
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultifunctional cellulose esters by olefin cross-metathesis and thiol-Michael addition†
Xiangtao
Meng
ab,
Shreya
Roy Choudhury
c and
Kevin J.
Edgar
*ab
aMacromolecules Innovation Institute, Virginia Tech, Blacksburg, VA 24061, USA. E-mail: kjedgar@vt.edu
bDepartment of Sustainable Biomaterials, Virginia Tech, Blacksburg, VA 24061, USA
cDepartment of Chemistry, Virginia Tech, Blacksburg, VA 24061, USA
First published on 25th April 2016
Abstract
Olefin cross-metathesis (CM) has been shown to be a versatile, mild, modular, and efficient approach to polysaccharide modification. One issue with regard to this approach is the susceptibility of the initial α,β-unsaturated CM derivatives to H-atom abstraction in the γ-position, followed by radical recombination that leads to insoluble, crosslinked products. In our original approach, we resolved this problem through removing the offending unsaturation by hydrogenation. In the current study, we describe a method to exploit these reactive conjugated olefins, by post-CM thiol-Michael addition, thereby appending additional functionality. CM substrates and thiols bearing various functional groups were combined and reacted, employing amine catalysis. Up to 100% conversion was achieved under proper conditions (e.g. catalyst and reaction time), with minimal side reactions observed. The combination of the two modular reactions creates versatile access to cellulose derivatives equipped with a wide diversity of functional groups.
Introduction
Thiol-Michael addition is the 1,4-nucleophilic conjugate addition of a thiol anion to an α,β-unsaturated carbonyl (hydrothiolation) usually in the presence of a base or nucleophile catalyst. In thiol-Michael reactions, the addition of thiols to electron-deficient carbon–carbon double bonds can be achieved rapidly, quantitatively, and orthogonally under mild conditions. Furthermore, unlike thiol–ene reactions in which radical–radical termination occurs from time to time due to the free radical nature of the chemistry, thiol-Michael addition reactions typically are not accompanied by significant side reactions.1 Because of these merits, the thiol-Michael addition is now thought of as a “click” reaction, and has been broadly implemented in polymer and polysaccharide synthesis and modification.2–5Typically, in order to introduce thiol-Michael reactions to polysaccharide modification, two major strategies have been used; (a) thiolation of the polysaccharide backbone to afford a polymeric thiol reagent, or (b) introduction of electron-deficient acceptor olefins to polysaccharides.6 Although both approaches have been extensively explored, polythiols are capable of forming intra- and intermolecular crosslinks via disulfide bond formation.7,8 Unless the networks are the desired products, this may become a problem in polymer preparation and storage. Attachment of an acceptor activated olefin to the polysaccharide, on the other hand, may avoid such problems, and such olefin functionality can be a handle either for crosslinking or “clicking” small molecules by thiol–ene/thiol-Michael reactions.9
Direct modification of cellulose derivatives bearing pendent alkenes has been reported by several groups. Schumann et al.10 and Meng et al.11 employed hydroboration–oxidation sequences to convert terminal olefins on cellulose ethers and esters, respectively, to ω-hydroxyl groups. Thiol–ene reactions have also received tremendous interest in cellulose functionalization.6 Researchers have taken advantage of its facility and simplicity to apply thiol–ene reactions to cellulose surface modification,12,13 homogeneous functionalization,14 and synthesis of crosslinked composites.15 Recently, the Edgar group successfully applied olefin CM to polysaccharide modification, in which cellulose esters and ethers with ω-unsaturated side chains were reacted with different CM partners such as acrylic acid,16 acrylates17,18 and acrylamides19 employing the Hoveyda–Grubbs 2nd generation Ru catalyst to afford a variety of side-chain functionalized cellulose derivatives under mild conditions. This is a mild, modular, versatile, and rapid method for polysaccharide post-modification. Several recent reviews6,20,21 on olefin metathesis and olefin cross-metathesis have described the utility of these methods for polymerization and other synthetic applications.
CM between ω-unsaturated polysaccharide side-chains and different CM partners affords α,β-unsaturated carboxylic acids, esters and amides. Given that α,β-unsaturated carbonyls are potentially good substrates for thiol-Michael addition, and since it is essential in any case to eliminate the α,β-unsaturation in order to stabilize the derivatives against radical-initiated dimerization, it is tempting to combine the CM reaction with the thiol-Michael addition. We hypothesize that, under mild conditions, a variety of activated olefin derivatives that result from modular CM reaction can react with a series of thiols via the thiol-Michael click reaction to provide polysaccharides bearing functional side-chains of even more diversity. Recently, Winkler and Meier22 reported the reaction of an α,β-unsaturated ester (from CM) with 2-mercaptoethanol or methyl thioglycolate via thiol-Michael addition to obtain AB- or AA-type of monomers for polyesterification. However, we are unaware of any such strategy being reported for either polymer post-modification or polysaccharide modification. We describe herein, our efforts to demonstrate proof of concept for such post-modification of CM products from cellulose esters.
Experimental section
Materials
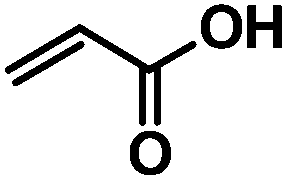
Cellulose acetate (CA-320S, Mn ∼ 38 kDa, DP ∼ 151 (data reported by supplier), DSAc ∼ 1.82 (measured by 1H-NMR spectroscopy)) was from Eastman Chemical Company. Triethylamine (TEA), 1,3-dimethyl-2-imidazolidinone (DMI), and dimethylsulfoxide (DMSO) were purchased from Acros Organics. Anhydrous tetrahydrofuran, acrylic acid, [2-(acryloyloxy)ethyl]trimethylammonium chloride (80 wt% in H2O), benzyl acrylate, 2,6-di-tert-butyl-4-methylphenol (butylated hydroxytoluene, or BHT), 2-hydroxyethyl acrylate, Hoveyda–Grubbs II generation catalyst, n-hexylamine (HA), 2-mercaptoethanol (2-ME), 3-mercaptopropionic acid (3-MPA), cysteamine (Cys), pent-4-enoyl chloride, and undec-10-enoyl chloride were purchased from Sigma Aldrich. Diethylene glycol monovinyl ether was purchased from TCI. DMI was dried over 4 Å molecular sieves. All other purchased reagents were used as received.Measurements
1H NMR spectra were acquired on Bruker Avance 500 spectrometers operating at 500 MHz. Samples were analyzed as solutions in DMSO-d6 (ca. 10 mg mL−1) at 25 °C in standard 5 mm o.d. tubes. Three drops of trifluoroacetic acid were added to shift the water peak in DMSO-d6 downfield from the spectral region of interest. 13C NMR and 1H–13C HSQC spectra were obtained on a Bruker Avance 500 MHz spectrometer with a minimum of 5000 scans in DMSO-d6 (ca. 50 mg mL−1) at 80 °C. To obtain the Tg values of the cellulosic polymers, DSC was performed on a TA Instruments Q100 apparatus or TA Discovery DSC using heat/cool/heat mode. Dry powders (ca. 5 mg) were loaded in Tzero™ aluminum pans. The scanning conditions were set as follows: each sample was equilibrated at 35 °C, and then heated to 150° at 20 °C min−1. The sample was then cooled at 100 °C min−1 to −50 °C. During the second heating cycle the sample was heated to 200 °C at 20 °C min−1. If the heat/cool/heat mode failed to give a clear transition, modulated DSC was performed as follows: each sample was equilibrated at −50 °C, the underlying ramp heating rate was 7 °C, the oscillation amplitude was ±1 °C, and oscillation period was 40 s. FTIR spectra were obtained on a Nicolet 8700 instrument. Size exclusion chromatography (SEC), if not otherwise specified, was performed on Agilent 1260 Infinity Multi-Detector SEC using DMAc with 0.05 M LiCl as the mobile phase (50 °C) with 3 PLgel 10 μm mixed-B 300 × 7.5 mm columns in series. A system of multiple detectors connected in series was used for the analysis. A multi-angle laser light scattering (MALS) detector (DAWN-HELEOS II, Wyatt Technology Corporation, Goleta, CA), operating at a wavelength of 658 nm and a refractive index detector operating at a wavelength of 658 nm (Optilab T-rEX, Wyatt Technology Corporation, Goleta, CA) provided online results. Data acquisition and analysis was conducted using Astra 6 software (Wyatt Technology Corporation, Goleta, CA). Monodisperse polystyrene standard (Mw ∼ 21k, Đ ∼ 1.02) was run first in every sample series for the purpose of calibration and confirmation.Preparation of cellulose acetate undec-10-enoate (CA-Un067)
Synthesis of CA-Un067 was performed following a procedure16,17 previously reported by our group. In detail, CA-320S (1.00 g, 4.19 mmol per AGU) was dissolved in DMI (30 mL), and the solution was heated to 90 °C in a three-neck round bottom flask equipped with condenser, with mechanical stirring under N2. Triethylamine (1.29 mL, 9.22 mmol, 2.2 equiv.) was added, and then 10-undecenoyl chloride (1.70 g, 8.36 mmol, 2.0 equiv.) was added dropwise. The resulting solution was stirred at 90 °C for 20 h. The reaction mixture was then filtered, and the filtrate was added to 300 mL 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 water/ethyl alcohol. The resulting precipitate was recovered by filtration, then redissolved in a minimal amount of CH2Cl2 and reprecipitated in hexane. The product was washed with hexane and dried under vacuum at 40 °C.
