
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ethynyl benziodoxolones: functional terminators for cell-penetrating poly(disulfide)s†
Paola
Morelli
ab,
Xavier
Martin-Benlloch
ab,
Romain
Tessier
ac,
Jerome
Waser
ac,
Naomi
Sakai
ab and
Stefan
Matile
*ab
aNational Centre of Competence in Research (NCCR) Chemical Biology, Switzerland Web: http://www.nccr-chembio.ch
bDepartment of Organic Chemistry, University of Geneva, Geneva, Switzerland. E-mail: stefan.matile@unige.ch; Web: http://www.unige.ch/sciences/chiorg/matile/
cLaboratory of Catalysis and Organic Synthesis, Ecole Polytechnique Fédérale de Lausanne (EPFL), Lausanne, Switzerland
First published on 29th April 2016
Abstract
Outperforming cell-penetrating peptides (CPPs), cell-penetrating poly(disulfide)s (CPDs) are attracting increasing interest. CPDs are accessible by ring-opening disulfide-exchange polymerization under mild conditions in neutral water. Initiation of the polymerization with thiols results in quantitative labeling of one CPD terminus with initiators of free choice. In contrast, labeling of the other terminus with iodoacetamides has so far been ineffective because of poor yields and the high excess of reagents needed. In this report, we introduce hypervalent iodine reagents as operational terminators of the synthesis of CPDs, also at high dilution. The power of the approach is exemplified with green-fluorescent initiators and ethynyl benziodoxolone terminators containing additional azides for CuAAC with red-fluorescent alkynes. The absorption spectra of the resulting CPDs demonstrate that stoichiometric application of ethynyl benziodoxolone terminators results in 46% incorporation. FRET between green-fluorescent initiators and red-fluorescent terminators demonstrates significant folding of CPDs in solution; it disappears upon reductive depolymerization. Substrates attached to the new termini are shown to enter into HeLa cells. Moreover, disappearance of FRET in the cytosol corroborated the reductive cleavage of CPDs upon internalization. Beyond the introduction of enthynyl benziodoxolones as operational terminators, these findings thus demonstrate also the compatibility of CuAAC with poly(disulfide)s and the usefulness of doubly-labeled CPDs for structural and mechanistic studies.
Introduction
Cell-penetrating poly(disulfide)s (CPDs)1–5 have been introduced recently to address a central challenge with cell-penetrating peptides (CPPs),6,7i.e., cytotoxicity. Like CPPs, CPDs are guanidinium-rich polymers. However, the peptide backbone of CPPs is replaced by a poly(disulfide).8–13 With dynamic covalent disulfide bonds in the backbone, a new way to enter cells opens up (Fig. 1).1,2 Approaching the cell surface, ion pairing of CPDs with anionic lipids in the membranes will be strengthened by the powerful proximity effect known from CPPs and other polycationic oligomers.7 However, at the same time, CPDs engage in dynamic covalent chemistry1,2,11,14 on the cell surface. Namely, disulfide exchange with exofacial thiols, present on the surface to protect against an oxidative environment,11 covalently attaches the CPDs to the cell surface. Presence and significance of this thiol-mediated uptake mechanism has been demonstrated by uptake inhibition with Ellman's reagent, which converts all exofacial thiols into disulfides.1,2 Once attached covalently to the surface of the cell, CPDs with insufficient activity, either too short or too hydrophobic, will be taken up by endocytosis and end up trapped in endosomes.1,4 Long enough CPDs, however, will move across the bilayer membrane through transient micellar pores.7 Arriving in the cytosol, CPDs will be destroyed by reductive depolymerization with glutathione. This