
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diffusion of single molecular and macromolecular probes during the free radical bulk polymerization of MMA – towards a better understanding of the Trommsdorff effect on a molecular level†
Jan Martin
Nölle
a,
Sebastian
Primpke
b,
Klaus
Müllen
c,
Philipp
Vana
b and
Dominik
Wöll
*ade
aFachbereich Chemie, Universität Konstanz, Universitätsstr. 10, D-78457 Konstanz, Germany
bInstitute of Physical Chemistry, Universität Göttingen, Tammannstr. 6, D-37077 Göttingen, Germany
cInstitute of Physical Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10-14, D-55128 Mainz, Germany
dZukunftskolleg, Universität Konstanz, Universitätsstr. 10, D-78457 Konstanz, Germany
eInstitute of Physical Chemistry, RWTH Aachen University, Landoltweg 2, D-52074 Aachen, Germany. E-mail: woell@pc.rwth-aachen.de
First published on 24th May 2016
Abstract
Free radical bulk polymerizations exhibit complex kinetics due to the viscosity increase during the polymerization process. Especially the termination rate constant can be strongly influenced by the mobility of polymer chains in the polymerization mixture. As a consequence an autoacceleration period, the so-called Trommsdorff effect, can be observed often. In order to investigate this behaviour on a nanoscopic scale, we directly visualized the mobility of molecules and macromolecules in polymerizing MMA solutions using a combination of highly sensitive fluorescence correlation spectroscopy and widefield fluorescence microscopy. For this purpose, rather monodisperse PMMA chains were synthesized by RAFT polymerization and fluorescence-labelled with perylenediimide derivatives. The behaviour of the different fluorescent probes could be related to their size and flexibility. Our studies show that diffusional heterogeneities must be accounted for when modeling bulk polymerization.
Introduction
Diffusion-controlled reactions play an essential role during polymerization processes, and not only influence polymerization kinetics but also the properties of the synthesized polymeric materials.1 In particular, at medium to high monomer conversions during free radical chain polymerizations with strong viscosity increase, the kinetics is significantly influenced. For that reason, many bulk polymerizations exhibit an autoacceleration period, also called the gel or Trommsdorff(-Norrish)2,3 effect at medium monomer conversion. It can be explained by a significant drop in the termination rate constant kt as a result of the decreasing number of encounters of two radical ends in the reaction medium of increasing viscosity.1 The magnitude of the Trommsdorff effect depends on the polymerization system and, for co-polymerizations, in a nonlinear way on the monomer ratio.4 The Trommsdorff effect can cause severe disadvantages for industrial scale polymerizations. As one possibility to reduce this effect, applying shear stress has been reported.5 Despite its technological importance and many advances and studies, a detailed understanding of the Trommsdorff effect is still missing.6 Thus, often simple empirical models with tunable parameters are used.7 However, only by including microscopic scale observations, the goal of a consistent overall picture can be reached. According to O'Neil et al., the temperature dependency of kt can be described by a free volume picture8 and its monomer conversion dependency can be related to monomer diffusion. In general, due to their high mobility, the shortest radical chains present in reasonable number determine the overall termination kinetics.9–13 The significance of the Trommsdorff effect is not restricted to simple polymerization systems, but can also rule the kinetics of polymerization induced phase separation.14 Recent advances in Monte Carlo methods allow even for a detailed modeling of the Trommsdorff effect by simulations of cross-linking polymerization reactions.15In our previous work, we achieved for the first time to directly observe the diffusion of fluorescent probes in polymerizing solutions and relate their motion to polymerization kinetics.16 In contrast to bulk methods, single molecule tracking allows for a determination of the distributions of diffusion coefficients.17–21 Thus, diffusional heterogeneities can be accessed and related to structural differences within the polymerizing mixture. We observed significant heterogeneity in single molecule motion during the autoacceleration period of the polymerization of methyl methacrylate, which is known to exhibit a strong Trommsdorff effect.22 Such a heterogeneous behaviour can also be found in microviscosity measurements using molecular rotors.23 In contrast, single molecule mobility during the styrene polymerization with only weak Trommsdorff effect was essentially homogeneous. Interestingly, already in 1977, Roschupkin et al. reported on the connection between autoacceleration of a polymerization and appearance of structural heterogeneity.24 These observations suggest the question how the appearance of diffusional heterogeneities influences the kinetics of polymerizations and how they can be included into the modeling of polymerization processes.