:50 water/ethyl alcohol. The resulting precipitate was recovered by filtration, then redissolved in a minimal amount of CH2Cl2 and reprecipitated in hexane. The product was washed with hexane and dried under vacuum at 40 °C.
A similar procedure was employed in the synthesis of cellulose acetate pent-4-enoate (CA-Pen079).
General procedure for olefin cross-metathesis reactions
The cross-metathesis reactions were performed following a procedure16,17 previously reported by our group. In detail, to a flask charged with cellulose derivative CA-Un067 or CA-Pen079 (100 mg, 1.0 equiv. olefin), 5 mg BHT and 5 mL anhydrous THF were added. After the reagents were completely dissolved, cross-metathesis partner (acrylic acid, benzyl acrylate, 2-hydroxylethyl acrylate; 20 equiv.) was added followed by the addition of Hoveyda–Grubbs Catalyst 2nd Generation (0.05 equiv. in 2 mL THF) via syringe. After stirring for 1 h under N2 at 40 °C, the reaction was stopped by adding 1–2 drops of diethylene glycol monovinyl ether or ethyl vinyl ether. The product was collected either by dialysis and freeze-drying, or by precipitating in H2O/ethanol followed by sufficient washing by H2O, then drying under vacuum at 40 °C.For the CM reaction with [2-(acryloyloxy)ethyl]trimethylammonium chloride), acetic acid was used as a solvent but otherwise the general procedure was used.
General procedure for thiol-Michael addition
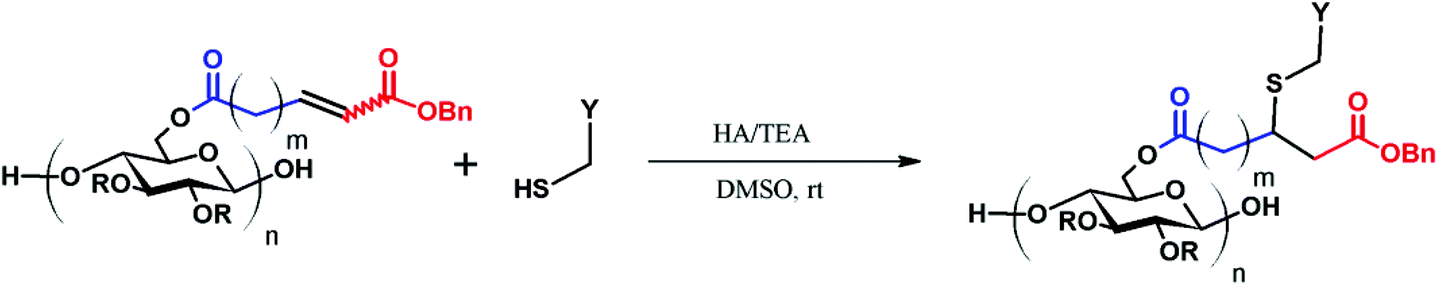
CM product (50 mg) was dissolved in 2 mL of DMSO in a 10 mL three-neck flask. After stirring under N2 for 30 min, the designated amount of a thiol species and a catalyst (either triethylamine or HA) was added and the solution was stirred for 5 h at room temperature. The mixture was then dialyzed against H2O for 3 d, and then the solid product was isolated by freeze-drying.Results and discussion
CM between an isolated, terminal olefin (Type 1 by the Grubbs classification23), and an electron deficient olefin (e.g. an acrylate, Type 2 by the Grubbs classification) leads to an α,β-unsaturated carboxylic acid derivative, which may serve as a substrate for thiol-Michael addition reaction. The polymer, already functionalized via CM, can thus be further modified with a secondary functional group, as shown in Fig. 1.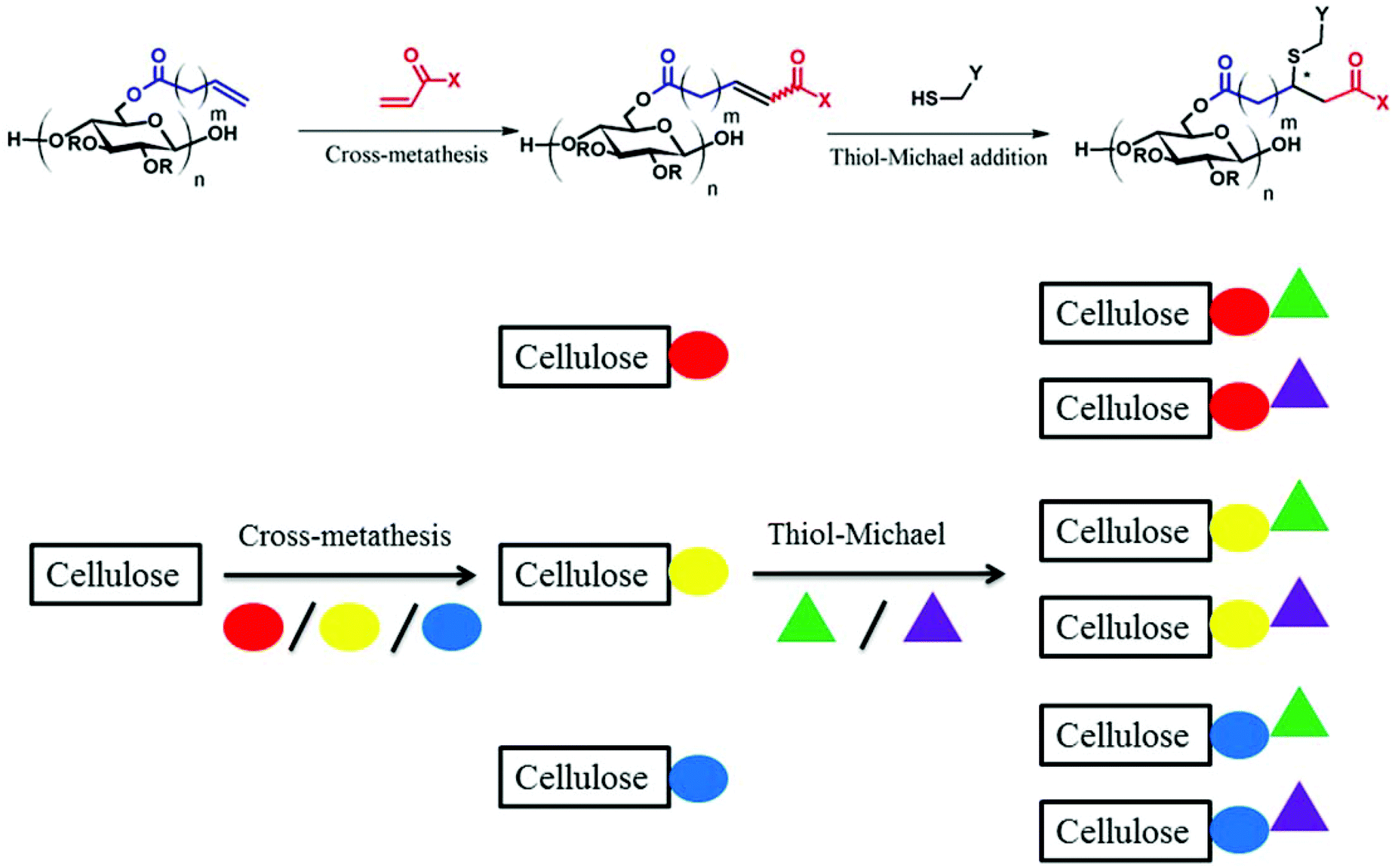 | ||
| Fig. 1 Schematic illustration of olefin cross-metathesis and thiol-Michael addition towards functionalized cellulose derivatives. The circles of different colors represent different functionalities introduced by CM, and the triangles of different colors represent different functionalities introduced by thiol-Michael addition. | ||