destruction of the transporter right after work liberates the substrate and eliminates toxicity. As depolymerization requires longer time with increasing length,4 fragments of long enough CPDs can escape into the nucleus and bind to the oligonucleotides in the nucleolus.1,3,4 This mechanistically new way of CPDs to enter cells has been probed in detail1,2,4 and applied to the delivery of small molecules such as fluorophores1 but also larger substrates such as proteins,3,5 antibodies,5 nanoparticles,5 and so on. Proteomics screens for targets of CPDs are ongoing and will be reported in due course.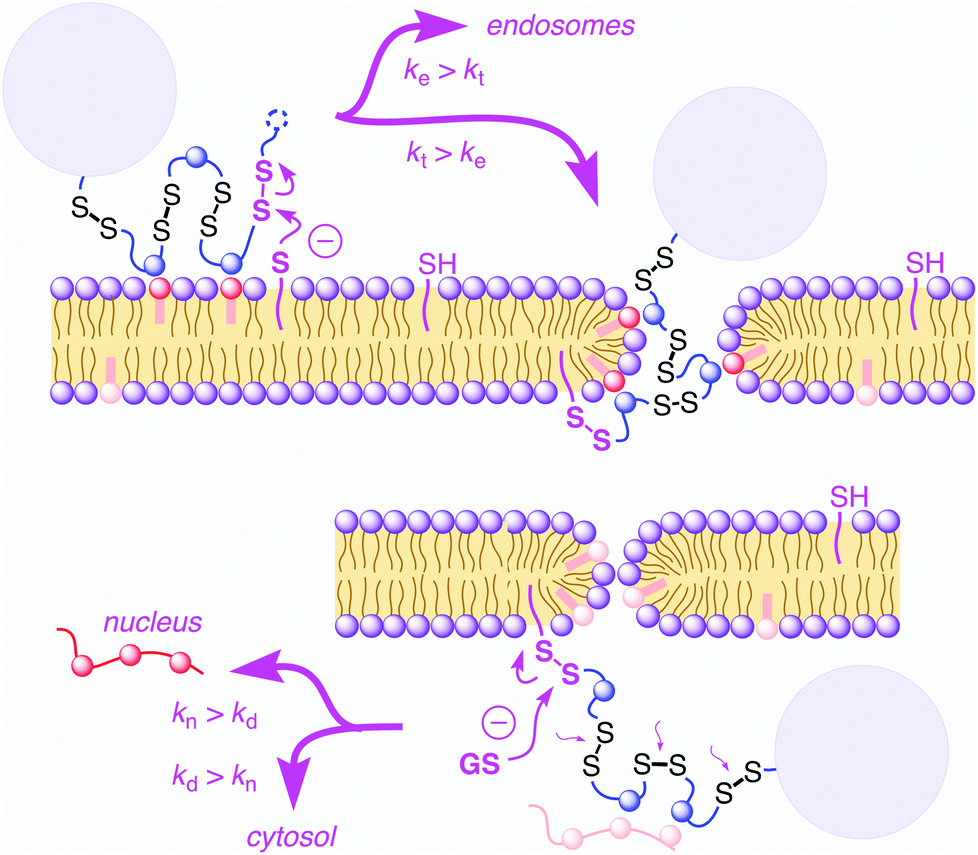 | ||
| Fig. 1 Cellular uptake of cell-penetrating poly(disulfide)s: supported by ion pairing of CPDs (blue) with anionic lipids (red), CPDs are attached covalently to the cell surface by disulfide exchange with exofacial thiols (magenta, top left). Slow translocation with less active CPDs (short, hydrophobic) results in endocytosis and endosomal trapping (ke > kt). Fast translocation through transient micellar pores (kt > ke, top right) followed by fast internal depolymerization with glutathione (GS−, kd > kn) results in cytosolic delivery of unmodified substrates (light blue circles, bottom). Slower internal depolymerization with longer CPDs results in binding to oligonucleotides (red) in the nucleoli (kn > kd). | ||
The synthesis of CPDs is based on substrate-initiated ring-opening disulfide-exchange polymerization (Fig. 2).12 Related to protein folding, this simple and reliable process has been developed originally in the context of the construction of multicomponent architectures on solid surfaces, particularly artificial photosystems.13 Transcribed to cellular uptake, the same process is initiated in solution using thiolated substrates of free choice as initiator.12 In this study, thiolated carboxyfluorescein 1 will be used as green-fluorescent initiator. Among several propagators reported, the conjugate 2 composed of lipoic acid and arginine has been most effective. Disulfide formation with the thiolate of initiator 1 opens the cyclic disulfide in propagator 2, releases the ring tension and generates a new thiolate for continuing polymerization.
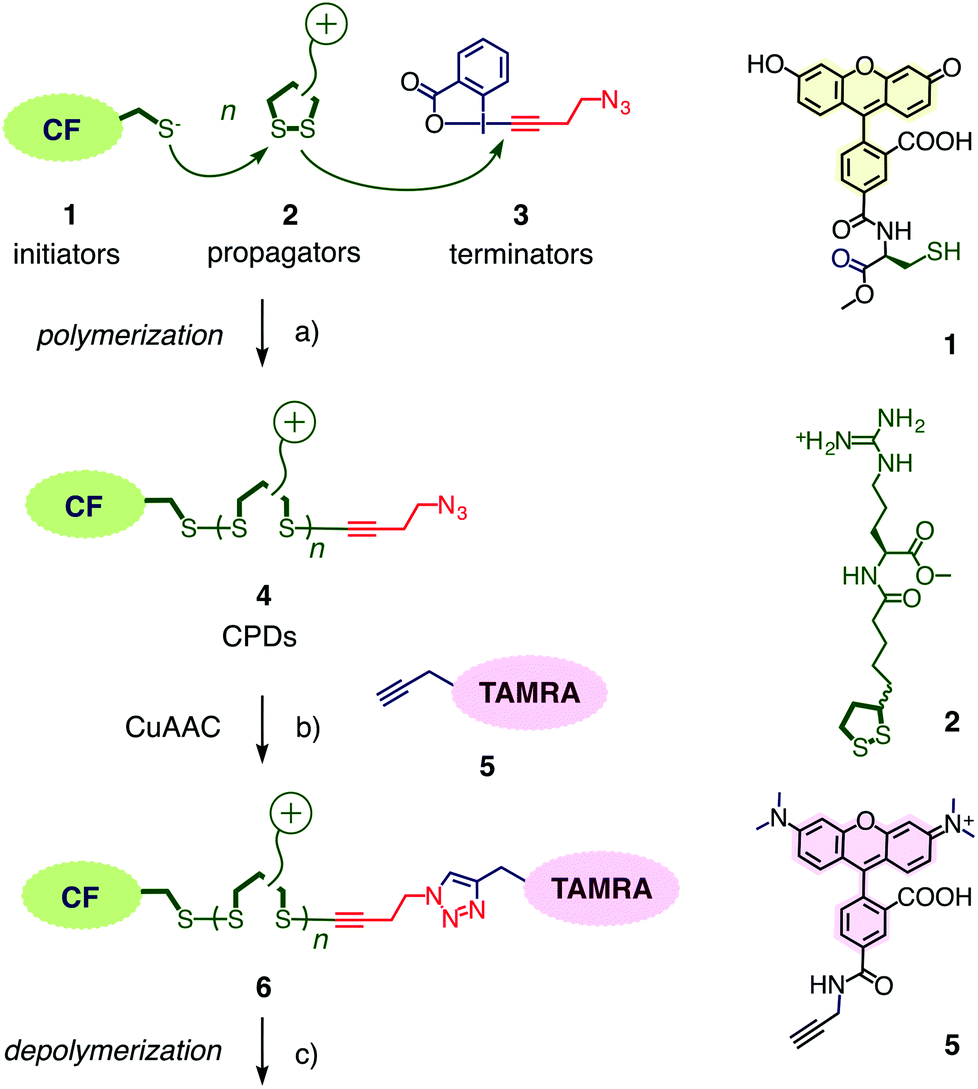 | ||
| Fig. 2 (a) Synthesis of cell-penetrating poly(disulfide) 4 by ring-opening disulfide-exchange polymerization, initiated with thiolate 1 and propagated with the strained cyclic disulfide 2 (pH 7, rt, 30 min). Termination with the new hypervalent iodine reagent 3 places an azide in polymer 4 (termination yield 46%, the nature of the termini of CPDs without azides is unknown). (b) The azide termini in 4 can be coupled with alkyne 5 (sodium ascorbate, CuSO4·5H2O, TBTA, rt, 17 h). (c) Reductive depolymerization can be followed by the disappearance of FRET from CF to TAMRA (using 100 mM DTT, pH 7, rt, 20 min). | ||
Substrate-initiated ring-opening disulfide-exchange polymerization occurs within minutes in neutral water, at ambient temperature.1 So far, iodoacetamides have been used to terminate the polymerization. However, these terminators were not very effective, incorporation yields remained far from quantitative, even at high concentrations. This poor performance of iodoacetamides is as such irrelevant but becomes problematic as soon as the termination is considered as an additional opportunity to covalently attach another substrate to the CPD. In this study, we introduce novel hypervalent iodine reagents15,16 that allow an efficient termination at stoichiometric concentrations, even at high dilution. These results are important because they provide a new and practical method to attach substrates to CPDs for their delivery into cells.
