, the mean particle diameters as measured from TEM images also demonstrated the same trend (Fig. 4b). Notably, number-average diameters Dn determined by TEM analysis are somewhat smaller than those measured by DLS due to the dry state of the analysis compared to solution analysis for DLS and hence the former technique does not fully account for the loose coronal PNAM domain. Furthermore, the change of the mean aggregation number Nagg of the micelles, upon addition of 8, was determined by SLS. The Mw values for both diblock copolymers were determined by multiplying their Mn (determined by end group analyses from 1H NMR spectroscopy) by the corresponding Mw/Mn values determined by SEC analyses. The Mw,mix values of the mixed micellar solutions were calculated according to the weighted sum of the copolymers in solutions. The mean aggregation number (Nagg) was then calculated by dividing the Mw,micelle determined for the micelles by the Mw,mix calculated for the mixed diblock copolymers. Using this method for Nagg determination by both SLS analysis and SAXS analyses showed a similar decrease of Nagg. Specifically, Nagg decreased significantly from 392 to 84 after adding 1 molar eq. of 8 and a slight decrease was observed with further addition of the complementary copolymer (Fig. 4c). Also, the core sizes Rcore of the micelles also showed the same trend (Table S2†). We propose that the increasing hydrophilic volume ratios (upon formation of the A:T complementary interactions) lead to smaller Nagg values,66 which also contributes to the reorganization to smaller micelles due to the increase of polymer curvature. Meanwhile, the average aggregation number of the micelles was significantly different from the expected value for a mixture of non-interacting micelles which could be calculated according to eqn (4), where c is the weight concentration of the copolymers in solution.67
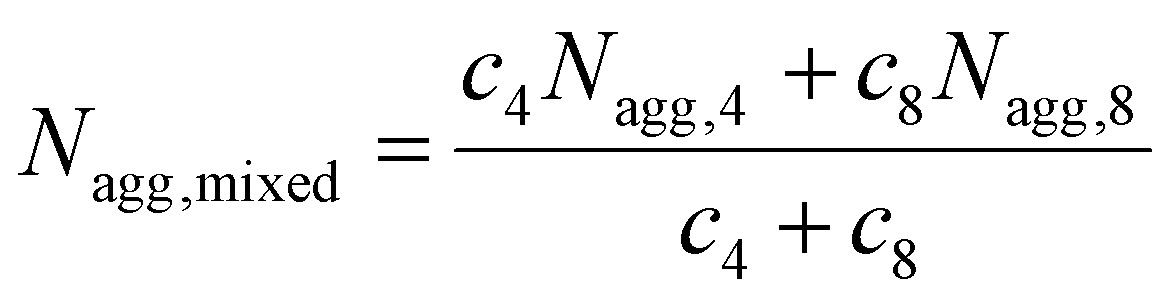 | (4) |
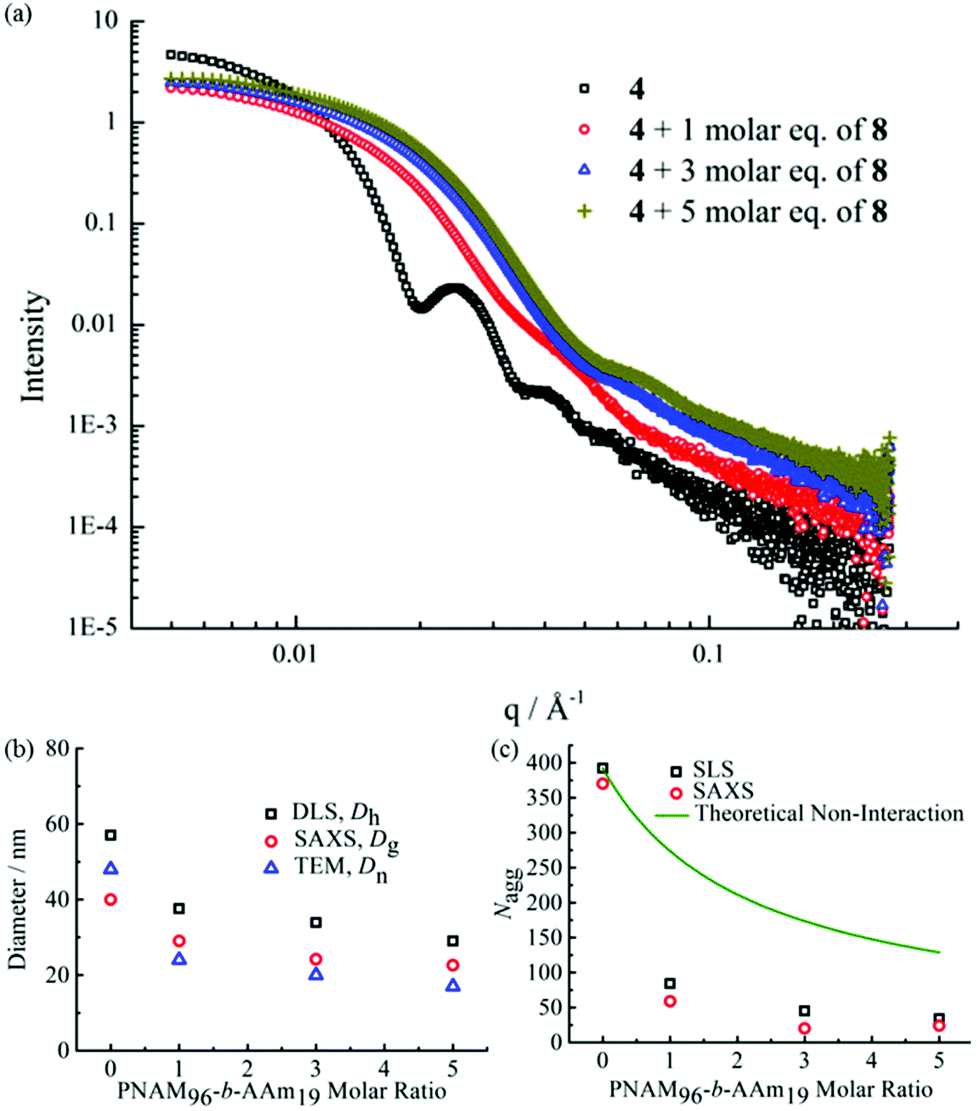 | ||
| Fig. 4 (a) SAXS experimental profiles of micellar solutions of 4 with 0, 1, 3 and 5 molar eq. of 8. (b) Mean particle diameters determined for micellar solutions of 4 with 0, 1, 3 and 5 molar eq. of 8 by SAXS (Dg), DLS (Dh), and TEM (Dn). (c) Mean aggregation number (Nagg) determined for micelle solutions of 4 with 0, 1, 3 and 5 molar eq. of 8 by SLS and SAXS and the theoretical aggregation number for a non-interacting mixture as calculated from eqn (4). | ||
