
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Triply responsive soft matter nanoparticles based on poly[oligo(ethylene glycol) methyl ether methacrylate-block-3-phenylpropyl methacrylate] copolymers†
Yiwen
Pei
ab,
Kevin
Jarrett
c,
Martin
Saunders
d,
Peter J.
Roth‡
ab,
Craig E.
Buckley
c and
Andrew B.
Lowe
*ab
aNanochemistry Research Institute (NRI), Curtin University, Kent Street, Bentley, Perth, WA 6102, Australia. E-mail: andrew.b.lowe@curtin.edu.au
bDepartment of Chemistry, Curtin University, Kent Street, Bentley, Perth, WA 6102, Australia
cDepartment of Physics and Astronomy, Curtin University, Kent Street, Bentley, Perth, WA 6102, Australia
dCentre for Microscopy, Characterisation and Analysis (CMCA), University of Western Australia, 35 Stirling Highway, Crawley, WA 6009, Australia
First published on 24th March 2016
Abstract
The stimulus-responsive properties of soft matter nanoparticles based on poly[oligo(ethylene glycol) methyl ether methacrylate-block-3-phenylpropyl methacrylate] (p(OEGMA-block-PPMA)) copolymers in methanol and ethanol are described. Methanolic synthesis, with 4-cyanopentanoic acid dithiobenzoate as the RAFT mediating agent, facilitates simple access to nanoparticles exhibiting the full range of common morphologies (spheres, worms and vesicles) simply by varying the copolymer composition (fixed average degree of polymerization (![[X with combining macron]](https://www.rsc.org/images/entities/i_char_0058_0304.gif) n) of the pOEGMA macro-CTA for variable n of the pPPMA block). Interestingly, we demonstrate that p(OEGMAx-block-PPMAy) nanoparticles are able to elicit three types of response to externally applied stimuli. These materials possess two distinct, but complementary, reversible thermal responses – one that results in an order–order transition, i.e. a morphological change, while the second is a reversible order–disorder transition based on upper critical solution temperature (UCST)-type behaviour associated with the pOEGMA coronal chains in the nanoparticles. Finally, we report the first example where specific p(OEGMA-block-PPMA) nanoparticles are shown to be sensitive to addition of an organobase – a response that is accompanied by an order–order, worm-to-sphere, morphology transition.
n) of the pOEGMA macro-CTA for variable n of the pPPMA block). Interestingly, we demonstrate that p(OEGMAx-block-PPMAy) nanoparticles are able to elicit three types of response to externally applied stimuli. These materials possess two distinct, but complementary, reversible thermal responses – one that results in an order–order transition, i.e. a morphological change, while the second is a reversible order–disorder transition based on upper critical solution temperature (UCST)-type behaviour associated with the pOEGMA coronal chains in the nanoparticles. Finally, we report the first example where specific p(OEGMA-block-PPMA) nanoparticles are shown to be sensitive to addition of an organobase – a response that is accompanied by an order–order, worm-to-sphere, morphology transition.
Introduction
The self-assembly of AB diblock copolymers, in a selective solvent, to give nanometre sized particles exhibiting a variety of morphologies is well documented.1–7 Traditionally, the preparation of such nanoparticles is achieved via the initial synthesis (under conditions in which all components are, and remain, soluble), isolation, purification and characterization of a well-defined parent block copolymer followed by a processing step inducing self-assembly. The processing step may involve direct addition of the block copolymer to a selective solvent, dialysis of the block copolymer initially dissolved unimerically in a non-selective solvent against a selective solvent or dilution of a concentrated sample of the block copolymer in a non-selective solvent with a selective solvent. While well-established, and perfectly valid protocols, such processing routes can be time consuming and may only facilitate preparation of nanoparticles at low concentrations (≤1.0 wt% is typical). Alternatively, AB diblock copolymers (or more complex architectural species) can be designed and prepared such that one, or both, blocks exhibit variable solvophilic/solvophobic characteristics. In such cases an external trigger such as a change in temperature,8,9 pH10,11 or electrolyte concentration