Results and discussion
Ethynyl benziodoxolones such as 3 have been introduced recently for the fast and highly chemoselective alkynylation of thiols (Fig. 2).15 Among other applications, they have been successfully used for the proteome-wide profiling of targets of cysteine-reactive probes and drugs.16 To terminate the polymerization of propagators 2 on initiator 1, ethynyl benziodoxolone 3 was selected because the resulting azide at one terminus of the resulting CPD 4 could be used to attach alkynylated substrates of free choice.16,17 The red-fluorescent TAMRA 5 was of interest for this purpose to quantify the efficiency of terminator 3 and to explore the usefulness of FRET to follow reductive CPD depolymerization.Initiator 1, propagator 2 and terminator 3 were synthesized as described.1,2,15 The alkynylated TAMRA 5 was accessible in one step from previously reported intermediates. Details can be found in the ESI.†
Polymers 4 were synthesized by ring-opening disulfide-exchange polymerization in neutral water at room temperature. To initiate the polymerization, the thiol in the green-fluorescent initiator 1 should be partially deprotonated. This thiolate then reacts with the strained disulfide in propagator 2, producing a disulfide bond and releasing ring tension together with a thiolate that reacts with the next propagator. After 30 minutes, the reactions were terminated by adding stoichiometric amounts of terminator 3. The obtained products were purified by gel-permeation chromatography (GPC). The example shown in Fig. 3a was characterized by Mw = 10.3 kDa, Mn = 9.9 kDa, and a resulting PDI = 1.04. Both length and polydispersity were very well reproducible and variable on demand.4
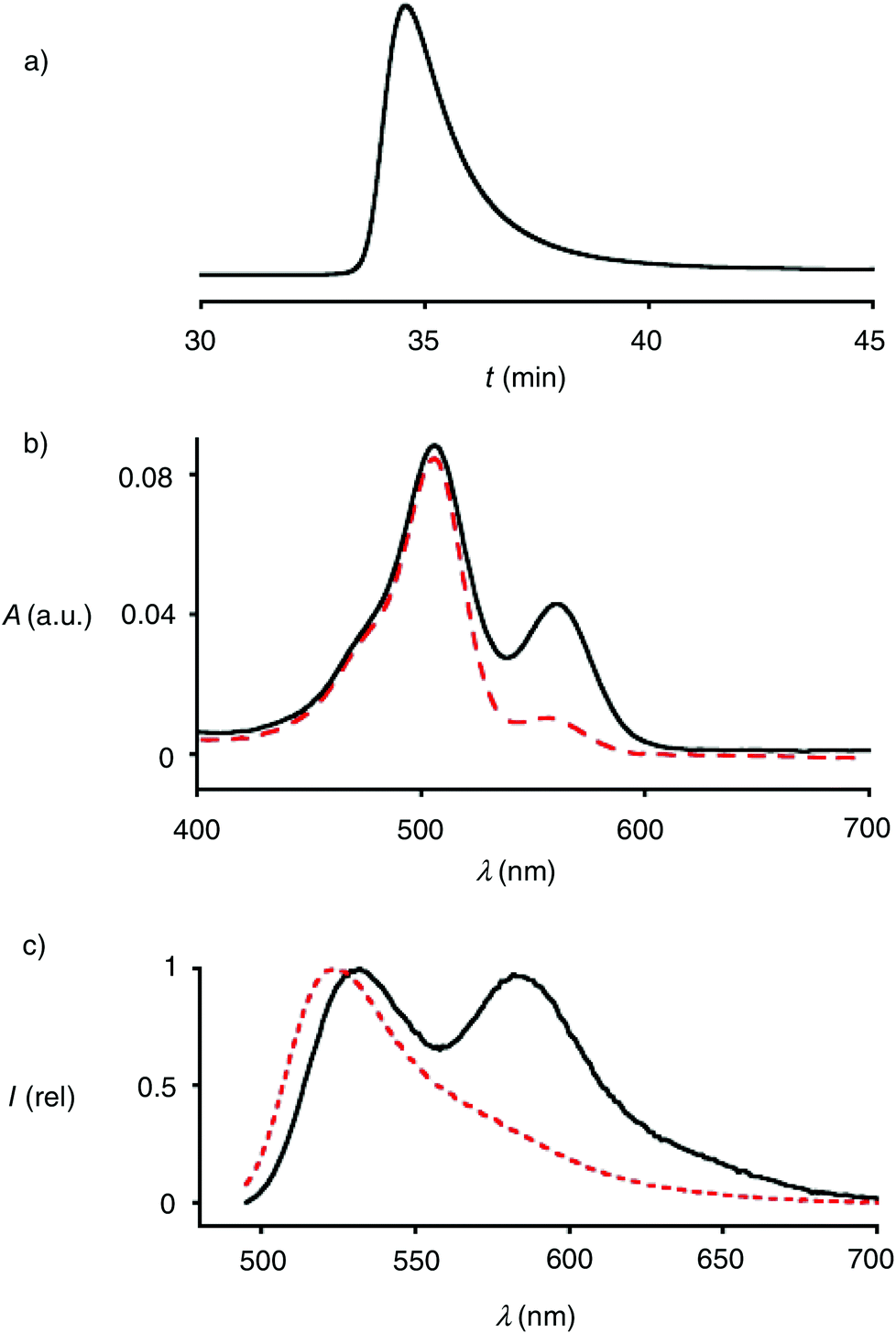 | ||
| Fig. 3 (a) GPC of polymer 4. (b) Absorption spectrum of polymer 6 (solid) and 9 (dashed) in TEOA buffer (1.0 M, pH 7.0). (c) Emission spectrum of polymer 6 in TEOA buffer (1.0 M, pH 7.0) before (solid) and after (dashed) incubation with 100 mM DTT (20 min, rt). | ||