With this paper, we lay the foundation for a better understanding of the Trommsdorff effect on a molecular level. We observed in situ the motion of molecular and macromolecular probes of different sizes during the free radical bulk polymerization of methyl methacrylate (MMA). All probes contain a perylene diimide (PDI) fluorophore. The size of the molecular probes is determined by substitution in the bay region, whereas a PMMA chain of low polydispersity was attached to the macromolecular probes. A combination of fluorescence correlation spectroscopy (FCS)25,26 and widefield fluorescence microscopy (WFM) allowed us to monitor diffusion over a range of eight orders of magnitude between 10−9 and 10−17 m2 s−1. The scaling of diffusion coefficients and the evolution of diffusional heterogeneities for the different probes could be analysed and related to their flexibility. Our results demonstrate the importance of single molecule studies for a detailed understanding of polymerization processes.
Results
We observed and compared the translational diffusion of two molecular and two macromolecular probes. As shown in Scheme 1, all probes contain a perylene diimide chromophore with four phenoxy substituents in the bay region. Their fluorescence spectra are shown in the ESI (Fig. S4†). The hydrodynamic radii of the probing dyes were determined in pure MMA solution, i.e. at zero monomer conversion, using FCS. Using the viscosity 0.584 mPa s of MMA and a reference diffusion coefficient of 4.14 × 10−10 m2 s−1 for Rhodamine 6G in water,27 we obtained hydrodynamic radii of 0.96 nm for PDI0, 3.17 nm for PDI3, 2.56 nm for PDI-PMMA13k and 10.3 nm for PDI-PMMA150k, respectively. In PDI3, the chromophore is embedded in a third generation dendrimer, resulting in a rather spherical shape of the probe.28 The macromolecular probes were synthesized using RAFT polymerization of methyl methacrylate and labelled via a thiole–ene-reaction with the maleimide functionalized perylene diimide (see also ESI†). The molecular weight of the macromolecular probes is 13.2 kg mol−1 for PDI-PMMA13k and 150.6 kg mol−1 for PDI-PMMA150k, respectively. The polydispersities of the labelled PMMA chains are 1.13 and 1.38. | ||
| Scheme 1 Structures of fluorescent probes used in this study: PDI0 represents a typical molecular probe, whereas the dendrimerised PDI3 is a compact molecular probe of large size. In contrast the macromolecular probes PDI-PMMA13k and PDI-PMMA150k, possess significant flexibility. | ||
A solution of MMA monomers with 1% of the thermal radical starter 2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile) (V70) and an appropriate amount of fluorescent probes in a subnanomolar concentration was polymerized at room temperature. The same polymerization system has previously been studied by fluorescence correlation spectroscopy, widefield fluorescence microscopy22 and fluorescence lifetime determination.23 The very low probe concentration does not influence polymerization kinetics of the formed material. The reaction may be divided into three temporal stages: the onset of the reaction, the Trommsdorff effect region of rapid acceleration and the glass effect region. The evolution of monomer conversion as a function of time can be found in a previous paper.23 From low to medium monomer conversion, we were able to measure diffusion coefficients of the different probes during polymerization by FCS. The results are presented in Fig. 1.
 | ||
| Fig. 1 Plot of the measured diffusion coefficients D of different probes (see legend) versus monomer conversion. The solid lines present fits according to the scaling law in eqn (1). For macromolecular probes, this fits fail at some point. From this point, the lines are drawn dashed. Fit values according to eqn (1) are presented in Table 1, and are carefully evaluated and discussed in section 6 of the ESI.† The evolution of conversion versus time can be found in Fig. S2 (see ESI†). | ||
For the molecular probes PDI0 and PDI3, the data can be fitted with the following scaling law:29
| log(D/D0) = −αξν | (1) |
Herein, D and D0 are the current diffusion coefficient and the diffusion coefficient at zero monomer conversion, respectively, ξ is the monomer conversion, and α and ν are scaling parameters. The values used for the fit in Fig. 1 are presented in Table 1.