for example, can be employed to induce self-assembly. Again, however, such systems are commonly limited to working with such stimuli responsive materials at relatively low concentrations.12 The use of stimuli responsive polymers as building blocks for soft matter nanoparticles in which unimolecular copolymer chains assemble upon application of a trigger is an example of a disorder–order transition. Such smart materials often exhibit this behaviour reversibly, i.e. order–disorder transitions are also common in such systems.The most commonly employed trigger for such reversible self-assembly is a change in temperature and is typically, but not exclusively, utilized in aqueous-based systems. Inducing self-assembly is usually accomplished by exploiting the lower critical solution temperature (LCST) characteristics of one of the building blocks with block copolymers containing poly(N-isopropylacrylamide) (PNIPAM) being the most widely studied in part due to its readily accessible LCST of 32 °C.13,14 Alternatively, block copolymers can undergo disorder–order transitions (or the opposite) due to upper critical solution temperature (UCST)-type behaviour.15 UCST-based triggers are not as widely reported as their LCST counterparts, especially in water, but are, arguably, as equally important and as potentially versatile when designing and preparing stimulus responsive materials.16–20
Less common in polymeric nanoparticle systems where the concentration, copolymer composition, and overall make-up of the system, are kept constant are order–order transitions. Examples of such transitions include, but are not limited to, reversible sphere-to-worm, worm-to-vesicle and sphere-to-vesicle morphology changes. For example, Abbas et al. detailed the stepwise change in nanoparticle morphology from spheres to worms to vesicles (S–W–V) in dilute solutions of poly(styrene-block-dimethylsiloxane) copolymers by employing mixtures of dialkyl phthalates to tune solvent selectivity. The reverse V–W–S transitions were accomplished by simple heating and were shown to be totally reversible.21
LaRue and co-workers reported thermally-induced morphology transitions in poly(styrene-block-isoprene) copolymers (for a fixed copolymer composition and concentration) in heptane between 25 and 40 °C, in which the solvent quality changed for both blocks upon heating. Room temperature worm nanoparticles rearranged to spheres upon heating and likewise, vesicles transformed to worms. These transitions were reversible although it is noted that the S–W transition required in excess of 36 days at 25 °C to achieve significant rearrangement to the worm morphology.22
Moughton and O'Reilly23 detailed a thermally induced S–V transition in a RAFT prepared block copolymer of NIPAM with tert-butyl acrylate (tBA) in which the R-group at the α-termini contained a cationic functionality. In aqueous media these AB diblock copolymers self-assembled to give spherical species, of narrow size distribution, with the polytBA block in the core, the NIPAM block in the corona and the surface decorated with the cationic, R group-derived functional groups. Heating above the LCST of the PNIPAM block, and thus formally changing the hydrophilic–hydrophobic balance, resulted in a S–V morphological transition. Adjusting the temperature to below the LCST of the PNIPAM block did lead to the reverse V–S transition but the reformed spherical species were poorly defined with a broad size distribution.
Recently there has been significant academic interest in polymerization-induced self-assembly24 and in particular the application of reversible addition–fragmentation chain transfer dispersion polymerization-based formulations (RAFTDP-PISA). RAFTDP-PISA is especially attractive since it allows for the preparation of polymeric nano-objects at high concentration (formulations ≥50 wt% are possible) in a fast and scalable fashion (enhanced kinetics compared to fully homogeneous syntheses are often observed especially in aqueous-based systems), yields nanoparticles exhibiting the full range of common morphologies (spheres, worms and vesicles) as well as more complex and transient species. Furthermore, it has now been documented in a wide range of media and is no more difficult to perform than a standard RAFT (co)polymerization and is thus readily implemented.