As revealed in our previous publications,16–19 CM in polysaccharide modification has a “click-like” profile and gives modular products. Using the procedure reported by Meng et al.17 cellulose esters with terminally olefinic side-chains of different chain lengths, i.e. cellulose acetate undec-10-enoate with DS of undecenoate 0.67 (CA-Un067) and cellulose acetate pent-4-enoate with DS of pentenoate 0.79 (CA-Pen079), were prepared and used as metathesis substrates. CM reaction of the starting cellulose esters with partners including acrylic acid (AA), benzyl acrylate (BA), and 2-hydroxyethyl acrylate (HEA) were performed to give CM products bearing a variety of functional groups, as substrates for subsequent thiol-Michael addition (Table 1, polymers 1, 2, 4, and 5). In order to ensure complete conversion of reactive Type I terminal olefins to CM products without competing self-metathesis, we used 20 eq. of small molecule acrylate per eq. terminal olefin (note however that the excess acrylate is unaltered by the CM conditions and could in principle be recycled).
|
|
||||
|---|---|---|---|---|
| Polymer # | Starting cellulose ester |
|
Product abbr.b | Conversionc |
| a 20 mol acrylate per mol CA-appended olefin. b Rules for polymer abbreviation are described in the reference section.24 c Conversions were calculated by 1H NMR. d As previously measured.17 | ||||
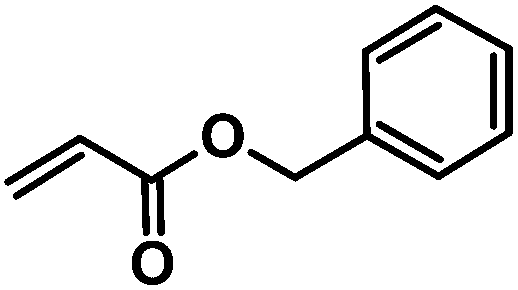
| 1 | CA-Un067 (m = 8) |
|
CA-Un067-BA | 100% |
| 2 | CA-Un067 |
|
CA-Un067-AA | 100%d |
| 3 | CA-Un067 |
|
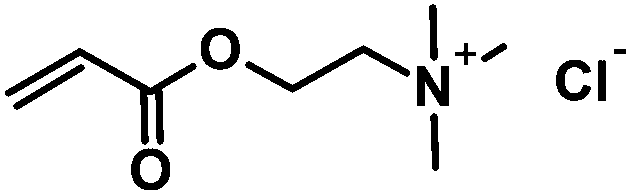
CA-Un067-TMA | 100% |
| 4 | CA-Un067 |
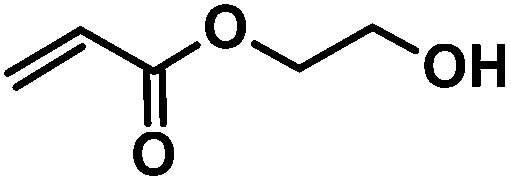
|
CA-Un067-HEA | 100%b |
| 5 | CA-Pen079 (m = 2) |
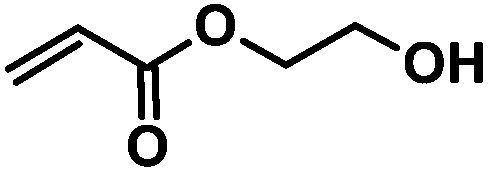
|
CA-Pen079-HEA | 100%b |
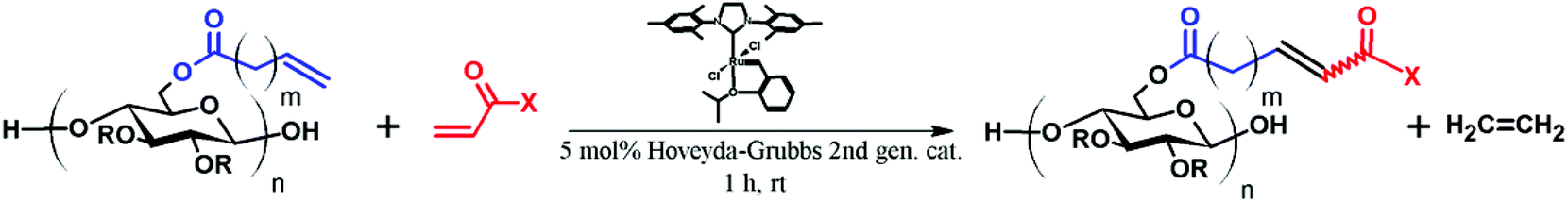
Positively charged cellulose ester (3) was also synthesized by CM reaction between CA-Un067 and CM partner [2-(acryloyloxy)ethyl]trimethylammonium chloride (TMACl, 80 wt% in H2O). As TMACl is not miscible with typical olefin metathesis solvents such as THF and dichloromethane, we employed acetic acid as the solvent in this reaction. Acetic acid was shown to be an effective cross-metathesis solvent in our previous study of CM reactions with acrylamides.19 As in the reactions with other CM partners, full conversion with TMACl was achieved after 1 h under the mild standard reaction conditions.