| PDI0 | PDI3 | PDI-PMMA13k | PDI-PMMA150k | |
|---|---|---|---|---|
| D 0/m2 s−1 | 3.76 × 10−10 | 1.17 × 10−10 | 1.48 × 10−10 | 0.36 × 10−10 |
| α | 63 ± 9 | 77 ± 15 | 424 ± 28 | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 ± 6800 700 ± 6800 |
| ν | 2.32 ± 0.09 | 1.95 ± 0.10 | 3.18 ± 0.03 | 4.70 ± 0.19 |
For the macromolecular probes PDI-PMMA13k and PDI-PMMA150k, the same scaling law can be applied.30 A fit, however, only works sufficiently well up to monomer conversions of 23% and 17%, respectively. At higher monomer conversions, the measured diffusion coefficients are higher than expected. The reason for this deviation from the scaling law can be explained by the flexibility of the macromolecular probes. They can more easily diffuse through meshes created by the growing polymer chains than rigid molecular probes of comparable size.
FCS is a suitable method to gain (average) diffusion coefficients for the low viscosity range. For medium monomer conversions, we switched to WFM which allows for the direct observation and tracking of the motion of single probe molecules. Since the monomer conversion during the autoacceleration period increases rather rapidly and our setup did not allow for simultaneous in situ measurement of monomer conversion by Raman spectroscopy, we were not able to determine the exact monomer conversion at each time of the WFM measurement. Since the exact time of the steepest point in the time-conversion plot of different experiments varies by some minutes, we can neither use the monomer conversions determined in a parallel experiment on the Raman setup. Therefore, only a rough estimate could be obtained and, instead of monomer conversion, we have chosen a reference time tR where all probes exhibit a comparable average diffusion coefficient of logD = −13.5 and similar distributions of diffusion coefficients (see third column of diagrams in Fig. 3). The FCS measurements in Fig. 1 can be used to estimate the corresponding monomer conversions to 44% for PDI-PMMA13k, 42% for PDI0, 33% for PDI-PMMA150k, and 32% for PDI3. Despite the fact that tR does not represent the same monomer conversion for the different probes, it allows for a comparison of the evolution of diffusion coefficients and their heterogeneities.
Movies recorded at different polymerization times within the autoacceleration period can be found in the ESI.† Representative trajectories obtained from single molecule tracking in these movies are presented in Fig. 2. For the connection of single molecule positions to trajectories (tracking), we applied our previously developed algorithm.18 Single molecule trajectories were analysed according to the maximum likelihood estimation as described in literature.31,32 Already from an inspection of these trajectories, significant differences between the four probes become obvious. In particular they differ for the period a few minutes before the reference time tR. At this stage only few trajectories could be recorded as a consequence of too rapid motion of most molecules in this low to medium monomer conversion range. As shown in Fig. 3, the distribution of the diffusion coefficients of molecules that can be tracked depends strongly on the probe. No trajectories are observed for PDI0. PDI3 and PDI-PMMA13k show a distribution centered around logD = −13.5 and PDI-PMMA150k additionally contains trajectories with basically immobile probes. Approaching tR, all probes reach a mobility which allows for their tracking. At tR, the number of trajectories is at a maximum. Molecules are sufficiently slow to be tracked, but due to their still rapid motion, new trajectories start rather frequently, trajectories end or trajectories restart. Unfortunately, to the best of our knowledge, there is no possibility to correct for this complication. At higher monomer conversions the number of trajectories decreases again since the number of molecules moving into and out of the focal plane decreases. At the same time, longer trajectories are obtained. Even though blinking events can cause the disappearance of probes for few frames, reliable tracking is possible since our tracking algorithm is adapted to such blinking events.18 The distributions of diffusion coefficients obtained 5 min after the reference time tR are very similar and present probes with low mobility which cannot be detected with the described WFM setup anymore. Here, apparent diffusion coefficients below 10−15 m2 s−1 can be biased by limited localization accuracy18 and the corresponding molecules will henceforth be defined as “immobile”.