Tuning micelle morphologies through complementary nucleobase interactions
Previous studies have demonstrated that polymer chain exchange is limited by the kinetic requirements of the system, which strongly depend on the length of the hydrophobic block.62,68 Clearly, a longer hydrophobic block, which would have a higher interfacial tension with the solvent, maybe sufficient to freeze the aggregates and prevent chain exchange. To explore this, a second block copolymer 5 with a longer hydrophobic domain was utilized. The same experiment was performed as described previously for 4. However in this second case distinct morphological changes were observed when 8 was added into the micelle solution of 5. This transition was examined by DLS, SAXS and TEM analysis. Fig. 5b showed the increase in hydrodynamic diameter from 57 nm to 89 nm which resulted from the addition of 5 molar eq. of 8 to a micellar solution of 5. Increasing the quantity of 8 from 10 and then to 20 molar eq. induced a decrease in the size of the micelles (Fig. 5b and S19†) to 25 nm by DLS analysis. To explore these changes in size as observed by light scattering the self-assembled micelles were also analyzed by dry-state TEM analysis. TEM images revealed that morphological transitions from spherical micelles to cylindrical micelles and then to smaller spherical micelles occurred upon increasing additions of polymer 8 to the micellar solution of 5. Only spherical micelles with a slight increase in size were observed by TEM after adding 1 molar eq. of 8, which is consistent with DLS analyses (Fig. 5c). Interestingly, long cylindrical micelles of ca. 300 nm in length and 20 nm in width were observed with 3 molar eq. or 5 molar eq. of 8, suggesting that cylindrical micelles were formed from the initially spherical micelles due to the addition of copolymer containing the complementary nucleobase functionality (Fig. 5d and e). It is also noteworthy that much smaller spherical micelles relative to those initially observed were produced once 10 eq. of 8 had been added (Fig. 5f). TEM image analysis showed the diameter was only around 24 nm. No new structures were formed when the amount of 8 was increased to 20 molar eq. (Fig. 5g). The interactions between the polymer 8 and micelles of 5 were also studied by 1H NMR spectroscopy. As before, an intensity decrease and disappearance signals attributable to adenine was observed, indicating the insertion of 8 into the micelles (Fig. S20†). Meanwhile, UV-vis