The variety of CM products synthesized was designed to explore the influences of functional group type and side-chain length upon the thiol-Michael addition. Functional groups like hydroxyl, carboxylic acid and trimethylammonium are of great interest especially in biomedical applications such as drug delivery,25 antimicrobial materials,26 and biomedical engineering,27 as they may enhance bioactivity and other performance attributes of polysaccharides. The benzyl group, on the other hand, is included since it may serve as a protecting group that enables introduction of additional functionality, so was grafted onto cellulose (CA-Un067-BA, 1) via CM in this study. Another impetus for including a benzyl acrylate substrate is that, unlike the 1H NMR spectra of other CM adducts, the benzyl ring peaks in the spectra of CA-Un067-BA and its Michael adducts are well separated from the cellulose backbone signals (e.g. see Fig. 3). As a result, the progress and conversion of the reactions can be easily monitored by comparing this peak integral with those of other corresponding peaks. For this reason, reactions with CA-Un067-BA were studied first.
Three thiol species: 2-mercaptoethanol (2-ME), 3-mercaptopropionic acid (3-MPA) and cysteamine (Cys), were selected as representative thiol-Michael partners, because they are uncharged, negatively charged, and positively charged at near-physiological pH values, respectively. Although a variety of types of thiol-Michael catalysts, and catalytic mechanisms including base- and nucleophile-catalyzed pathways have been proposed,1 in the current study we chose to focus only on TEA and HA, arguably representing primary catalysis by base and nucleophile, respectively (Fig. 2).
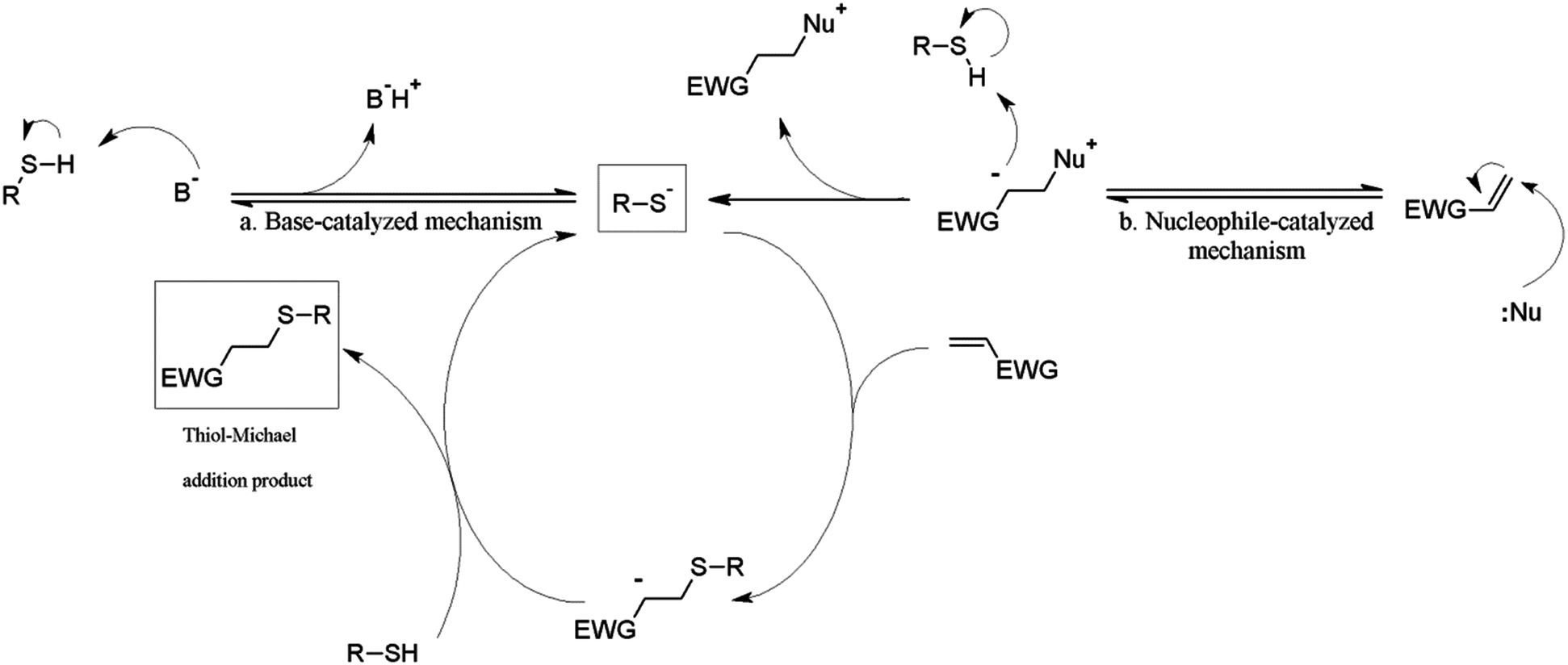 | ||
| Fig. 2 Base- and nucleophile-catalyzed mechanisms of the thiol-Michael addition reaction. | ||
HA proved to be more effective in catalyzing the thiol-Michael reaction of CA-Un067-BA with both 2-ME and 3-MPA (Table 2). With 2 eq. of 2-ME and 0.3 eq. of HA, complete conversion of olefin to thiol-Michael adduct (Table 2, polymer 7, CA-Un067-BA-2ME) was achieved, as indicated by the complete disappearance of olefin proton signals at 5.87 and 6.92 ppm in the 1H NMR spectrum in Fig. 3, while the other related proton signals designated H3, H4, H5, and H* appeared at 3.47, 2.61, 2.57 and 2.99 ppm. Carbon NMR spectra also showed complete conversion (ESI Fig. S1 and S2†). Reaction completion was also confirmed by FTIR spectroscopy, by disappearance of the alkene C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch at 1653 cm−1. It should be noted that the 1,4-conjugate addition is not expected to be chirally selective under the conditions described here, and thus in the afforded Michael adduct CA-Un067-BA-2ME, carbon * is expected to be racemic, assuming no net influence from the distant chiral centers of the cellulose backbone.28
C stretch at 1653 cm−1. It should be noted that the 1,4-conjugate addition is not expected to be chirally selective under the conditions described here, and thus in the afforded Michael adduct CA-Un067-BA-2ME, carbon * is expected to be racemic, assuming no net influence from the distant chiral centers of the cellulose backbone.28
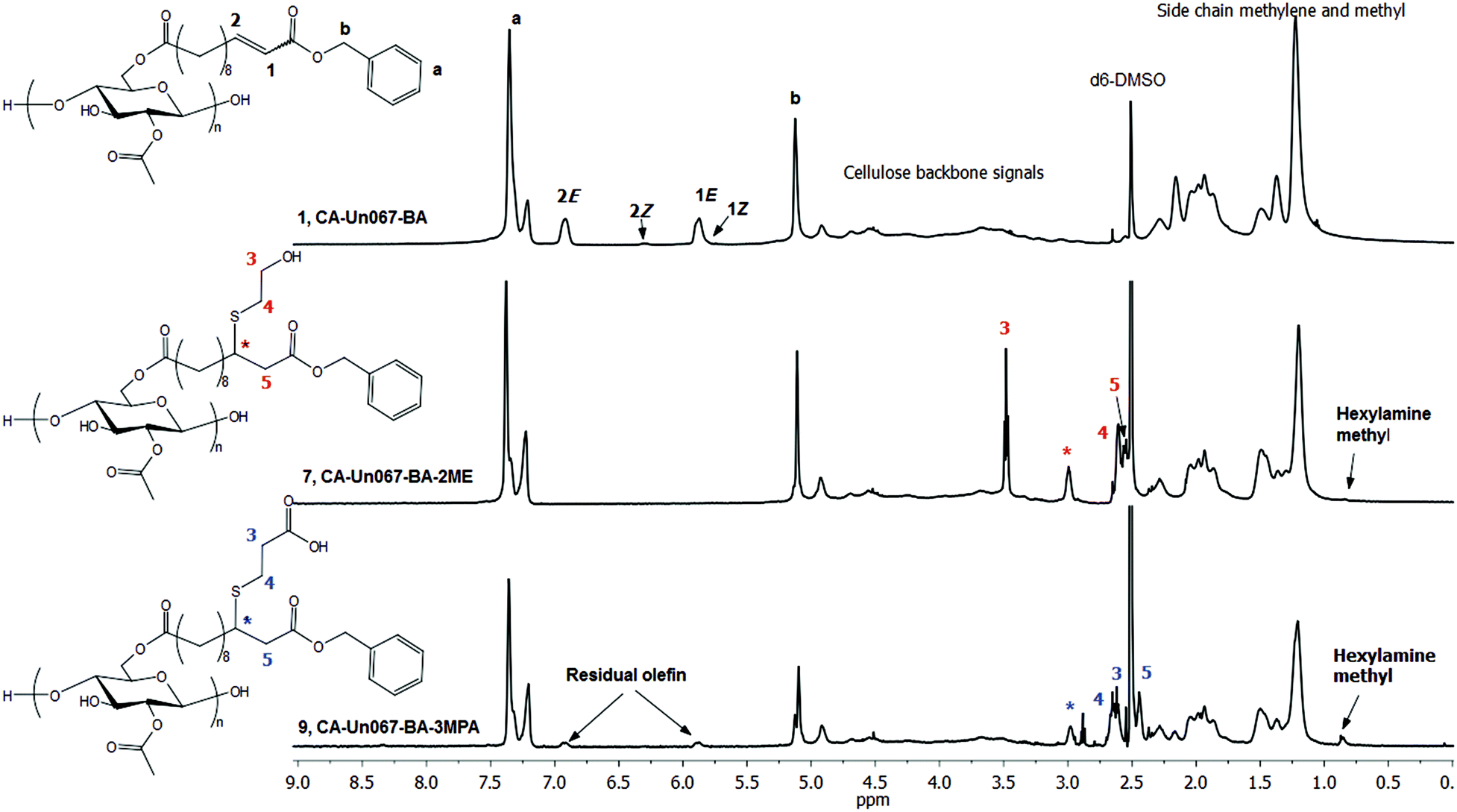 | ||
| Fig. 3 1H NMR spectra of CA-Un067-BA and its thiol-Michael products with 2-ME (CA-Un067-BA-2ME) and 3-MPA (CA-Un067-BA-3MPA). | ||
|
|
|||||
|---|---|---|---|---|---|
| Polymer # | Thiol | Catalyst | Time (h) | Conversiona (%) | Side reaction |
| a Conversions were calculated by 1H NMR. b Conversion not available due to crosslinking. | |||||
| 6 | 2-ME (3 eq.) | TEA (6 eq.) | 12 | 35 | No |
| 7 | 2-ME (2 eq.) | HA (0.3 eq.) | 5 | 100 | Minimal |
| 8 | 3-MPA (9 eq.) | TEA (18 eq.) | 12 | 50 | No |
| 9 | 3-MPA (2 eq.) | HA (3 eq.) | 5 | 87 | Yes |
| 10 | Cys (6 eq.) | TEA (3 eq.) | 12 | NAb | Cross-linked |
| 11 | Cys (2 eq.) | HA (0.3 eq.) | 5 | NAb | Cross-linked |