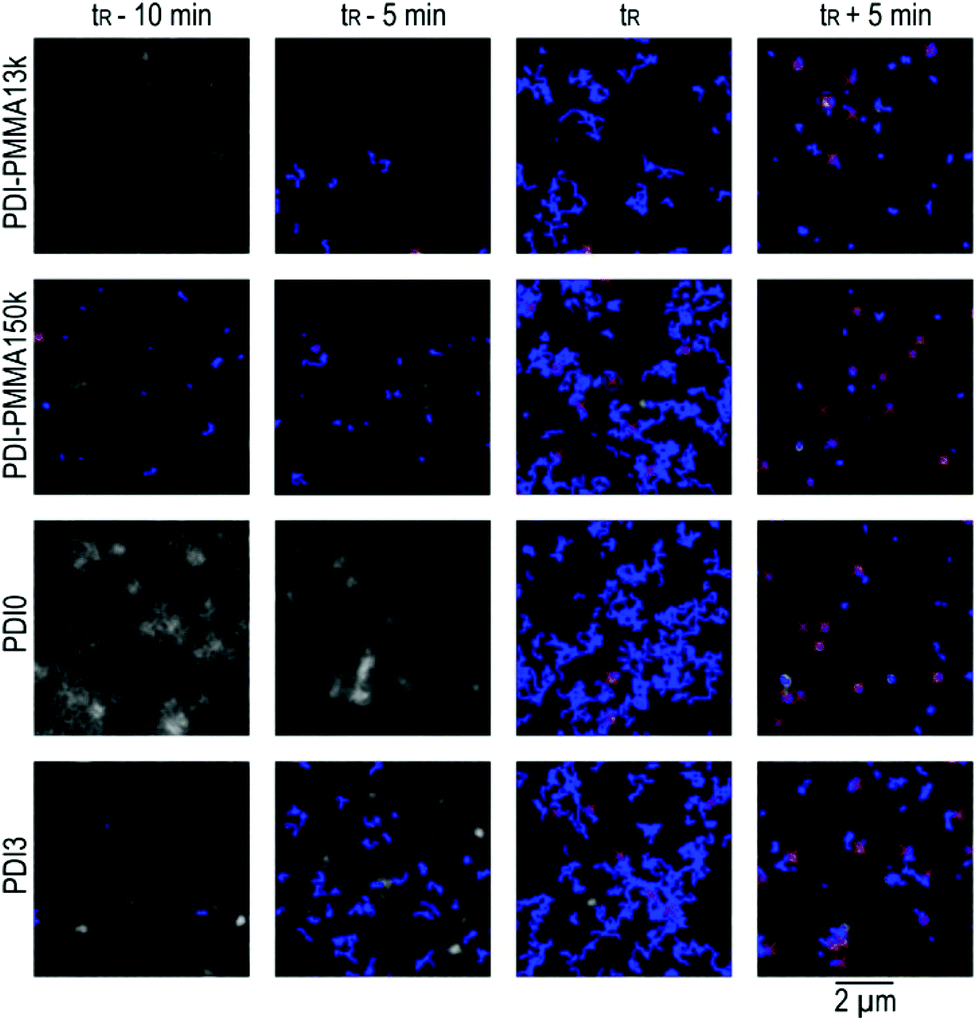 | ||
| Fig. 2 Representative widefield fluorescence microscopy images of a movie series shown in the ESI.† Different fluorescent probes (see labels of the rows) in the corresponding reaction mixture are shown at different points in time (see labels of the columns). Red crosses indicate single molecule localizations in the corresponding frame. Trajectories are presented as blue lines. | ||
 | ||
| Fig. 3 Evolution of the diffusion coefficients in the Trommsdorff effect region. The occurrence of logarithmic diffusion coefficients are plotted for molecular and macromolecular dyes (rows) at different points in time (columns) during the bulk polymerization of MMA. Only the diffusion coefficients of the molecules that could be tracked are shown. The distributions at additional times and a statistical characterization of all distributions can be found in section 1 and 2 of the ESI.† | ||
Discussion
Diffusion during the free radical bulk polymerization of MMA was found to evolve in a significantly different way for the investigated molecular and macromolecular probes. At low monomer conversion and the concomitant low viscosity, all probes follow a stretched exponential scaling law. Proceeding to the medium monomer conversion range, it could be shown that macromolecular probes deviate from this scaling at rather low monomer conversion and move significantly faster. For the short chain length probe PDI-PMMA13k, this deviation appeared at 17%, whereas it was found at 23% for the long chain length probe PDI-PMMA150k. This behaviour is caused by the higher flexibility of macromolecular probes which possess a lot of conformational freedom to sneak through meshes formed by the polymer chains in the polymer solution. In contrast, the molecular dyes are rather rigid so that the mesh size more critically determines their motion. Thus, PDI3 which possesses an only slightly larger hydrodynamic radius than PDI-PMMA13k (3.17 nm versus 2.56 nm as measured in monomer solution) follows the stretched exponential scaling law up to at least 30% monomer conversion. In a previous paper, we have estimated that the mesh size reaches the diameter of PDI3 already at a monomer conversion of ca. 13%.22 Interestingly, within the same monomer conversion range the deviation of the similarly sized PDI-PMMA13k from the initial scaling appears. At this point, macroscopic (bulk-)viscosity looses its validity to describe molecular motion, and nanoviscosity starts to rule. The transition from macroscopic to microscopic viscosity has been discussed by Hołyst and coworkers,33,34 and is also known for denser polymer systems close to their glass transition.35,36 Our results show that the observed viscosity is not only length-scale dependent, but also influenced by the flexibility of the probe. The more flexible macromolecules can adapt their conformation to mesh sizes which would be too small for rigid probes. The latter can only move after expansion of the temporally fluctuating meshes.Turning towards medium monomer conversions, the motion of probes becomes sufficiently slow to be observed and tracked by widefield fluorescence microscopy where we developed powerful tools to analyse single molecule diffusion.18 As already described in the results part, especially in the monomer conversion range between 20% and 40% the probes diffuse significantly heterogeneously. In particular, WFM allows for analysing diffusion coefficient distributions and, thus, to visualize and analyse heterogeneities of single molecule motion in the polymerization system. Especially for the largest probe PDI-PMMA150k, we observed molecules becoming immobile already at monomer conversions of ca. 25%. Even