measurements also suggested that interactions between complementary nucleobases in the micellar core and added 8 were taking place (Fig. S21†). This data alongside the nanostructure characterization data suggests that micelles of differing morphologies could be generated through complementary nucleobase interactions just by changing the corresponding molar ratios added to the assemblies.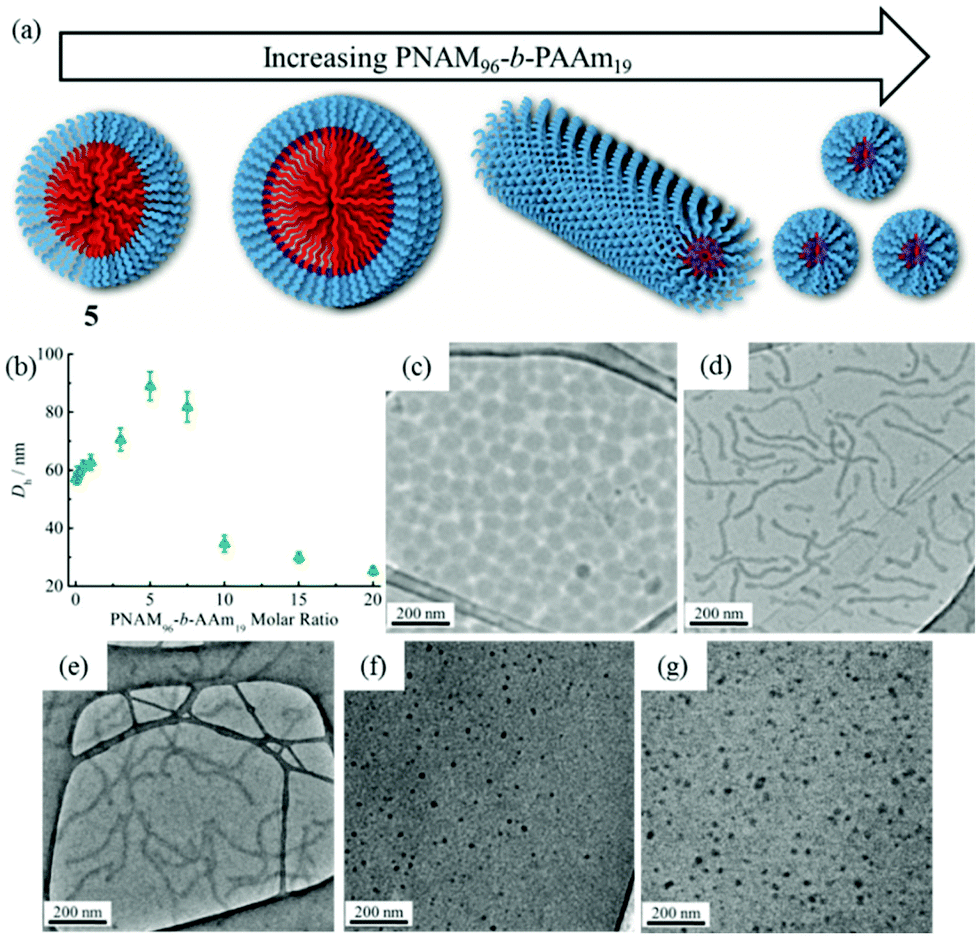 | ||
| Fig. 5 Morphological transitions induced by the interactions between 8 and micelles of 5. (a) Schematic of the morphological transition through hydrogen-bonding interactions between 8 and micelles of 5; (b) DLS hydrodynamic diameters (Dh) of micelles of 5 with increasing molar ratios of 8; (c–g) TEM images of micelles of 5 with 1, 3, 5, 10 and 20 molar eq. of 8. | ||
Further SAXS analyses also confirmed that transitions from spherical micelles to cylindrical micelles and then to smaller spherical micelles were taking place (Fig. 6). A slight increase in spherical micellar size was observed, which is strongly indicative of the insertion of 8 into the original micelles (Table S3†). Interestingly, morphological changes occurred as 3 molar eq. or 5 molar eq. of 8 were added. Long cylinders were formed, which was also confirmed by increase in the s parameter from SAXS analyses and which was also consistent with TEM imaging (Table S3 and Fig. S22†). Compared with micelles containing moderate hydrophobic blocks, the interactions between complementary nucleobase copolymers could thermodynamically favor the formation of smaller spherical micelles due to the decreased interfacial tension and corona-chain repulsion. However, the much longer hydrophobic block, which had a higher energy barrier for chain exchange, confined the mobility of the micellar copolymer. More importantly, the micelle of 5 with a larger core provided more space and surface area for the inserted copolymers compared with the nanostructure formed from 4 (Table S1†). Hence, a slight increase in the micelle size could, to some extent, relieve the corona-chain repulsion through increasing the volume of the corona. In response to the resulting entropic penalty, and in order to reduce the total free energy of the system, a morphological transition from spheres to smaller diameter cylinders occurred after adding 3 molar eq. of 8, which led to a reduction in both corona-chain repulsion and core-chain stretching. Only smaller spherical micelles were formed after