We noticed that the proton NMR spectrum of CA-Un067-BA-2ME contained a small peak at 0.86 ppm, in the area where we would expect the methyl group of HA to resonate. This was an indication of possible side reactions with HA. Li et al.29 have reported that primary amines may compete with thiols in the Michael addition reaction, leading to amine adducts, especially when the amine is in large excess. We conducted reactions with higher ratios of HA to activated olefin (olefin/2-ME/HA ratio equals to 1/6/6, Fig. S4†), and indeed observed a larger peak at 0.86 ppm. A primary amine acts as both base and nucleophile, and thus can catalyze both base and nucleophile hydrothiolation pathways. From this point of view, it is not unexpected to observe the 1,4-conjugate addition of HA to α,β-unsaturated carbonyls, both as a means to initiate thiol-Michael addition, and also as a side reaction in which amine competes with thiol. A preliminary kinetic study (Fig. 5) employing the same conditions as in polymer 7 (olefin/2-ME/HA ratio 1/2/0.3) showed that the double bonds were consumed within 5 h, while a small peak at 0.86 ppm appeared in each spectrum from the 45 min sample to the 300 min sample. However, it is interesting that after all of the double bonds had been consumed at 300 min, the resonance at 0.86 ppm continued to grow. Clearly, nucleophilic attack of HA at the β carbons of α,β-unsaturated carbonyls may not be the only side reaction. As the cellulose derivative has many ester linkages, amide-ester exchange is a likely source of side reactions.30 If the amine attacks the acrylate ester linkage to form the corresponding acrylamide, this new linkage would be retained in the cellulosic product and observed spectroscopically. Primary amine attack at the alkanoate ester linkages (e.g. acetate in CA-Un067) would result in loss of the alkanoate to form a small molecule amide (e.g. n-hexyl acetamide in the case of CA-Un067 with hexylamine catalyst), that might or might not be isolated along with the cellulosic product. Another possibility would be retro thiol-Michael addition catalyzed by the amine, followed by aza-Michael addition of HA to the regenerated α,β-unsaturation. However, we feel that this is at best a minor side reaction pathway, as no loss of the thio substituent was observed by proton NMR.
When TEA was used as the catalyst (Table 2, polymer 6, olefin/2-ME/TEA ratio equals to 1/6/6), only 35% olefin conversion was observed, while no by-products were observed (proton NMR, Fig. S5†). This observation is consistent with observations by previous investigators. Li et al.29 observed that TEA-catalyzed thiol-Michael reactions proceeded with no noticeable side reactions, while use of primary amine catalysts like HA led to amine addition side products. Chan et al.31 compared several thiol-Michael catalysts including HA and TEA, and found that TEA catalysis affords a significantly lower rate constant for thiol-Michael addition than does HA. The authors of both studies attributed these observations to the greater nucleophilicity of the primary HA vs. the tertiary TEA amine (while the pKa values of the two amines are similar (10.75 and 10.56 respectively)), and the resultant difference in catalytic mechanisms.
When 3-MPA was the thiol for 1,4-conjugate addition to CA-Un067-BA (Table 2, polymer 8 and 9), we observed results similar to those with 2-ME: HA is a more efficient catalyst than TEA. The carboxylic acid of 3-MPA can form salts with the amine catalyst, and thereby interfere with both base- and nucleophile-catalyzed additions. To overcome these issues, we explored use of a molar excess of amine vs. 3-MPA, intending to overwhelm the deleterious effects of the carboxylic acid functionality. Using TEA catalyst, only 50% of the available double bonds underwent hydrothiolation even when up to 18 eq. of TEA and 9 eq. of 3-MPA were employed, though no side reaction was observed (Fig. S6†). Employing HA as catalyst (olefin/3-MPA/HA mole ratio equals to 1/2/3) improved the conversion to a gratifying 87%, as indicated by the amount of residual olefin signal in the proton NMR spectrum (Fig. 3). However, the HA methyl peak at 0.86 ppm was also evident in the 1H NMR spectrum (Fig. 3), and an amide II band around 1566 cm−1 in the FTIR spectrum (Fig. 4) provided further evidence of side reactions involving incorporation of HA into the product.