The incorporation of terminator 3 into polymers 4 was readily demonstrated by CuAAC (copper(I)-catalyzed alkyne–azide cycloaddition)17 with alkynylated TAMRA 5. The absorption spectra of polymers 4 showed the maximum of TAMRA (λmax = 556 nm, εmax = 91.0 mM−1 cm−1) on the bathochromic side of that of CF (λmax = 494 nm, εmax = 83.0 mM−1 cm−1, Fig. 4b, solid). Assuming quantitative CuAAC,17 the absorbance of polymer 6 at these two maxima calibrated against the respective extinction coefficients indicated that terminator 3 was incorporated with an excellent yield of 46%. This yield was impressive considering that terminator 3 was applied in stoichiometric amounts at a comparably low concentration around 4.0 mM.
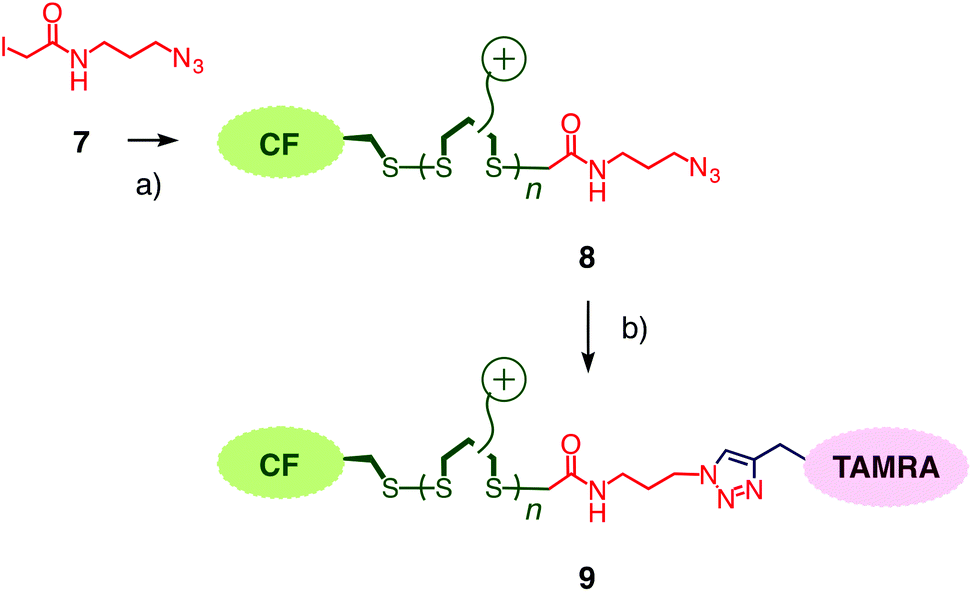 | ||
| Fig. 4 (a) Incorporation of control terminator 7 into CF-initiated polymer 8 (1, 2, pH 7.0, rt, 30 min, termination yield 11%, the nature of the termini of CPDs without azides is unknown). (b) CuAAC with TAMRA 5 (sodium ascorbate, CuSO4·5H2O, TBTA, rt, 17 h) to afford doubly-labeled polymer 9. | ||
The emission spectra of the doubly-labeled polymer 6 revealed the occurrence of FRET (Fig. 4c, solid). Upon excitation of the CF donor at 488 nm, emission from the green-fluorescent donor and the red-fluorescent acceptor was roughly equal. Judged from Mn = 9.9 kDa and a propagator weight of 378 Da, polymers 6 contained n ∼ 24 monomers, i.e., a degree of polymerization DP ∼ 24. With an estimated length l ∼ 6 Å per monomer, this approximated to l ∼ 14.4 nm of polymers 4 at full length. This is far beyond the Förster radius R0 = 55 Å of the donor–acceptor pair.18 This rough estimation suggested that in water, CPDs are folded down beyond half of their full length.
In the presence of 100 mM DTT, the FRET emission disappeared within minutes (Fig. 3c, dashed). This rapid response demonstrated that the observed FRET indeed originates from doubly-labeled monomers and not from aggregates. Detailed polymerization kinetics as a function of the length of the CPDs have been reported previously.4 In studies on cellular uptake of 4, the additional presence of CPDs without terminal TAMRA will not influence results that are based on TAMRA emission (see below).