adding 10 molar eq. of 8. Further increasing the quantity of 8 only led to smaller spherical micelles and no new nanostructures were formed. This transition was similar to the micelles formed from copolymers with moderate hydrophobic blocks. When more complementary copolymers interacted through hydrogen-bonding with the long hydrophobic core, the interfacial tension was further decreased with higher hydrophilic volume ratios, leading to a lower energy barrier for chain exchange. Smaller spherical micelles were, therefore, formed to reduce the increased corona-chain repulsion introduced through the insertion of the complementary copolymer. Compared with the morphology or size change of micellar nanostructures induced by external stimuli such as temperature, pH or salts, the unique complementarity of nucleobases (A and T) can be utilized to progressively tailor micellar sizes and shapes just by varying the relative molar ratios. Complementary nucleobase interactions between copolymers with A and T can change the dynamics and stabilities of nanostructures to afford a new method for control over nanostructure size and morphology changes.
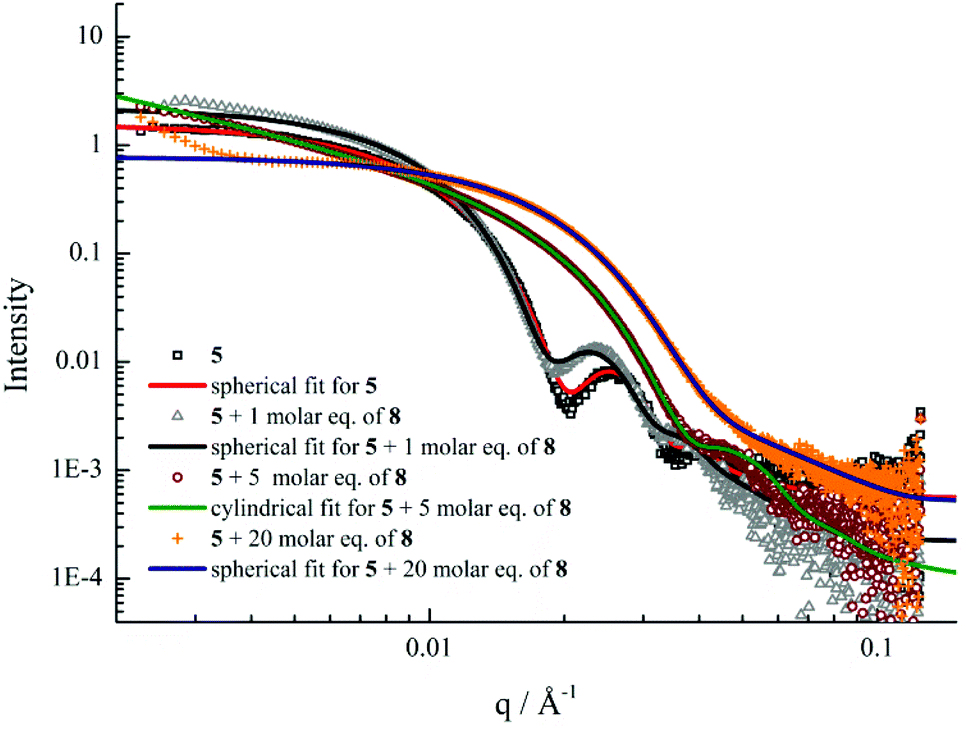 | ||
| Fig. 6 SAXS experimental profiles and fittings of micellar solutions of 5 with 0, 1, 5 and 20 molar eq. of 8. | ||
Conclusions
In summary, a series of nucleobase-containing synthetic block copolymers were prepared via RAFT polymerization. Monodisperse spherical micelles were formed through direct dissolution or solvent switch methods and characterized by DLS and TEM. The interactions between these micelles and amphiphilic copolymers with complementary nucleobases were exploited to trigger size and morphology changes in the nanostructures. The insertion of the complementary copolymers into the micelles was analyzed by 1H NMR spectroscopy and nucleobase interactions confirmed by UV-vis spectroscopy. For micelles with moderate hydrophobic TAm blocks, a significant decrease in micellar sizes with increased quantities of complementary copolymers was observed and characterized by DLS, TEM and SAXS. Both SAXS and SLS confirmed a significant decrease in Nagg upon the addition of complementary copolymer. Low interfacial tension of hydrophobic blocks with moderate length enabled micelle reorganization to smaller spherical micelles by reducing the corona-chain repulsion and core-chain stretching. Furthermore, for the micelle with a much longer hydrophobic block, morphological