Recent interest in RAFTDP-PISA has been driven, to a large extent, by work from the groups of Armes,25–34 Pan,35–42 Zhang43–52 and more recently others.53–69 Several recent mini-reviews/highlights summarize the current state-of-the-art in this increasingly popular area of research.70–73 Of particular relevance to the work detailed herein is the fact that certain reversible, stimulus-induced, order–order, morphological transitions have been reported by Armes74–76 and by us53,55 and it appears such behaviour might be a fairly ubiquitous feature in many RAFTDP formulations. For example, in an aqueous RAFTDP-PISA formulation based on glycerol monomethacrylate (GMA, permanently hydrophilic block) with 2-hydroxypropyl methacrylate (HPMA) Blanazs et al.77 reported that the formulation giving a material with an average composition of p(GMA54-block-HPMA140) yielded a pure worm nanoparticle phase and presented, macroscopically, as a soft gel due to worm nano-object entanglements. Interestingly, cooling a solution of these worm nanoparticles from ambient to 5 °C resulted in a macroscopic change to a free flowing solution which was demonstrated to be due to a nanoscale W–S morphology transition. Such gelation–degelation behaviour was shown to be fully reversible. In related work, we reported complementary reversible gelation phenomena in AB diblock copolymers of 2-(dimethylamino)ethyl methacrylate (DMAEMA) with 3-phenylpropyl methacrylate (PPMA) in EtOH.53 Specifically, a p(DMAEMA20-block-PPMA47) copolymer existed at ambient temperature as a pure worm phase and presented as a gel due to the same worm nanoparticle entanglements. In this system, and in contrast to the aqueous formulation noted above, heating to 70 °C resulted in a rapid degelation and formation of a free flowing solution. This phenomenon was also due to the same fundamental W–S order–order transition and was likewise fully reversible with gelation occurring within a few minutes upon cooling. We have also observed identical reversible gelation, and associated morphology changes, in non-polar media (n-tetradecane and n-octane) in block copolymer worm nanoparticles based on poly(stearyl methacrylate) with PPMA.55,59
While a change in temperature represents a straightforward approach for effecting such order–order transitions it is possible to accomplish similar morphology transitions employing alternative triggers. Armes and co-workers reported that a p(GMA56-block-HPMA155) block copolymer, prepared by aqueous RAFTDP-PISA with a carboxylic acid functional RAFT CTA, presented as a soft gel at ambient temperature at 10 wt% under mildly acidic conditions. As with their previous GMA–HPMA materials this sample underwent degelation and an order–order transition upon cooling. However, similar reversible degelation was also observed upon raising the pH of an ambient temperature gel ‘solution’ from 3.5 to 6.0 via the addition of NaOH.78 The observed degelation was again due to a W–S morphology change. This pH-induced transition was attributed to the ionization of the carboxylic acid groups located at the α-termini of the block copolymer chains (and therefore at the outer periphery of the nanoparticles). A similar, NaOH-induced V–W transition was also reported for nanoparticles formed from a p(GMA43-block-HPMA200) copolymer in aqueous media.
Building on our previous reports of RAFTDP-PISA formulations in alcoholic media with PPMA as comonomer, herein we detail the stimulus responsive properties of soft matter nano-objects based on oligo(ethylene glycol) methyl ether methacrylate (OEGMA) with PPMA in ethanol and methanol. We demonstrate that the p(OEGMA-block-PPMA)-based nanoparticles are responsive to three triggers – two different and distinct thermal processes (one reversible order–order transition and one reversible order–disorder transition) as well as a relatively unique organobase-induced order–order transition. We believe this is the first example of RAFTDP-PISA prepared nanoparticles that possess three different stimulus responsive properties and is the first example in which an organoamine, in a non-aqueous environment, has been employed to effect an order–order transition.
Experimental
All reagents were purchased from the Aldrich Chemical Company at the highest available purity and used as received unless noted otherwise. 3-Phenylpropyl methacrylate (PPMA) was purchased from Monomer–Polymer and Dajac Labs. 2,2′-Azobis(isobutyronitrile) (AIBN) was purified by recrystallization twice from methanol and then stored in a freezer until needed. Oligo(ethylene glycol) methyl ether methacrylate (OEGMA) (average![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n = 300) and PPMA were purified by passing through a basic Al2O3 column to remove the inhibitor and acidic impurities prior to use. 4-Cyanopentanoic acid dithiobenzoate (CPADB) was prepared according to a procedure described elsewhere.79
n = 300) and PPMA were purified by passing through a basic Al2O3 column to remove the inhibitor and acidic impurities prior to use. 4-Cyanopentanoic acid dithiobenzoate (CPADB) was prepared according to a procedure described elsewhere.79
Synthesis of poly[oligo(ethylene glycol) methyl ether methacrylate] macro-CTA via RAFT homopolymerization
Below is a general procedure for the RAFT homopolymerization of OEGMA mediated by CPADB.A solution containing OEGMA (10.0 g, 3.33 × 10−2 mol), CPADB (1.86 × 10−1 g, 6.67 × 10−4 mol), AIBN (2.18 × 10−2 g, 1.33 × 10−4 mol), and acetonitrile (30.0 mL) was added to a reaction vial equipped with a magnetic stir bar. The reaction vessel was sealed and the reaction mixture purged with Argon for 30 min prior to being placed in a preheated oil bath at 70 °C. Polymerization was allowed to proceed for 4 h after which it was halted by exposure to air while cooling in an ice/water bath. The pOEGMA homopolymer(s) were isolated by precipitation into a mixture of diethyl ether and petroleum spirit (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) followed by filtration and dried overnight prior to NMR spectroscopic and SEC analysis.