To compare results with hypervalent iodine terminators 3 with conventional terminators, iodoacetamide 7 with an additional azide was prepared (Fig. 4). Polymers 8 were synthesized from initiator 1 and propagators 2 under the conditions used to prepare polymer 4, i.e., neutral water, room temperature, thirty minutes. Also the conditions for the termination of polymer 4 with iodoacetamide 7 were kept identical to the stoichiometric termination of polymer 4 with ethynyl benziodoxolone 3. CuAAC of polymer 8 with TAMRA 5 gave the doubly-labeled polymer 9. Assuming quantitative CuAAC, the absorption spectrum of polymer 9 revealed that the termination with iodoacetamide 7 was as ineffective as expected (Fig. 3b, dashed). The identified yield of 11% with iodoacetamide terminators 7 was 4 times less than the 46% with the new ethynyl benziodoxolone terminators 3. This significant difference identified ethynyl benziodoxolones as terminators of choice for the efficient labelling of CPDs under stoichiometric conditions at high dilution. Moreover, the clearly different incorporation of TAMRA into polymers 4 and 8 confirmed that terminal CPD labelling is not determined by the yield of CuAAC. This finding demonstrated that CuAAC is compatible with poly(disulfide)s, despite the presence of disulfides at high effective molarity that could conceivably inactivate the copper catalyst.
Cellular uptake of the doubly-labeled polymer 6 was determined using confocal laser scanning microscopy (CLSM). Images were taken after 15 min incubation of polymer 6 at low concentration (500 nM) with HeLa Kyoto cells. The emission of CF detected at 517 ± 18 nm upon excitation at 488 nm indicates the location of CF fluorophore without attached TAMRA. It was found mostly in the nucleoli, seen as the characteristic cluster of larger bright areas, and the cytosol, seen as the characteristic diffuse overall fluorescence of the cells (Fig. 5a). Drastically different pictures were obtained by detecting the TAMRA emission at 610 ± 40 nm upon excitation of CF (Fig. 5b). The observed highly localized punctate spots indicated that FRET active, intact CF–CPD–TAMRA transporters 6 reside mainly in endosomes and, perhaps, lysosomes. Rare occurrence of faint emission in nucleoli coincided with the emission in CF channel (Fig. 5a), and thus could be assigned to the fluorescence of CF itself, which emits weakly within this detection window (Fig. 3c). The image obtained by detecting TAMRA emission upon TAMRA excitation appeared similar to the sum of the two earlier pictures (Fig. 5c and d). This was as expected because all TAMRA fluorophores can be visualized here.
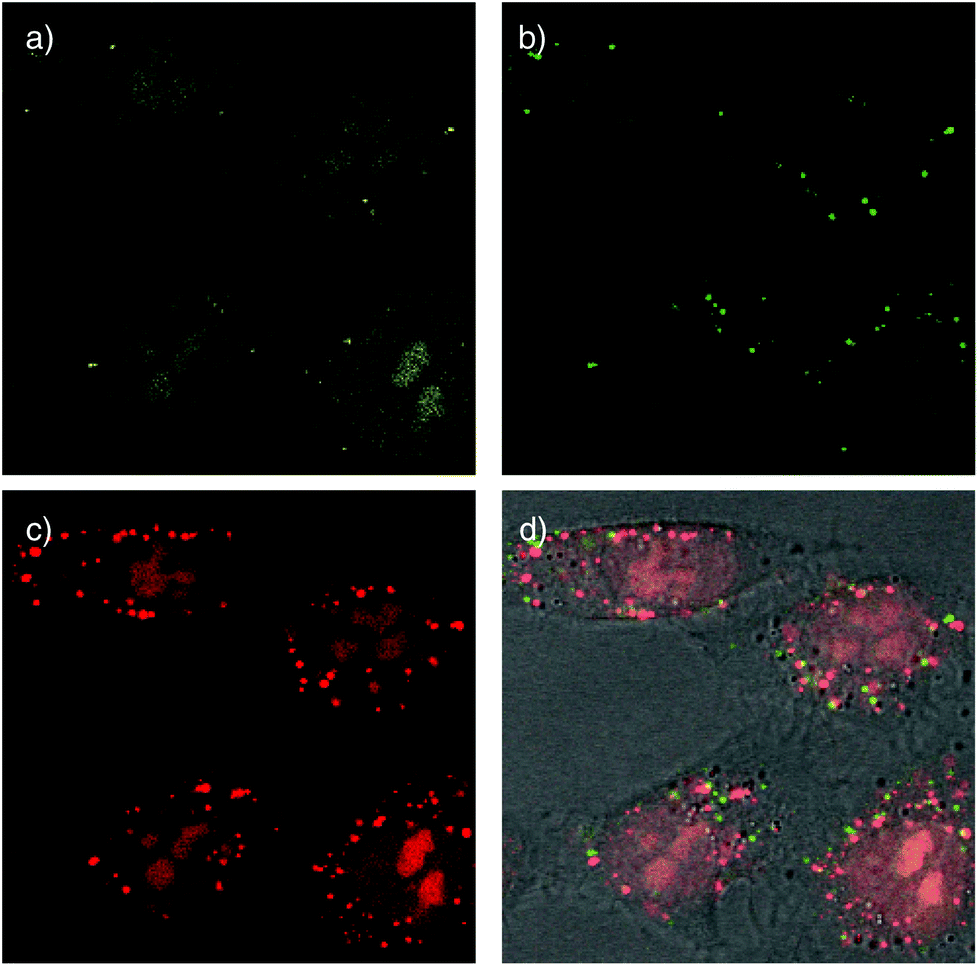 | ||
| Fig. 5 CLSM images of HeLa Kyoto cells after 15 min incubation with 500 nM of polymer 6 at 37 °C in Leibovitz's medium. The emission of CF was detected at 517 ± 18 nm (a) and TAMRA at 610 ± 40 nm (b, c) upon excitation of CF at 488 nm (a, b) and TAMRA at 561 nm (c). The images (b) and (c) are merged with the bright-field image in (d). | ||
Compared to TAMRA emission from FRET (green, Fig. 5b), TAMRA emission from TAMRA excitation was overall much stronger, also in the endosomes (red, Fig. 5c). This difference supported that most CPDs are cleaved at least once already in the endosomes. This observation was consistent with the thiol-mediated uptake mechanism (Fig. 1), in which disulfide exchange between cell-surface thiolates and disulfides in the backbone of the CPDs cleaves them in two. Evidence for thiol-mediated uptake of CPDs from inhibition experiments has been reported previously.1 The rare occurrence of intact CF–CPD–TAMRA conjugates in endosomes (Fig. 5b) should then originate from a different mechanism. The most likely mechanism is the classical ion-pairing based one, as similar punctate spots are often found with conventional CPPs.7c
Alternatively, the overall dominance of emission from acceptor excitation (TAMRA, red, Fig. 5c) over emission from FRET (green, Fig. 5b) in endosomes/lysosomes could also originate from decreasing FRET from the pH-sensitive CF with decreasing pH from early endosomes to late endosomes and lysosomes. The occurrence of CF emission from endosomes has been supported previously with co-localization experiments.2 Processed at different intensities, the weak red emission from TAMRA excitation that necessarily corresponds to the weak green (FRET) emission in Fig. 5b was below threshold in the more intense image in Fig. 5c and thus mostly absent in the green emission from endosomes in the merged images in Fig. 5d.