transitions from spheres to cylinders and then to smaller spheres could be achieved with increased quantities of the complementary copolymer. Swollen and slightly larger spherical micelles were formed with initial addition of PNAM-b-AAm due to a high energy barrier for chain exchange of the long hydrophobic block. Through reducing corona-chain repulsion and core-chain stretching, long cylinders were produced followed by disassembly into smaller spherical micelles with further addition of the complementary copolymer. We propose that complementary nucleobase interactions in synthetic polymers provide a new stimulus to access different nanostructure sizes and morphologies as desired for applications such as delivery vehicles or catalytic nanoreactors.Acknowledgements
The authors thank the University of Warwick, China Scholarship Council (Z. H.), NSF and EPSRC, and the ERC for research funding.Notes and references
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CAS.
- Y.-Y. Won, H. T. Davis and F. S. Bates, Science, 1999, 283, 960–963 CrossRef CAS PubMed.
- Z. Li, E. Kesselman, Y. Talmon, M. A. Hillmyer and T. P. Lodge, Science, 2004, 306, 98–101 CrossRef CAS PubMed.
- H. Cui, Z. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647–650 CrossRef CAS PubMed.
- X. Wang, G. Guerin, H. Wang, Y. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644–647 CrossRef CAS PubMed.
- S. Jain and F. S. Bates, Science, 2003, 300, 460–464 CrossRef CAS PubMed.
- P. Yang, L. P. D. Ratcliffe and S. P. Armes, Macromolecules, 2013, 46, 8545–8556 CrossRef CAS.
- S. E. Mastroianni, J. P. Patterson, R. K. O'Reilly and T. H. Epps III, Soft Matter, 2013, 9, 10146–10154 RSC.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- D. A. Christian, A. Tian, W. G. Ellenbroek, I. Levental, K. Rajagopal, P. A. Janmey, A. J. Liu, T. Baumgart and D. E. Discher, Nat. Mater., 2009, 8, 843–849 CrossRef CAS PubMed.
- J. R. Howse, R. A. L. Jones, G. Battaglia, R. E. Ducker, G. J. Leggett and A. J. Ryan, Nat. Mater., 2009, 8, 507–511 CrossRef CAS PubMed.
- M. Massignani, C. LoPresti, A. Blanazs, J. Madsen, S. P. Armes, A. L. Lewis and G. Battaglia, Small, 2009, 5, 2424–2432 CrossRef CAS PubMed.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- M. Antonietti and S. Förster, Adv. Mater., 2003, 15, 1323–1333 CrossRef CAS.
- Y. Geng, P. Dalhaimer, S. Cai, R. Tsai, M. Tewari, T. Minko and D. E. Discher, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef CAS PubMed.