:1, v/v) followed by filtration and dried overnight prior to NMR spectroscopic and SEC analysis.
RAFT dispersion polymerization of 3-phenylpropyl methacrylate with polyOEGMAx in alcoholic media
Below is a general procedure for the RAFTDP of PPMA with a polyOEGMA28 (pOEGMA28) macro-CTA in methanol. All RAFTDPs were performed in a similar fashion.A solution containing PPMA (1.65 × 10−1 g, 8.07 × 10−4 mol), AIBN (3.78 × 10−4 g, 2.30 × 10−6 mol), pOEGMA28 macro-CTA (1.0 × 10−1 g, 1.15 × 10−5 mol) and anhydrous methanol (1.34 mL) was added to a vial equipped with a magnetic stir bar. The reaction vessel was sealed and the solution purged with Argon for 15 min prior to being placed in a preheated oil bath at 70 °C. Polymerization was allowed to proceed for 24 h after which it was halted by exposure to air while cooling in an ice/water bath. Block copolymer was isolated by precipitation into a large excess of diethyl ether and petroleum spirit mixture (1:1, v/v) followed by filtration and dried overnight prior to NMR spectroscopic and size exclusion chromatographic analyses.
Size exclusion chromatography (SEC)
SEC analysis was performed on a Shimadzu modular system consisting of a 4.0 mm × 3.0 mm Phenomenex Security Guard™ Cartridge guard column and two linear phenogel columns (103 and 104 Å pore size) in tetrahydrofuran (THF) operating at a flow rate of 1.0 mL min−1 and 40 °C using a RID-20A refractive index detector, a SPD-M20A prominence diode array detector and a miniDAWN TREOS multi-angle static light scattering (MALS) detector. The system was also calibrated with a series of narrow molecular weight distribution poly(methyl methacrylate) standards with molecular weights ranging from 2.8 to 220 kg mol−1. Chromatograms were analysed by Lab Solutions GPC software.NMR spectroscopy
NMR spectra were recorded on a Bruker 400 MHz spectrometer. 1H NMR spectra were recorded in deuterated chloroform (CDCl3). Residual CHCl3 (δ = 7.26 ppm) was utilized as the internal reference signal. Variable temperature NMR characterization analyses were performed in fully deuterated methanol (CD3OD) or ethanol (CD3CD2OD) using a Bruker 400 spectrometer (1H, 400 MHz) with 128 scans averaged per spectrum. For measurements made in d4-methanol, the pulse program employing solvent suppression at a 1H chemical shift of δ = 3.3 ppm was utilized to eliminate the solvent peak. A series of 1H NMR spectra of block copolymer p(OEGMA28-block-PPMA69) (0.5 wt%) was recorded in d4-methanol covering the temperature range 25–63 °C. The block copolymer p(OEGMA27-block-PPMA36) (0.5 wt%) in d6-ethanol was first heated from −5 to 70 °C and cooled back to ambient temperature at a rate of 0.2 °C min−1. Both samples were allowed to equilibrate at each temperature for at least 10 min prior to measurements.Dynamic light scattering and zeta-potential measurements
Dynamic light scattering (DLS) measurements were performed utilizing a Malvern Instrument Zetasizer Nano Series instrument (laser power = 4.0 mW, wavelength = 633 nm, detection angle = 173°) at 25 °C. For sample preparation, 50 μL of the parent RAFTDP solution was diluted with 1.45 mL of methanol or ethanol and the solution stirred for 10 min prior to filtration through 0.45 μm PTFE filters. For temperature-dependent DLS analysis, samples were heated and allowed to equilibrate at each temperature for at least 10 min prior to measurements. Electrophoresis studies were conducted on 0.07 wt% copolymer solutions utilizing the same Malvern Zetasizer Nano Series instrument. Samples for electrophoretic analysis were prepared by adding the organobase 1,8-diazabicycloundec-7-ene (DBU) to the parent RAFTDP solutions, followed by equilibration overnight under continuous stirring and dilution with methanol prior to measurements.Turbidity measurements