Taken together, these preliminary results demonstrate that cargos attached on both ends of CPDs can reach the cytosol and nucleoli, but only after at least partial cleavage of the CPD (Fig. 5a and c). Intact full length CPDs occur rarely and only in endosomes (Fig. 5b). These findings were consistent with the mechanism of thiol-mediated uptake (Fig. 1). Moreover, they provided experimental evidence as direct as possible in support of the occurrence of intracellular reductive depolymerisation of CPDs.
Conclusion
Cell-penetrating poly(disulfide)s (CPDs) offer a conceptually innovative way to enter into cells, driving the idea of covalent delivery to completion. Strategies have been developed previously to introduce substrates of free choice (probes, drugs, proteins, etc.) on the side of the initiator, either by substrate-initiated ring-opening disulfide-exchange polymerization, or by biotin/streptavidin biotechnology. So far, substrates could not be effectively introduced at the side of the terminators because iodoacetamides did not react well. In this report, hypervalent ethynyl benziodoxolones are introduced as terminators of disulfide-exchange polymerization that operate efficiently. Covalent attachment of functional terminators is shown to occur with 46% yield even under stoichiometric conditions at relatively high dilution. More favourable conditions will easily afford higher yields, if desired. The functional terminators are then compatible with diverse conjugation methods for quantitative substrate loading, including the already confirmed streptavidin/biotin technology or the here introduced CuAAC. Iodoacetamide controls give only 11% terminator incorporation under identical conditions.Synthetic access to doubly-labeled CPDs is demonstrated with the attachment of a FRET pair. In preliminary results, both ends of doubly-labeled CPDs are shown to enter into cells although they are cut in two at the outer surface during thiol-mediated uptake. Synthetic access to doubly-labeled CPDs further demonstrates that CuAAC is compatible with poly(disulfide)s. This finding is not trivial considering possible copper binding to disulfides and most attractive considering possible applications to side-chain modifications or substrate conjugation. Studies along these lines are ongoing and will be reported in due course.
Acknowledgements
We thank A. Roux, G. Gasparini and N. Chuard for access to and assistance with cell culture, the NMR and the Sciences Mass Spectrometry (SMS) platforms for services, and the University of Geneva, the EPFL, the European Research Council (ERC Advanced Investigator), the Swiss National Centre of Competence in Research (NCCR) Chemical Biology, the NCCR Molecular Systems Engineering and the Swiss NSF for financial support.Notes and references
- G. Gasparini, E.-K. Bang, G. Molinard, D. V. Tulumello, S. Ward, S. O. Kelley, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 6069–6074 CrossRef CAS PubMed.
- G. Gasparini, G. Sargsyan, E.-K. Bang, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2015, 54, 7328–7331 CrossRef CAS PubMed.
- G. Gasparini and S. Matile, Chem. Commun., 2015, 51, 17160–17162 RSC.
- N. Chuard, G. Gasparini, A. Roux, N. Sakai and S. Matile, Org. Biomol. Chem., 2015, 13, 64–67 CAS.
- (a) J. Fu, C. Yu, L. Lu and S. Q. Yao, J. Am. Chem. Soc., 2015, 137, 12153–12160 CrossRef CAS PubMed; (b) C. J. McKinlay, R. M. Waymouth and P. A. Wender, J. Am. Chem. Soc., 2016, 138, 3510–3517 CrossRef CAS PubMed.