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- A. Choucair and A. Eisenberg, Eur. Phys. J. E, 2003, 10, 37–44 CrossRef CAS PubMed.
- Y. Zheng and H. T. Davis, Langmuir, 2000, 16, 6453–6459 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- W. Cai, W. Wan, C. Hong, C. Huang and C. Pan, Soft Matter, 2010, 6, 5554–5561 RSC.
- Y. Pei and A. B. Lowe, Polym. Chem., 2014, 5, 2342–2351 RSC.
- Y. Cai, K. B. Aubrecht and R. B. Grubbs, J. Am. Chem. Soc., 2011, 133, 1058–1065 CrossRef CAS PubMed.
- A. O. Moughton, J. P. Patterson and R. K. O'Reilly, Chem. Commun., 2011, 47, 355–357 RSC.
- K. Wei, L. Su, G. Chen and M. Jiang, Polymer, 2011, 52, 3647–3654 CrossRef CAS.
- Y. Tao, K. Satoh and M. Kamigaito, Macromol. Rapid Commun., 2011, 32, 226–232 CrossRef CAS PubMed.
- M. Akashi, H. Takada, Y. Inaki and K. Takemoto, J. Polym. Sci., Polym. Chem. Ed., 1979, 17, 747–757 CrossRef CAS.
- Y. Kang, A. Lu, A. Ellington, M. C. Jewett and R. K. O'Reilly, ACS Macro Lett., 2013, 2, 581–586 CrossRef CAS.
- A. Khan, D. M. Haddleton, M. J. Hannon, D. Kukulj and A. Marsh, Macromolecules, 1999, 32, 6560–6564 CrossRef CAS.
- C. R. South and M. Weck, Macromolecules, 2007, 40, 1386–1394 CrossRef CAS.
- P. K. Lo and H. F. Sleiman, J. Am. Chem. Soc., 2009, 131, 4182–4183 CrossRef CAS PubMed.
- E. Stahl, T. G. Martin, F. Praetorius and H. Dietz, Angew. Chem., Int. Ed., 2014, 53, 12735–12740 CrossRef CAS PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- Z. Li, Y. Zhang, P. Fullhart and C. A. Mirkin, Nano Lett., 2004, 4, 1055–1058 CrossRef CAS.
- Y. Dong, Z. Yang and D. Liu, Acc. Chem. Res., 2014, 47, 1853–1860 CrossRef CAS PubMed.
- P. J. Milnes, M. L. McKee, J. Bath, L. Song, E. Stulz, A. J. Turberfield and R. K. O'Reilly, Chem. Commun., 2012, 48, 5614–5616 RSC.
- M. L. McKee, P. J. Milnes, J. Bath, E. Stulz, R. K. O'Reilly and A. J. Turberfield, J. Am. Chem. Soc., 2012, 134, 1446–1449 CrossRef CAS PubMed.
- R. McHale and R. K. O'Reilly, Macromolecules, 2012, 45, 7665–7675 CrossRef CAS.
- F. Ilhan, T. H. Galow, M. Gray, G. Clavier and V. M. Rotello, J. Am. Chem. Soc., 2000, 122, 5895–5896 CrossRef CAS.
- R. J. Thibault, P. J. Hotchkiss, M. Gray and V. M. Rotello, J. Am. Chem. Soc., 2003, 125, 11249–11252 CrossRef CAS PubMed.
- H. S. Bazzi and H. F. Sleiman, Macromolecules, 2002, 35, 9617–9620 CrossRef CAS.
- H. J. Spijker, A. J. Dirks and J. C. M. van Hest, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4242–4250 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- R. McHale, J. P. Patterson, P. B. Zetterlund and R. K. O'Reilly, Nat. Chem., 2012, 4, 491–497 CrossRef CAS PubMed.
- Y. Kang, A. Pitto-Barry, A. Maitland and R. K. O'Reilly, Polym. Chem., 2015, 6, 4984–4992 RSC.
- Y. Kang, A. Pitto-Barry, H. Willcock, W. Quan, N. Kirby, A. M. Sanchez and R. K. O'Reilly, Polym. Chem., 2015, 6, 106–117 RSC.
- Y. Tao, Y. Yang, D. Shi, M. Chen, C. Yang and X. Liu, Polymer, 2012, 53, 1551–1557 CrossRef CAS.