Turbidity measurements were performed on a Crystal16 purchased from Technobis Crystallization Systems. Samples for analysis were prepared by slowly adding 50 μL of the parent RAFTDP solution to 1.45 mL of ethanol under stirring. The solubility studies were conducted by heating the sample solutions from −5 to 70 °C with a heating rate of 0.2 °C min−1 followed by cooling to −5 °C at a cooling rate of 0.2 °C min−1 after holding the temperature at 70 °C for 20 min. During the heating/cooling cycles all samples were visually monitored under continuous stirring to facilitate interpretation of the observed transmittance. The baseline was corrected to 100% transmittance utilizing a clear empty glass vial. The upper critical solution temperature, TUCSTcp, was recorded as the temperature at a transmittance of 50%.Transmission electron microscopy (TEM)
2-Dimensional (2D) TEM analyses were conducted on a JEOL 2100 transmission electron microscope operated at 120 kV. Samples for analysis were prepared by the slow addition of 50 μL of the parent RAFTDP solution to 1.45 mL of methanol or ethanol under stirring. The copolymer solution (0.7 wt%) was dropped onto the top of a copper grid (ProSciTech) or a carbon-coated copper grid (SPI Suppliers) and allowed to contact for 1 min. Excess solution was wicked off using a filter paper. To stain the nano-objects uranyl acetate (0.2 wt%) solution was soaked on the sample-loaded grid for 30 s and then carefully blotted to remove excess stain solution.For TEM sample preparation at high temperature all materials and equipment were placed in a hot oven at 63 °C for 10 min. The parent RAFTDP solution (20 wt%) was kept under continuous stirring at 63 °C for 10 min and then diluted to 0.7 wt% with warm anhydrous methanol (at the same temperature as the copolymer sample) prior to TEM sample preparation. The TEM grid was then stained as described above.
For tomography measurements, polymer samples were prepared and stained as described above. TEM tomography analyses were performed on a Titan G2 80-200 TEM/STEM instrument operated at 200 kV. The images were collected at sample-tilting angles ranging from −70° to 70° in 2° intervals. The series of 2D images were reconstructed to the 3D image based on the filtered back projection method.
Results and discussion
In a recent paper we described the detailed synthesis and characterization of AB diblock copolymers based on oligo(ethylene glycol) methyl ether methacrylate (OEGMA) with 3-phenylpropyl methacrylate (PPMA), and their corresponding nanoparticles in methanol, Scheme 1.80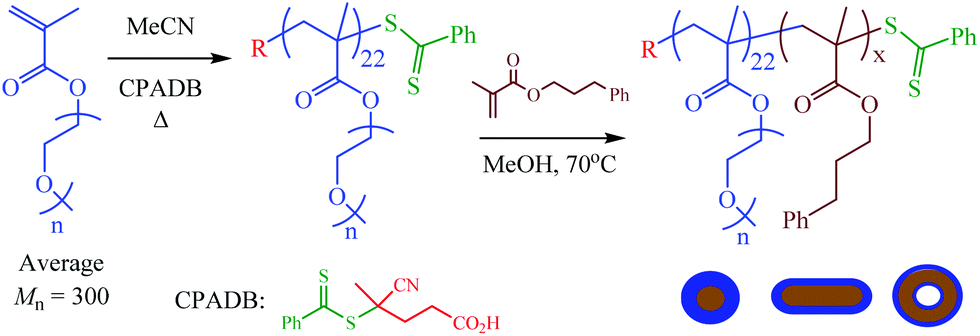 | ||
| Scheme 1 General approach for the preparation of the poly[oligo(ethylene glycol) methyl ether methacrylate-block-3-phenylpropyl methacrylate]-based nanoparticles by methanolic RAFT dispersion polymerization with polymerization-induced self-assembly. | ||
This is a versatile RAFTDP-PISA formulation and allows for ready access to nanoparticles with the full range of common morphologies all of which can be obtained as pure phases under appropriate conditions. These particular block copolymers were inspired by our previous work with PPMA-based RAFTDP-PISA formulations53,55,57 as well as the recent report from Lovett et al.78 regarding water-based pH-sensitive block copolymer nano-objects containing reversibly ionizable carboxylic acid groups.