- (a) E. Bartolami, C. Bouillon, P. Dumy and S. Ulrich, Chem. Commun., 2016, 52, 4257–4273 RSC; (b) M. Li, S. Schlesiger, S. K. Knauer and C. Schmuck, Angew. Chem., Int. Ed., 2015, 54, 2941–2944 CrossRef CAS PubMed; (c) B. M. deRonde, A. Birke and G. N. Tew, Chem. – Eur. J., 2015, 21, 3013–3019 CrossRef CAS PubMed; (d) R. Brock, Bioconjugate Chem., 2014, 25, 863–868 CrossRef CAS PubMed; (e) G. A. Eggimann, E. Blattes, S. Buschor, R. Biswas, S. M. Kammer, T. Darbre and J. L. Reymond, Chem. Commun., 2014, 50, 7254–7257 RSC; (f) C. Bechara and S. Sagan, FEBS Lett., 2013, 587, 1693–1702 CrossRef CAS PubMed; (g) S. Futaki, H. Hirose and I. Nakase, Curr. Pharm. Des., 2013, 19, 2863–2868 CrossRef CAS PubMed; (h) E. Lei, M. P. Pereira and S. O. Kelley, Angew. Chem., Int. Ed., 2013, 52, 9660–9663 CrossRef CAS PubMed; (i) H. Tang, L. Yin, K. H. Kim and J. Cheng, Chem. Sci., 2013, 4, 3839–3844 RSC; (j) M. Lnoue, W. Tong, J. D. Esko and Y. Tor, ACS Chem. Biol., 2013, 8, 1383–1388 CrossRef PubMed; (k) F. Sgolastra, B. M. Deronde, J. M. Sarapas, A. Som and G. N. Tew, Acc. Chem. Res., 2013, 46, 2977–2987 CrossRef CAS PubMed; (l) C. Ornelas-Megiatto, P. R. Wich and J. M. Fréchet, J. Am. Chem. Soc., 2012, 134, 1902–1905 CrossRef CAS PubMed; (m) I. Nakase, H. Akita, K. Kogure, A. Gräslund, Ü. Langel, H. Harashima and S. Futaki, Acc. Chem. Res., 2012, 45, 1132–1139 CrossRef CAS PubMed; (n) E. Koren and V. P. Torchilin, Trends Mol. Med., 2012, 18, 385–393 CrossRef CAS PubMed; (o) J. S. Appelbaum, J. R. LaRochelle, B. A. Smith, D. M. Balkin, J. M. Holub and A. Schepartz, Chem. Biol., 2012, 19, 819–830 CrossRef CAS PubMed; (p) B. R. McNaughton, J. J. Cronican, D. B. Thompson and D. R. Liu, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6111–6116 CrossRef CAS PubMed; (q) E. M. Kolonko, J. K. Pontrello, S. L. Mangold and L. L. Kiessling, J. Am. Chem. Soc., 2009, 131, 7327–7333 CrossRef CAS PubMed; (r) A. Hennig, G. J. Gabriel, G. N. Tew and S. Matile, J. Am. Chem. Soc., 2008, 130, 10338–10344 CrossRef CAS PubMed; (s) J. M. Gump and S. F. Dowdy, Trends Mol. Med., 2007, 13, 443–448 CrossRef CAS PubMed; (t) J. Fernandez-Carneado, M. Van Gool, V. Martos, S. Castel, P. Prados, J. De Mendoza and E. Giralt, J. Am. Chem. Soc., 2005, 127, 869–874 CrossRef CAS PubMed; (u) Y. Fillon, J. Anderson and J. Chmielewski, J. Am. Chem. Soc., 2005, 127, 11798–11803 CrossRef CAS PubMed; (v) P. Zhou, M. Wang, L. Du, G. W. Fisher, A. Waggoner and D. H. Ly, J. Am. Chem. Soc., 2003, 125, 6878–6879 CrossRef CAS PubMed; (w) A. O. Frankel and C. O. Pabo, Cell, 1988, 55, 1189–1193 CrossRef CAS PubMed; (x) M. Green and P. M. Lowenstein, Cell, 1988, 55, 1179–1188 CrossRef CAS PubMed.
- (a) N. Sakai and S. Matile, J. Am. Chem. Soc., 2003, 125, 14348–14356 CrossRef CAS PubMed; (b) J. B. Rothbard, T. C. Jessop, R. S. Lewis, B. A. Murray and P. A. Wender, J. Am. Chem. Soc., 2004, 126, 9506–9507 CrossRef CAS PubMed; (c) T. Takeuchi, M. Kosuge, A. Tadokoro, Y. Sugiura, M. Nishi, M. Kawata, N. Sakai, S. Matile and S. Futaki, ACS Chem. Biol., 2006, 1, 299–303 CrossRef CAS PubMed; (d) D. Schmidt, Q. X. Jiang and R. MacKinnon, Nature, 2006, 444, 775–779 CrossRef CAS PubMed; (e) M. Whitney, E. N. Savariar, B. Friedman, R. A. Levin, J. L. Crisp, H. L. Glasgow, R. Lefkowitz, S. R. Adams, P. Steinbach, N. Nashi, Q. T. Nguyen and R. Y. Tsien, Angew. Chem., Int. Ed., 2013, 52, 325–330 CrossRef CAS PubMed; (f) H. D. Herce, A. E. Garcia and M. C. Cardoso, J. Am. Chem. Soc., 2014, 136, 17459–17467 CrossRef CAS PubMed; (g) K. Fujisawa, M. Humbert-Droz, R. Letrun, E. Vauthey, T. A. Wesolowski, N. Sakai and S. Matile, J. Am. Chem. Soc., 2015, 137, 11047–11056 CrossRef CAS PubMed; (h) G. Gasparini, E.-K. Bang, J. Montenegro and S. Matile, Chem. Commun., 2015, 51, 10389–10402 RSC.
- E.-K. Bang, M. Lista, G. Sforazzini, N. Sakai and S. Matile, Chem. Sci., 2012, 3, 1752–1763 RSC.
- (a) T.-I. Kim and S. W. Kim, React. Funct. Polym., 2011, 71, 344–349 CrossRef CAS PubMed; (b) M. Piest and J. F. J. Engbersen, J. Controlled Release, 2011, 155, 331–340 CrossRef CAS PubMed; (c) C. R. Drake, A. Aissaoui, O. Argyros, M. Thanou, J. H. G. Steinke and A. D. Miller, J. Controlled Release, 2013, 171, 81–90 CrossRef CAS PubMed; (d) H. Zeng, H. C. Little, T. N. Tiambeng, G. A. Williams and Z. Guan, J. Am. Chem. Soc., 2013, 135, 4962–4965 CrossRef CAS PubMed; (e) S. Son, R. Namgung, J. Kim, K. Singha and J. W. Kim, Acc. Chem. Res., 2012, 45, 1100–1112 CrossRef CAS PubMed; (f) M. Balakirev, G. Schoehn and J. Chroboczek, Chem. Biol., 2000, 7, 813–819 CrossRef CAS PubMed; (g) P. K. Hashim, K. Okuro, S. Sasaki, Y. Hoashi and T. Aida, J. Am. Chem. Soc., 2015, 137, 15608–15611 CrossRef CAS PubMed.