- R.-S. Lee, K.-Y. Peng, S.-W. Wang and Y.-Z. Li, Polym. J., 2014, 46, 710–721 CrossRef CAS.
- H. Kuang, S. Wu, Z. Xie, F. Meng, X. Jing and Y. Huang, Biomacromolecules, 2012, 13, 3004–3012 CrossRef CAS PubMed.
- M. Garcia, K. Kempe, D. M. Haddleton, A. Khan and A. Marsh, Polym. Chem., 2015, 6, 1944–1951 RSC.
- T. Schnitzler and A. Herrmann, Acc. Chem. Res., 2012, 45, 1419–1430 CrossRef CAS PubMed.
- T. G. Edwardson, K. M. Carneiro, C. K. McLaughlin, C. J. Serpell and H. F. Sleiman, Nat. Chem., 2013, 5, 868–875 CrossRef CAS PubMed.
- T. R. Wilks, J. Bath, J. W. de Vries, J. E. Raymond, A. Herrmann, A. J. Turberfield and R. K. O'Reilly, ACS Nano, 2013, 7, 8561–8572 CrossRef CAS PubMed.
- F. E. Alemdaroglu, K. Ding, R. Berger and A. Herrmann, Angew. Chem., Int. Ed., 2006, 45, 4206–4210 CrossRef CAS PubMed.
- M. Kwak, A. J. Musser, J. Lee and A. Herrmann, Chem. Commun., 2010, 46, 4935–4937 RSC.
- M.-P. Chien, A. M. Rush, M. P. Thompson and N. C. Gianneschi, Angew. Chem., Int. Ed., 2010, 49, 5076–5080 CrossRef CAS PubMed.
- J. P. Patterson, A. M. Sanchez, N. Petzetakis, T. P. Smart, T. H. Epps III, I. Portman, N. R. Wilson and R. K. O'Reilly, Soft Matter, 2012, 8, 3322–3328 RSC.
- P. V. Konarev, V. V. Volkov, A. V. Sokolova, M. H. J. Koch and D. I. Svergun, J. Appl. Crystallogr., 2003, 36, 1277–1282 CrossRef CAS.
- S. Kline, J. Appl. Crystallogr., 2006, 39, 895–900 CrossRef CAS.
- G. Gody, T. Maschmeyer, P. B. Zetterlund and S. Perrier, Nat. Commun., 2013, 4, 2505–2513 Search PubMed.
- L. Sun, N. Petzetakis, A. Pitto-Barry, T. L. Schiller, N. Kirby, D. J. Keddie, B. J. Boyd, R. K. O'Reilly and A. P. Dove, Macromolecules, 2013, 46, 9074–9082 CrossRef CAS.
- T. Nicolai, O. Colombani and C. Chassenieux, Soft Matter, 2010, 6, 3111–3118 RSC.
- L. Zhang and A. Eisenberg, Macromolecules, 1999, 32, 2239–2249 CrossRef CAS.
- R. Thomas, Gene, 1993, 135, 77–79 CrossRef CAS PubMed.
- J. P. Patterson, M. P. Robin, C. Chassenieux, O. Colombani and R. K. O'Reilly, Chem. Soc. Rev., 2014, 43, 2412–2425 RSC.
- D. B. Wright, J. P. Patterson, A. Pitto-Barry, P. Cotanda, C. Chassenieux, O. Colombani and R. K. O'Reilly, Polym. Chem., 2015, 6, 2761–2768 RSC.
- D. B. Wright, J. P. Patterson, A. Pitto-Barry, A. Lu, N. Kirby, N. C. Gianneschi, C. Chassenieux, O. Colombani and R. K. O'Reilly, Macromolecules, 2015, 48, 6516–6522 CrossRef CAS.
- D. B. Wright, J. P. Patterson, N. C. Gianneschi, C. Chassenieux, O. Colombani and R. K. O'Reilly, Polym. Chem., 2016, 7, 1577–1583 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details for nucleobase monomers and CTA, 1H NMR spectra, SEC traces of the macro-CTA and nucleobase-containing diblock copolymers, and DLS, SAXS, TEM, 1H NMR and UV-vis characterization of the interactions between PNAM-b-PAAm and PNAM-b-PTAm. See DOI: 10.1039/c6py00716c |
| This journal is © The Royal Society of Chemistry 2016 |