Thermally responsive properties of p(OEGMA28-block-PPMAx) copolymer nano-objects: temperature-induced order–order transitions in MeOH
We initially prepared a series of p(OEGMA28-block-PPMAx) copolymers in methanol in which the average degree of polymerization,n, of the PPMA block was varied from 36 to 83. Within this series of block copolymers the p(OEGMA28-block-PPMA69) and p(OEGMA28-block-PPMA71) materials yielded nano-objects with pure worm morphologies at ambient temperature, as determined by TEM, and presented macroscopically as soft gels. However, at the polymerization temperature these systems existed as free flowing solutions. Consistent with previous reports from our group53,55,59 this difference in macroscopic presentation was shown to be due to a fundamental change in nanoparticle morphology, Fig. 1, at these two different temperatures.
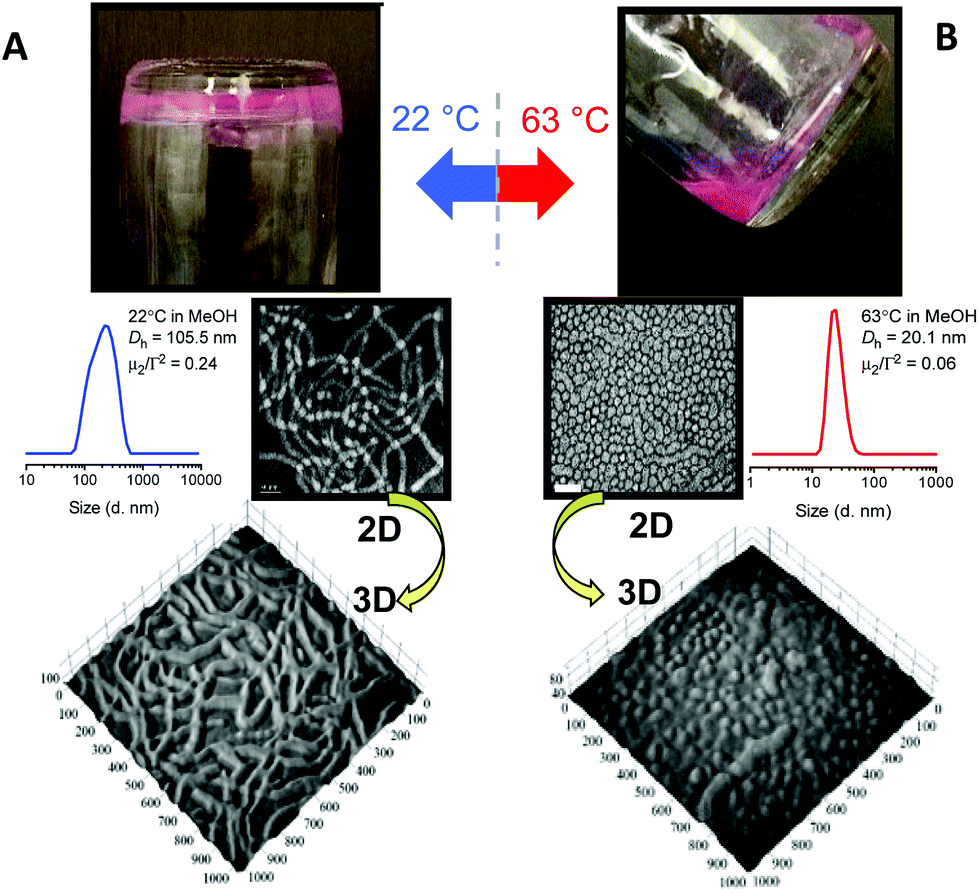 | ||
| Fig. 1 (A) Digital picture of the macroscopic presentation of the p(OEGMA28-block-PPMA69) copolymer worm nanoparticles at 22 °C with representative 2D and 3D TEM images below and the DLS-measured nanoparticle size distribution, and (B) the same sample after heating to 63 °C highlighting the change in macroscopic presentation with the 2D/3D TEM images of the new, predominantly, spherical nanoparticles and the concomitant change in the DLS nanoparticle size distribution. | ||
For the p(OEGMA28-block-PPMA69) species at 22 °C TEM clearly highlights the cylindrical, or worm, nature of the nanoparticles. The volumetric uniformity of the worm nanoparticles was also confirmed from the tomographic image. DLS gave a sphere-equivalent hydrodynamic diameter of 105.5 nm with a corresponding DLS polydispersity (μ2/Γ2) of 0.24. In contrast, heating briefly to 63 °C resulted in a morphological transition from the worm phase to a near-pure spherical phase (mixture of predominantly spheres and a few short ‘oligomeric’ worms) as judged by TEM and also verified by tomographic imaging. DLS gave an average hydrodynamic diameter of these species of 20.1 nm with a corresponding μ2/Γ2 of 0.06. Such temperature induced reversible worm–sphere order–order transitions, whether induced upon heating or cooling, have emerged as a rather common feature in many RAFTDP-PISA formulations and will likely be observed in many more future formulations.