- (a) N. K. P. Samuel, M. Singh, K. Yamaguchi and S. L. Regen, J. Am. Chem. Soc., 1985, 107, 42–47 CrossRef CAS; (b) D. Basak, R. Kumar and S. Ghosh, Macromol. Rapid Commun., 2014, 35, 1340–1344 CrossRef CAS PubMed; (c) L. Monnereau, M. Nieger, T. Muller and S. Bräse, Adv. Funct. Mater., 2014, 24, 1054–1058 CrossRef CAS; (d) G. A. Barcan, X. Zhang and R. M. Waymouth, J. Am. Chem. Soc., 2015, 137, 5650–5653 CrossRef CAS PubMed; (e) D. J. Hansen, I. Manuguerra, M. B. Kjelstrup and K. V. Gothelf, Angew. Chem., Int. Ed., 2014, 53, 14415–14418 CrossRef CAS PubMed.
- (a) A. G. Torres and M. J. Gait, Trends Biotechnol., 2012, 30, 185–190 CrossRef CAS PubMed; (b) D. Oupický and J. Li, Macromol. Biosci., 2014, 14, 908–922 CrossRef PubMed; (c) T. Li and S. Takeoka, Int. J. Nanomed., 2014, 9, 2849–2861 Search PubMed; (d) I. D. Alves, A. Walran, C. Bechara and S. Sagan, Curr. Protein Pept. Sci., 2012, 13, 658–671 CrossRef CAS PubMed; (e) M. M. Fretz, N. A. Penning, S. Al-Taei, S. Futaki, T. Takeuchi, I. Nakase, G. Storm and A. T. Jones, Biochem. J., 2007, 403, 335–342 CrossRef CAS PubMed; (f) E. P. Feener, W. C. Shen and H. J. P. Ryser, J. Biol. Chem., 1990, 265, 18780–18785 CAS; (g) L. Brulisauer, M. A. Gauthier and J.-C. Leroux, J. Controlled Release, 2014, 195, 147–154 CrossRef CAS PubMed; (h) C. Wu, S. Wang, L. Brülisauer, J.-C. Leroux and M. A. Gauthier, Biomacromolecules, 2013, 14, 2383–2388 CrossRef CAS PubMed.
- (a) E.-K. Bang, G. Gasparini, G. Molinard, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2013, 135, 2088–2091 CrossRef CAS PubMed; (b) E.-K. Bang, S. Ward, G. Gasparini, N. Sakai and S. Matile, Polym. Chem., 2014, 5, 2433–2441 RSC.
- (a) M. Lista, J. Areephong, N. Sakai and S. Matile, J. Am. Chem. Soc., 2011, 133, 15228–15231 CrossRef CAS PubMed; (b) N. Sakai and S. Matile, J. Am. Chem. Soc., 2011, 133, 18542–18455 CrossRef CAS PubMed; (c) H. Hayashi, A. Sobczuk, A. Bolag, N. Sakai and S. Matile, Chem. Sci., 2014, 5, 4610–4614 RSC; (d) K.-D. Zhang and S. Matile, Angew. Chem., Int. Ed., 2015, 54, 8980–8983 CrossRef CAS PubMed.
- (a) J. Li, P. Nowak and S. Otto, J. Am. Chem. Soc., 2013, 135, 9222–9239 CrossRef CAS PubMed; (b) J.-M. Lehn, Top. Curr. Chem., 2011, 322, 1–32 CrossRef; (c) S. Zarra, D. M. Wood, D. A. Roberts and J. R. Nitschke, Chem. Soc. Rev., 2015, 44, 419–432 RSC; (d) A. Herrmann, Chem. Soc. Rev., 2014, 43, 1899–1933 RSC; (e) Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC; (f) A. Wilson, G. Gasparini and S. Matile, Chem. Soc. Rev., 2014, 43, 1948–1962 RSC; (g) X. Wu, Z. Li, X.-X. Chen, J. S. Fossey, T. D. James and Y.-B. Jiang, Chem. Soc. Rev., 2013, 42, 8032–8048 RSC; (h) S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC; (i) H. H. Jo, R. Edupuganti, L. You, K. N. Dalby and E. V. Anslyn, Chem. Sci., 2015, 6, 158–164 RSC; (j) A. G. Campaña, D. A. Leigh and U. Lewandowska, J. Am. Chem. Soc., 2013, 135, 8639–8645 CrossRef PubMed; (k) J. F. Teichert, D. Mazunin and J. W. Bode, J. Am. Chem. Soc., 2013, 135, 11314–11321 CrossRef CAS PubMed; (l) K. D. Okochi, Y. Jin and W. Zhang, Chem. Commun., 2013, 49, 4418–4420 RSC; (m) B. Icli, E. Solari, B. Kilbas, R. Scopelliti and K. Severin, Chem. – Eur. J., 2012, 18, 14867–14874 CrossRef CAS PubMed; (n) L. Rocard, A. Berezin, F. De Leo and D. Bonifazi, Angew. Chem., Int. Ed., 2015, 54, 15739–15743 CrossRef CAS PubMed; (o) S. Hagihara, H. Tanaka and S. Matile, J. Am. Chem. Soc., 2008, 130, 5656–5657 CrossRef CAS PubMed; (p) C. Wang, M. R. Krause and S. L. Regen, J. Am. Chem. Soc., 2015, 137, 664–666 CrossRef CAS PubMed.
- R. Frei, M. D. Wodrich, D. P. Hari, P. A. Borin, C. Chauvier and J. Waser, J. Am. Chem. Soc., 2014, 136, 16563–16573 CrossRef CAS PubMed.
- D. Abegg, R. Frei, L. Cerato, D. P. Hari, C. Wang, J. Waser and A. Adibekian, Angew. Chem., Int. Ed., 2015, 54, 10852–10857 CrossRef CAS PubMed.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS; (b) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064 CrossRef; (c) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC; (d) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed; (e) L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS; (f) J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Kluwer Academic/Plenum Publishers, New York, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed procedures and results for all reported experiments. See DOI: 10.1039/c6py00562d |
| This journal is © The Royal Society of Chemistry 2016 |