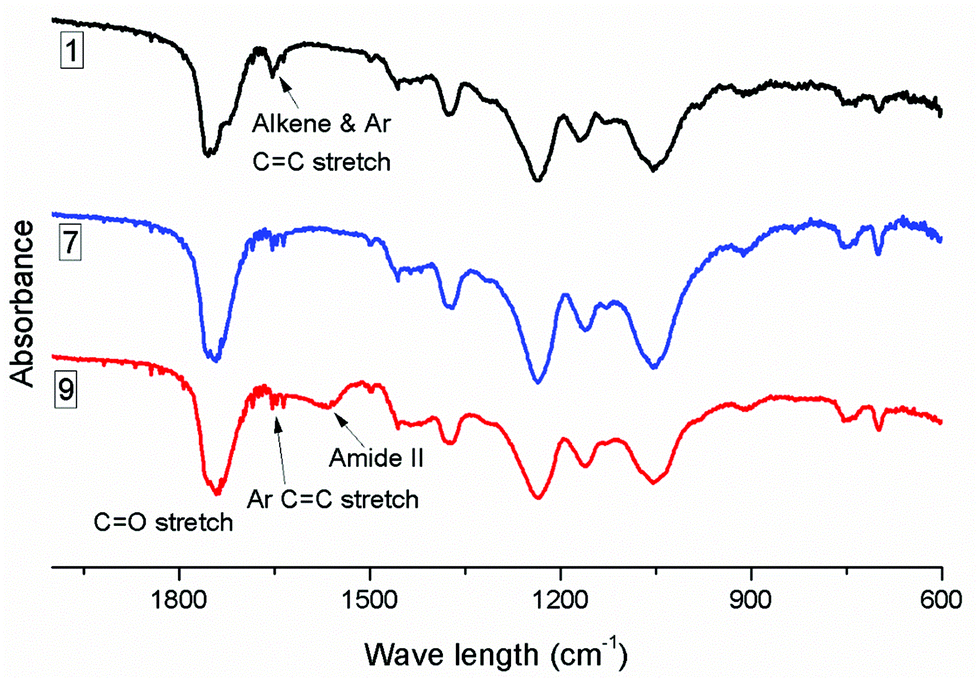 | ||
| Fig. 4 FTIR spectra of CA-Un067-BA (1) and its thiol-Michael products with 2-ME (CA-Un067-BA-2ME, 7) and with 3-MPA (CA-Un067-BA-3MPA, 9). | ||
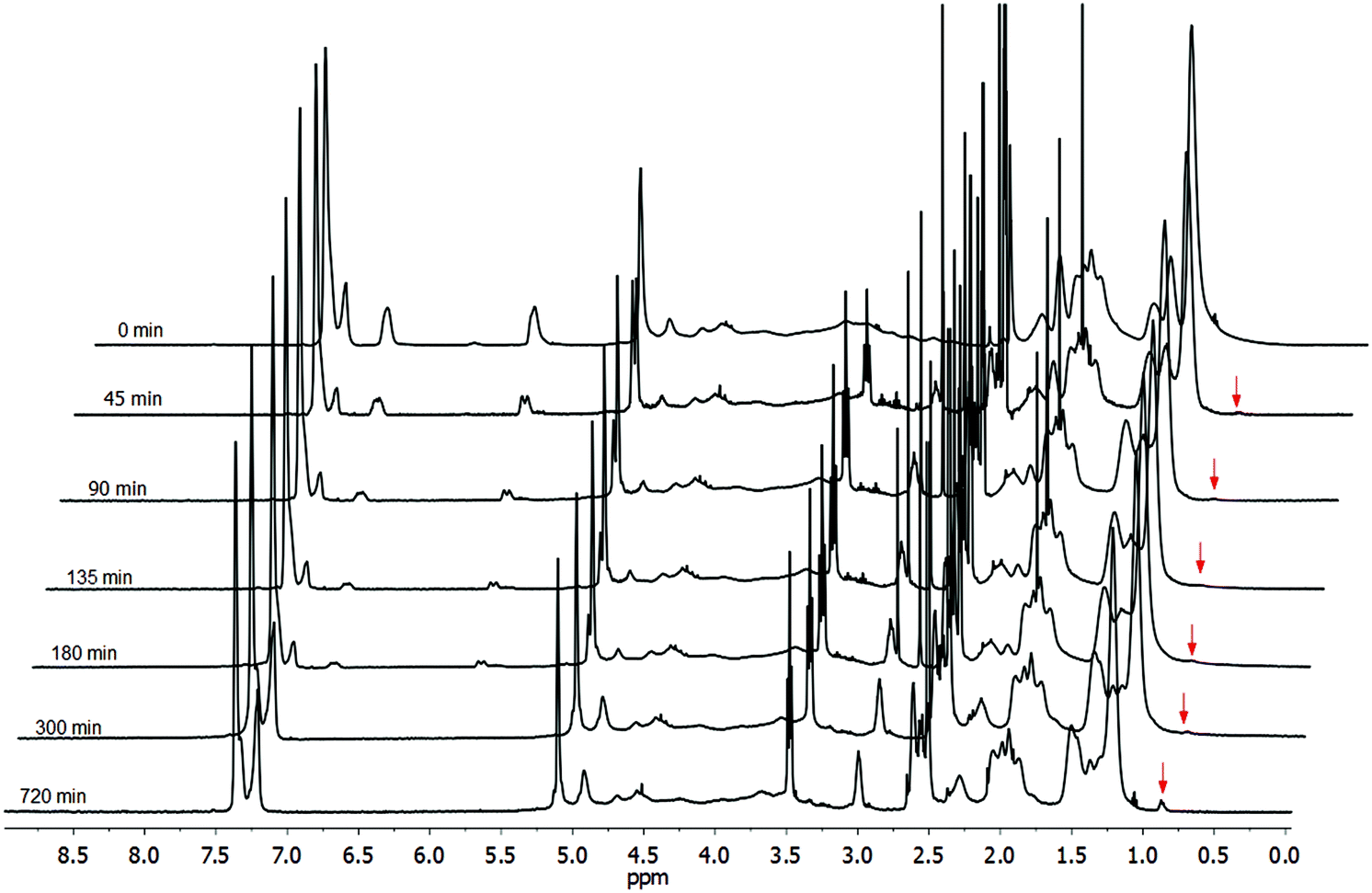 | ||
| Fig. 5 1H NMR spectra for thiol-Michael adducts of 2-ME with CA-Un067-BA under HA catalysis (olefin/2-ME/HA mole ratio equals to 1/2/0.3). Small portions of reaction mixture were withdrawn at each reaction time, precipitated, washed and dried before NMR spectra were recorded. | ||
We also examined the potential of a thiol that also bears amine functionality (cysteamine, Cys) as a thiol-Michael partner, due to the intriguing possibility that amine groups could impart to the polymer useful biological activity, for example antibacterial26 properties. However, in the systems we investigated using either CA-Un067-BA or other substrates in Table 1, cross-linked products were obtained. For certain reactions, even if the samples were soluble at the end of the reaction, after dialysis and freeze-drying the afforded products could no longer dissolve even in polar aprotic solvents such as DMSO and DMF. By analogy to the side reactions observed when using HA as catalyst, it is likely that aza-Michael reaction and/or ester-amide exchange are responsible for the observed crosslinking. It should be noted that for substrates that do not possess ester linkages (e.g. CM products that result from reaction of cellulose ω-unsaturated ethers with Type II olefin partners other than esters32,33) ester-amide interchange cannot be a problem, and therefore crosslinking may not be an issue, since any observed aza-Michael reaction could be circumvented or suppressed.1 Therefore in these cases it may well be practical and efficient to use amine-bearing thiols like cysteamine as thiol-Michael partners. Having demonstrated successful conjugate addition of neutral and anionic thiols to the activated olefins of CA-Un067-BA, we wished to explore the scope of the reaction with regard to CM product terminal functionality. We investigated CM products bearing carboxylic acid, hydroxyl, and trimethylammonium functionalities. Not unexpectedly, the acid derivative CA-Un067-AA (2) did not react with the thiols under either TEA or HA catalysis. Presumably the negatively charged carboxylate inhibits formation of Michael adducts through either base or nucleophile mechanism due to electrostatic repulsion between the carboxylate and the nearby developing negative charge upon conjugate addition in the position α to the carboxyl. In contrast, and consistent with this explanation, the ester CM products CA-Un067-TMA, CA-Un067-HEA, and CA-Pen079-HEA (3, 4, and 5 in Table 1) underwent successful thiol-Michael addition reaction with both 2-ME and 3-MPA (Table 3). HA was an effective catalyst for all of these systems. High conversions (up to 100%) were achieved in most reactions, although minor HA-involved side reactions are observed, especially when reacting with 3-MPA, which requires excess catalyst (Fig. S10 and S12†).