The driving force for such changes in morphology have been rationalized in terms of changes in the relative volume fraction of the core and coronal blocks and the impact this has on the dimensionless packing parameter, p. The changes in relative volume of the two blocks is a direct result of the temperature-dependent solvation with, generally, a greater effect being observed on the ‘solvophobic’ core chains. Fortunately, changes in the average degree of solvation of either the core and/or coronal chains can be qualitatively examined by 1H NMR spectroscopy as we, and others, have previously demonstrated.53,55,59Fig. 2 gives the general structure of the p(OEGMA28-block-PPMAx) copolymers synthesized and a series of 1H NMR spectra recorded from 25 to 63 °C in d4-methanol. Isotopic effects of this deuterated solvent compared to non-deuterated methanol were presumed to be negligible and d4-methanol was assumed to be a suitable model solvent to elucidate molecular events observed in non-deuterated methanolic solutions.
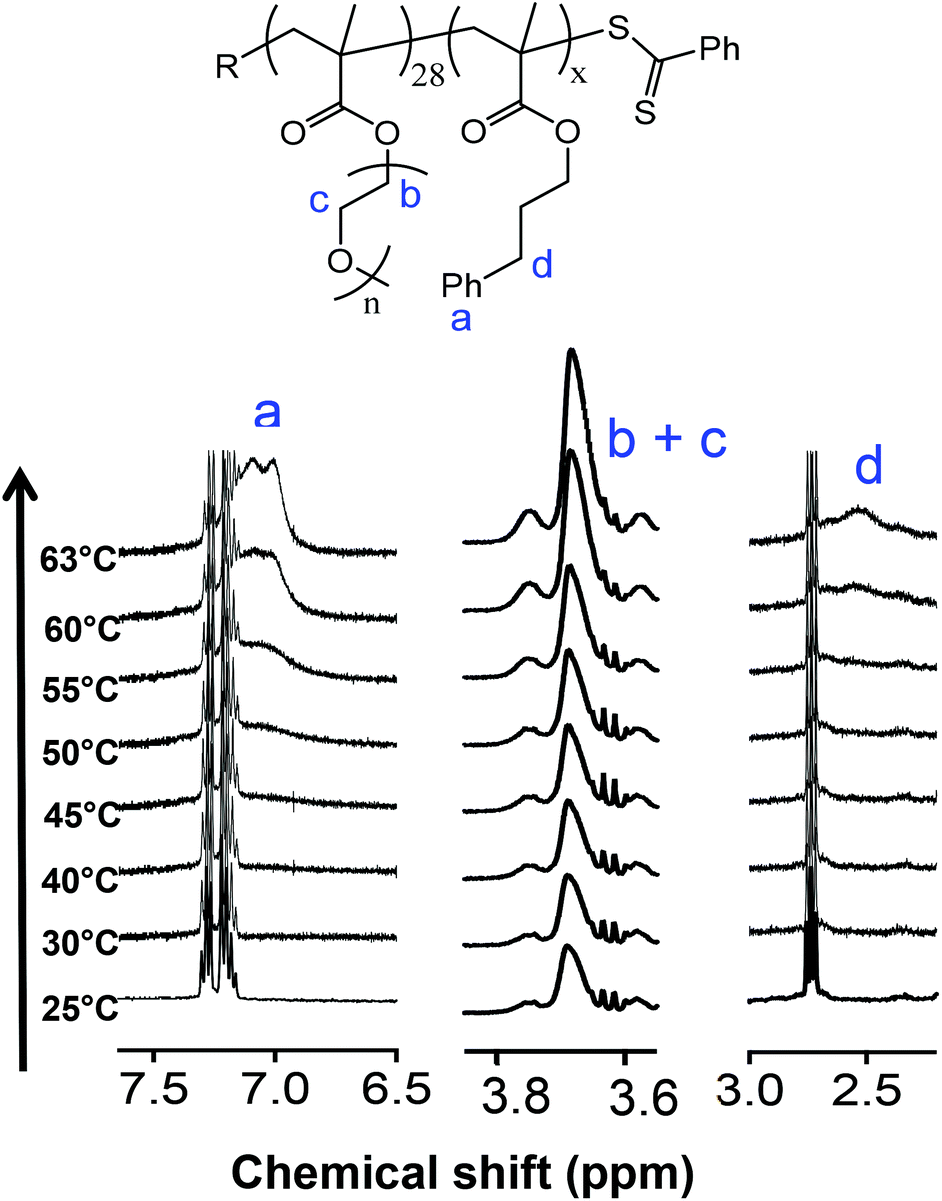 | ||
| Fig. 2 General structure of the p(OEGMA28-block-PPMAx) copolymers (top) and a series of 1H NMR spectra covering the temperature range 25–63 °C (bottom) highlighting the increase in solvation of key functionality associated with both the pOEGMA coronal and pPPMA core blocks with increasing temperature. Spectra were recorded in d4-methanol. | ||
Consider first the signals between δ = 7.5 and 6.5 ppm, labelled a, the range in which signals associated with the Ph group of the pPPMA block would be expected. At low temperature there is little-to-no indication that these aromatic groups are solvated. This is consistent with a solvophobic species in a selective solvent in an assembled state, i.e. the pPPMA blocks residing in a desolvated core of a nanoparticle. Any evidence of a signal associated with the aromatic species remains absent until the temperature reaches ca. 50 °C at which point signals attributed to the Ph side groups begin to appear. The intensity of these signals continues to increase as the temperature is raised further and are relatively pronounced at the final temperature of 63 °C, the critical temperature associated with the order–order morphology transition. This change in solvation of the pPPMA block with increasing temperature is also evident when examining the signals labelled d which are associated with the benzylic groups of the pPPMA block (δ = ca. 3.0–2.0 ppm). However, unlike the Ph signals we do not observe any clear increase in the intensity of these signals