though the number of these immobilized probes is low (a fraction of <10%), this observation demonstrates the appearance of high density polymer regions. Such a deceleration and immobilization of probes is, much less pronounced, also observed for PDI3 and PDI-PMMA13k. At the current stage we, unfortunately, cannot determine the size or structure of these regions, and only can assume that polydisperse microgels are formed37 due to limited solubility of PMMA in MMA. The mesh size of these regions, however, can be estimated to be in a size range around the diameter of ca. 5 nm, the size of the probes PDI3 and PDI-PMMA13k which in some cases can be immobilized. The smallest dye PDI0 is too fast to be captured. The consequences of the described heterogeneities in polymerization kinetics have, to the best of our knowledge, never been addressed. The polymerization rate in regions of higher polymer density could be locally enhanced due to the restricted mobility of the radicals of growing macromolecules. As a result, the overall polymerization rate of the entire system can significantly be determined by such domains. This argumentation is also in agreement with previous studies where we found that significant diffusional heterogeneities can be found in a MMA polymerization system with pronounced Trommsdorff effects, whereas a styrene polymerization system showed neither heterogeneities nor a strong Trommsdorff effect.22
At medium to high monomer conversions, the probes immobilize on the time and length scales at which we are able to detect single molecule diffusion. No significant differences in the heterogeneity in this phase were found.
Conclusions
In summary, we observed the translational diffusion of single molecular and macromolecular probes of different sizes, all of them bearing a perylene diimide fluorophore. Fluorescence correlation spectroscopy allowed for a comparison of the scaling of the diffusion coefficients with increasing monomer conversion. Initially, all probes followed a stretched exponential scaling. However, at monomer conversions below 20%, when the diameter of the probes becomes similar to the mesh size of the polymer, the diffusion coefficient of the macromolecular probes decreases in a less pronounced fashion than the one of rigid probes. This behaviour can be attributed to the flexibility of the polymer chains. Despite their flexibility, a fraction of the macromolecular probes becomes immobilized at monomer conversions below 30%, a range where the molecular probes are still moving rather rapidly. The observed heterogeneity in diffusion coefficients of the macromolecular probes points to a structural heterogeneity of the polymerizing solution which can have significant influence on the termination rate constant and, therefore, on the strength of the Trommsdorff effect. Future studies are planned to investigate this phenomenon in more detail and to connect it with polymerization kinetics.Experimental
Polymerizations
Between 1 and 3 drops of a solution of the corresponding perylenediimide derivative in spectroscopically pure toluene were placed in the reaction vessel, and the toluene was evaporated. 3 mL of MMA and 1% w/w of the radical starter V70 (Wako Chemicals) were added, so that the probe concentration was in a sub-nanomolar range. The reaction mixture was homogenized by vortex shaking and degassed by three pump–freeze–thaw cycles. In a glovebox, approx. 1 ml of the polymerization mixture was added to specially designed, closed glass vessels for microscopy. In these vessels, the mixture was polymerized at room temperature under nitrogen atmosphere.Fluorescence correlation spectroscopy