| Cpd. | CM producta, polymer # | Thiol (eq.) | Catalyst (eq.) | Time (h) | Conversionb (%) | Side reactionc |
|---|---|---|---|---|---|---|
| a Rules for polymer abbreviation are described in the reference section.24 b Conversions were calculated by proton NMR (ESI). c Extent of side reaction was evaluated qualitatively by proton NMR; quantitative determination not possible due to signal overlap. | ||||||
| 12 | CA-Un067-TMA, 3 | 2-ME (6) | TEA (6) | 12 | 100 | No |
| 13 | CA-Un067-TMA, 3 | 3-MPA (9) | TEA (18) | 12 | 100 | No |
| 14 | CA-Un067-HEA, 4 | 2-ME (6) | TEA (6) | 12 | 92 | No |
| 15 | CA-Un067-HEA, 4 | 2-ME (3) | HA (2) | 5 | 100 | Minimal |
| 16 | CA-Un067-HEA, 4 | 3-MPA (9) | TEA (18) | 12 | 65 | No |
| 17 | CA-Un067-HEA, 4 | 3-MPA (3) | HA (6) | 5 | 83 | Yes |
| 18 | CA-Pen079-HEA, 5 | 2-ME (6) | TEA (3) | 12 | >95 | No |
| 19 | CA-Pen079-HEA, 5 | 3-MPA (5) | TEA (10) | 12 | >95 | No |
TEA was a more efficient catalyst for these other ester substrates than in reactions with CA-Un067-BA. Differences in nucleophilicity and therefore potentially mechanisms of catalysis of the two amines (HA and TEA) are key factors affecting catalyst efficiency and the extent of side reactions.31 However, for catalyst efficiency, we believe that other factors such as the ability of the thiol anion to access the β carbon (e.g., steric factors) may also influence reactivity (the electron withdrawing ability of the particular ester group may also influence polymer reactivity towards conjugate addition, for example by reducing olefin electron density and stabilizing enolate intermediates, but such influences were not clearly important in the current study). CA-Un067-BA has long hydrophobic chains and hydrophobic benzyl rings, which provide a rather hydrophobic milieu for the β carbon. It could be that it is easier for Et3N or the Et3N+RS− salt to access the β-carbon of the benzoate ester than for the more hydrophilic HA or HA+RS− salt. On the other hand, the other CA-based CM product substrate esters, e.g. polymers 3, 4 and 5, have hydrophilic ester substituents, making it easier for the more hydrophilic HA or HA+RS− salt to access the β carbon. At this point the mechanistic explanation is hypothetical, and more data will be required to refute or confirm this hypothesis. Considering that the use of HA leads to some extent of undesired side reactions, and TEA does not, TEA is a more suitable catalyst for thiol-Michael reactions of polymers 3, 4 and 5.
Table 4 shows the glass transition temperatures and molecular weights of the thiol-Michael products. For certain samples, an endothermic transition at lower temperature was observed, which we attributed to segmental movement of the side chain (β relaxation).34 While the transition temperatures differ somewhat from sample to sample, it is difficult to identify a specific structure–property trend. The Tg values are likely affected by multiple factors including side chain length, nature of the terminal functional groups, and specific interactions. Molecular weight data for samples that were charged and/or bore carboxylic acid groups could not be obtained, due to strong interaction between the samples and the SEC column packing. Nevertheless, no significant change in degree of polymerization (DP) was observed according to SEC analysis of the neutral polymers, indicating that the mild thiol-Michael reaction conditions did not cause significant chain scission.
| Polymer # | Abbreviationa | T g (β relaxation) (°C) | M n (kDa)/DP | Đ |
|---|---|---|---|---|
| a Rules for polymer abbreviation are described in the reference section.24 b Dispersity. c Polymer molecular weight data unavailable due to polymer aggregation and/or polymer-column interaction. d Not observed. | ||||
| 1 | CA-Un067-BA | 99 (0) | 59.6/135 | 1.7 |
| 7 | CA-Un067-BA-2ME | 84 (1) | 76.4/155 | 2.5 |
| 9 | CA-Un067-BA-3MPA | 111 (11) | NAc | NA |
| 3 | CA-Un067-TMA | 116 (N.O.d) | NA | NA |
| 12 | CA-Un067-TMA-2ME | 103 (N.O.) | NA | NA |
| 13 | CA-Un067-TMA-3MPA | 88 (N.O.) | NA | NA |
| 4 | CA-Un067-HEA | 87 (10) | 50.6/123 | 1.9 |
| 15 | CA-Un067-HEA-2ME | 108 (10) | 49.7/108 | 2.0 |
| 17 | CA-Un067-HEA-3MPA | 89 (6) | NA | NA |
| 5 | CA-Pen079-HEA | 125 (N.O.) | 52.9/142 | 1.9 |
| 18 | CA-Pen079-HEA-2ME | 59 (−5) | 66.8/154 | 1.9 |
| 19 | CA-Pen079-HEA-3MPA | 63 (N.O.) | NA | NA |
Conclusions
In this study, we present the first examples of elaboration of α,β-unsaturated cellulose esters prepared from CM products by post-CM modification using thiol-Michael addition. The modular characteristics of CM and the “click” nature of thiol-Michael reaction have made both reactions powerful tools in organic and polymer chemistry. The combination of these two powerful reactions for modification of the polysaccharide cellulose in this study creates new methodology for preparation of derivatives of much greater functional diversity, including the option for possessing multiple functional groups, than by using either of these reactions alone. CM reactions initially provide reactive, electron deficient olefins (e.g. α,β-unsaturated esters) that are prone to crosslinking and/or degradation due to abstraction of the labile γ-hydrogens by radical initiators. Therefore these reactive olefins must in any case be eliminated in order to stabilize the CM products for any useful purpose. We show here that these olefins may be excellent substrates for thiol-Michael addition under the right conditions and with the right thiol partner. To be a good substrate for thiol-Michael addition, the olefin should be electron-deficient and the functionality in its vicinity should not interfere with the hydrothiolation process (as does CO2H, for example). The catalyst plays an important role in thiol-Michael addition. The tertiary amine triethylamine causes no observable side reactions but is sometimes less effective, while the primary amine catalyst n-hexylamine is sometimes more effective, but may promote side reactions by ester-amide exchange or possibly by aza-Michael addition. Amine-containing thiols (e.g. cysteamine) afford crosslinked products with ester CM substrates due to these same side reactions, while other thiol partners showcased in the current study give discrete products with high conversions. Overall, the disadvantage of the reactive conjugated olefin in the initial CM products is thus turned into an advantage, by employing it as the vehicle for introduction of further useful functionality. The double functionalization thus enabled by the combined CM/thiol-Michael addition approach is a strategy that could be of high value for targeting,35 pro-drug synthesis,36 visualization,37 and any number of other uses. This double modification approach showcased herein also has the potential to be adapted to many other polysaccharides, as well as to even broader areas of polymer synthesis and modification.Acknowledgements
We thank the Eastman Chemical Company for their kind donation of the commercial cellulose esters used in this work. We thank the National Science Foundation for partially funding this work through Grant DMR-1308276, the Institute for Critical Technologies and Applied Science at Virginia Tech for facility and financial support, and the Macromolecules and Interfaces Institute of Virginia Tech for educational support.References
- D. P. Nair, M. Podgorski, S. Chatani, T. Gong, W. X. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724 CrossRef CAS.