A cw-laser with Gaussian beam profile (Cobolt Samba 532 nm) was expanded by a telescope and attenuated to 100 μW by neutral density filters. It was coupled into a microscope (Zeiss Axiovert 200) and focused into the sample by an objective (Zeiss Plan-FLUAR 100×/1.45 Oil). The focus was located in solution approx. 10 μm above the coverslip surface. Fluorescence light was collected through the same objective and separated from scattered excitation light by a beamsplitter (Chroma z532RDC). A pinhole (50 μm diameter) was used to block out-of-focus light. Remaining scattered light was blocked by a Notch filter (Semrock NF01-532U-25) and a longpass filter (Semrock LP568). The emission light was subsequently divided by a 50/50 beam splitter (Melles Griot 03BSC007) and was focused onto two avalanche photodiodes (Perkin Elmer SPCM-AQR-14 SPAD). To prevent APD afterpulses, a shortpass filter (Chroma 680SP-2P8) was placed in front of one APD. The signal was processed by a HydraHarp 400 device using the SymPhoTime software (Picoquant GmbH). Each measurement was performed for 120 s with a detector count rate of approx. 5000 counts per second.Fluorescence microscopy
A Cobolt Jive laser (561 nm, 75 mW) was coupled into a multi-mode glass fiber (NA = 0.22 ± 0.02) which was shaken at 2000 rpm by a VWR vortexer to temporally average out interference patterns. The output of the glass fiber end was coupled into a DMI6000B microscope (Leica Microsystems GmbH) in epi-mode and projected onto the sample with a HCX PL APO 100×/1.40–0.70 oil immersions objective (Leica). The fluorescence light was separated from the scattered and reflected excitation light using a dichroic mirror (z561/660rpc, Chroma Technology Corp.) and a longpass filter (RazorEdge LP568 RU, AHF Analysentechnik Tübingen). Using two photo objectives (Nikkor 28 mm f2.8, Nikkor 85 mm f2.0), the sample plane was imaged onto a Andor Ixon DV887DCS-BV EMCCD camera with a chip cooled to −80 °C. Widefield movies with an integration time of 100 ms per image were recorded from the time when the polymerization mixture reached an appropriate viscosity. The measured axial position was approx. 8 μm above the cover slip surface. For each measurement, a new lateral position was chosen to minimize bleaching effects.Synthesis of labelled PMMA chains
The controlled synthesis of PDI fluorophores containing PMMA chains can be found in the ESI.†Acknowledgements
D. W. thanks the Zukunftskolleg of the University of Konstanz for financial and administrative support. We want to thank Prof. Michael Buback (University of Göttingen) for fruitful discussions. K. M. thanks the Gutenberg Research College of the Johannes Gutenberg Universität Mainz.Notes and references
- C. Barner-Kowollik and G. T. Russell, Prog. Polym. Sci., 2009, 34, 1211–1259 CrossRef CAS.
- E. Trommsdorff, H. Kohle and P. Lagally, Makromol. Chem., 1948, 1, 169–198 CrossRef CAS.
- R. G. Norrish and R. R. Smith, Nature, 1942, 150, 336–337 CrossRef CAS.
- K. F. O'Driscoll and J. Huang, Eur. Polym. J., 1990, 26, 643–647 CrossRef.
- M. Cioffi, A. C. Hoffmann and L. P. B. M. Janssen, Polym. Eng. Sci., 2001, 41, 595–602 CAS.
- D. S. Achilias, Macromol. Theory Simul., 2007, 16, 319–347 CrossRef CAS.
- J. S. Sangwai, S. A. Bhat, S. Gupta, D. N. Saraf and S. K. Gupta, Polymer, 2005, 46, 11451–11462 CrossRef CAS.
- G. A. O'Neil, M. B. Wisnudel and J. M. Torkelson, Macromolecules, 1998, 31, 4537–4545 CrossRef.
- G. T. Russell, R. G. Gilbert and D. H. Napper, Macromolecules, 1992, 25, 2459–2469 CrossRef CAS.
- B. O'Shaughnessy and J. Yu, Macromolecules, 1994, 27, 5067–5078 CrossRef.
- G. T. Russell, R. G. Gilbert and D. H. Napper, Macromolecules, 1993, 26, 3538–3552 CrossRef CAS.
- B. O'Shaughnessy and J. Yu, Macromolecules, 1994, 27, 5079–5085 CrossRef.
- P. Derboven, D. R. D'hooge, M.-F. Reyniers, G. B. Marin and C. Barner-Kowollik, Macromolecules, 2015, 48, 492–501 CrossRef CAS.
- T. Ozaki, T. Koto, T. V. Nguyen, H. Nakanishi, T. Norisuye and Q. Tran-Cong-Miyata, Polymer, 2014, 55, 1809–1816 CrossRef CAS.
- M. Lang, A. John and J.-U. Sommer, Polymer, 2016, 82, 138–155 CrossRef CAS.
- D. Wöll, H. Uji-i, T. Schnitzler, J. Hotta, P. Dedecker, A. Herrmann, F. C. De Schryver, K. Müllen and J. Hofkens, Angew. Chem., Int. Ed., 2008, 47, 783–787 CrossRef PubMed.
- K. H. T. Ba, T. A. Everett, T. Ito and D. A. Higgins, Phys. Chem. Chem. Phys., 2011, 13, 1827–1835 RSC.
- D. Wöll, C. Kölbl, B. Stempfle and A. Karrenbauer, Phys. Chem. Chem. Phys., 2013, 15, 6196–6205 RSC.
- B. Shuang, J. Chen, L. Kisley and C. F. Landes, Phys. Chem. Chem. Phys., 2014, 16, 624–634 RSC.
- M. J. Saxton, Nat. Methods, 2008, 5, 671–672 CrossRef CAS PubMed.
- T. Cordes and S. A. Blum, Nat. Chem., 2013, 5, 993–999 CrossRef CAS PubMed.
- B. Stempfle, M. Dill, M. Winterhalder, K. Müllen and D. Wöll, Polym. Chem., 2012, 3, 2456–2463 RSC.
- J. M. Nölle, C. Jüngst, A. Zumbusch and D. Wöll, Polym. Chem., 2014, 5, 2700–2703 RSC.
- V. P. Roschupkin, B. V. Ozerkovskii, Y. B. Kalmykov and G. V. Korolev, Polym. Sci. U.S.S.R., 1977, 19, 809–819 CrossRef.
- D. Wöll, RSC Adv., 2014, 4, 2447–2465 RSC.
- K. Koynov and H.-J. Butt, Curr. Opin. Colloid Interface Sci., 2012, 17, 377–387 CrossRef CAS.
- C. B. Müller, A. Loman, V. Pacheco, F. Koberling, D. Willbold, W. Richtering and J. Enderlein, EPL, 2008, 83, 46001 CrossRef.
- J. Q. Qu, J. Y. Zhang, A. C. Grimsdale, K. Müllen, F. Jaiser, X. H. Yang and D. Neher, Macromolecules, 2004, 37, 8297–8306 CrossRef CAS.
- G. D. J. Phillies, Macromolecules, 1995, 28, 8198–8208 CrossRef CAS.
- G. D. J. Phillies, Macromolecules, 1986, 19, 2367–2376 CrossRef CAS.
- D. Montiel, H. Cang and H. Yang, J. Phys. Chem. B, 2006, 110, 19763–19770 CrossRef CAS PubMed.
- B. Shuang, C. P. Byers, L. Kisley, L.-Y. Wang, J. Zhao, H. Morimura, S. Link and C. F. Landes, Langmuir, 2013, 29, 228–234 CrossRef CAS PubMed.
- R. Hołyst, A. Bielejewska, J. Szymanski, A. Wilk, A. Patkowski, J. Gapinski, A. Zywocinski, T. Kalwarczyk, E. Kalwarczyk, M. Tabaka, N. Ziebacz and S. A. Wieczorek, Phys. Chem. Chem. Phys., 2009, 11, 9025–9032 RSC.
- K. Sozański, A. Wiśniewska, T. Kalwarczyk and R. Hołyst, Phys. Rev. Lett., 2013, 111, 228301 CrossRef PubMed.
- P. K. Dixon, S. R. Nagel and D. A. Weitz, J. Chem. Phys., 1991, 94, 6924–6925 CrossRef CAS.
- F. Brochard Wyart and P. G. De Gennes, Eur. Phys. J. E, 2000, 1, 93–97 CrossRef.
- N. Ide and T. Fukuda, Macromolecules, 1999, 32, 95–99 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py00590j |
| This journal is © The Royal Society of Chemistry 2016 |