- A. Lv, Z.-L. Li, F.-S. Du and Z.-C. Li, Macromolecules, 2014, 47, 7707 CrossRef CAS.
- L. Ding, J. Qiu and Z. Zhu, Macromol. Rapid Commun., 2013, 34, 1635 CrossRef CAS PubMed.
- C. O. Bounds, R. Goetter, J. A. Pojman and M. Vandersall, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 409 CrossRef CAS.
- C. O. Bounds, J. Upadhyay, N. Totaro, S. Thakuri, L. Garber, M. Vincent, Z. Huang, M. Hupert and J. A. Pojman, ACS Appl. Mater. Interfaces, 2013, 5, 1643 CAS.
- X. Meng and K. J. Edgar, Prog. Polym. Sci., 2016, 53, 52 CrossRef CAS.
- L. Maleki, U. Edlund and A.-C. Albertsson, Biomacromolecules, 2015, 16, 667 CrossRef CAS PubMed.
- K. Peng, C. Cui, I. Tomatsu, F. Porta, A. H. Meijer, H. P. Spaink and A. Kros, Soft Matter, 2010, 6, 3778 RSC.
- W. M. Gramlich, I. L. Kim and J. A. Burdick, Biomaterials, 2013, 34, 9803 CrossRef CAS PubMed.
- K. Schumann, A. Pfeifer and T. Heinze, Macromol. Symp., 2009, 280, 86 CrossRef CAS.
- X. Meng, E. A. York, S. Liu and K. J. Edgar, Carbohydr. Polym., 2015, 133, 262 CrossRef CAS PubMed.
- P. Tingaut, R. Hauert and T. Zimmermann, J. Mater. Chem., 2011, 21, 16066 RSC.
- G.-L. Zhao, J. Hafrén, L. Deiana and A. Córdova, Macromol. Rapid Commun., 2010, 31, 740 CrossRef CAS PubMed.
- Y. Wang, T. Heinze and K. Zhang, Nanoscale, 2016, 8, 648 RSC.
- H. Rosilo, E. Kontturi, J. Seitsonen, E. Kolehmainen and O. Ikkala, Biomacromolecules, 2013, 14, 1547 CrossRef CAS PubMed.
- X. Meng, J. B. Matson and K. J. Edgar, Biomacromolecules, 2014, 15, 177 CrossRef CAS PubMed.
- X. Meng, J. B. Matson and K. J. Edgar, Polym. Chem., 2014, 5, 7021 RSC.
- Y. Dong and K. J. Edgar, Polym. Chem., 2015, 6, 3816 RSC.
- X. Meng and K. J. Edgar, Carbohydr. Polym., 2015, 132, 565 CrossRef CAS PubMed.
- R. H. Grubbs, Tetrahedron, 2004, 60, 7117 CrossRef CAS.
- S. J. Connon and S. Blechert, Angew. Chem., Int. Ed., 2003, 42, 1900 CrossRef CAS PubMed.
- M. Winkler and M. A. R. Meier, Green Chem., 2014, 16, 3335 RSC.
- A. K. Chatterjee, T. L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360 CrossRef CAS PubMed.
- The rules for polymer abbreviation/nomenclature in this manuscript are as follows. For terminally olefinic cellulose esters CA-Un067 and CA-Pen079, CA denotes cellulose acetate DSAc = 1.82; Un denotes undec-10-enoate, and Pen denotes pent-4-enoate, while the numbers 067 and 079 indicate DS of each olefinic side chain. For cross-metathesis products, e.g. CA-Un067-XX, CA-Un067 indicates the starting cellulose ester, while XX stands for the CM partner used in the reaction e.g. AA denotes acrylic acid and BA is benzyl acrylate. For thiol-Michael addition products, e.g. CA-Un067-BA-YY, CA-Un067-BA indicates that the thiol-Michael substrate used is from CM product CA-Un067-BA, while YY stands for the thiol donor employed e.g. 3MPA denotes 3-mercaptopropionic acid.
- G. A. Ilevbare, H. Liu, J. Pereira, K. J. Edgar and L. S. Taylor, Mol. Pharmaceutics, 2013, 10, 3392 CrossRef CAS PubMed.
- X. Meng, R. Xing, S. Liu, H. Yu, K. Li, Y. Qin and P. Li, Int. J. Biol. Macromol., 2012, 50, 918 CrossRef CAS PubMed.
- S. N. Pawar and K. J. Edgar, Biomacromolecules, 2011, 12, 4095 CrossRef CAS PubMed.
- M. A. Bonache, C. Cativiela, M. T. García-López and R. González-Muñiz, Tetrahedron Lett., 2006, 47, 5883 CrossRef CAS.
- G.-Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1196 RSC.
- N. Liu and W. Baker, Adv. Polym. Technol., 1992, 11, 249 CrossRef CAS.
- J. W. Chan, C. E. Hoyle, A. B. Lowe and M. Bowman, Macromolecules, 2010, 43, 6381 CrossRef CAS.
- N. Pahimanolis, P. Kilpeläinen, E. Master, H. Ilvesniemi and J. Seppälä, Carbohydr. Polym., 2015, 131, 392 CrossRef CAS PubMed.
- Y. Dong, L. I. Mosquera-Giraldo, L. S. Taylor and K. J. Edgar, Biomacromolecules, 2016, 17, 454 CrossRef CAS PubMed.
- W. G. Glasser, G. Samaranayake, M. Dumay and V. Davé, J. Polym. Sci., Part B: Polym. Phys., 1995, 33, 2045 CrossRef CAS.
- P. M. Kharkar, M. S. Rehmann, K. M. Skeens, E. Maverakis and A. M. Kloxin, ACS Biomater. Sci. Eng., 2016, 2, 165 CrossRef CAS.
- T. Kuhnt, A. Herrmann, D. Benczedi, C. Weder and E. J. Foster, RSC Adv., 2014, 4, 50882 RSC.
- C. Yin, F. Huo, J. Zhang, R. Martinez-Manez, Y. Yang, H. Lv and S. Li, Chem. Soc. Rev., 2013, 42, 6032 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py00539j |
| This journal is © The Royal Society of Chemistry 2016 |