until some 10 °C higher than in the case of the aromatic species. While they are present at 63 °C this difference in behaviour suggests a stepwise process for solvation of the pPPMA block. We should also note that while these key signals associated with the pPPMA block are clearly evident at elevated temperature the pPPMA block does not become fully solvated. If this were the case, heating would simply result in molecular dissolution and the disappearance of any aggregates, whether they be worms or spheres. This observed behaviour is consistent, however, with the core of the worm nanoparticles becoming increasingly solvent swollen with increasing temperature as the pPPMA block becomes, at least partially, solvated, and thus induces an overall change in the volume fraction of core-building component. We note that Armes et al.76 have argued that such processes are due to surface plasticization since a significant change in the core volume alone would actually favour a worm-to-vesicle transition (assuming transitions are governed only by those factors defined in the packing parameter) rather than the observed worm-to-sphere morphology change. However, their argument does not consider solvation effects on l (the length of the core forming block), how surface plasticization could directly impact a nor how changes in solvation could impact features such as the dynamic nature of the particles and, therefore, possible changes in physical attributes such as the aggregation number.
Importantly, however, these NMR experiments also indicated a change in the relative solvation of the coronal pOEGMA block with increasing temperature (and presumably an associated change in a), see signals labelled b and c. As such, rationalizing observed morphology changes becomes more difficult since both blocks in this system are undergoing changes in relative solvation as a function of increasing, or decreasing, temperature. While the oligo(ethylene oxide) side chains are solvated at ambient temperature an increase in the temperature also results in an increase in the intensity of the signals associated with these side groups and thus a change in the volume fraction of the coronal chains. These observations are consistent with the reported UCST-type behaviour of pOEGMA in aliphatic alcohols and are discussed and highlighted in more detail below. However, at this point we note that in methanol the actual UCST associated with pOEGMA is below ambient (ca. −10 °C)16 and thus the resulting nanoparticles prepared in methanolic RAFTDP formulations exist as stable dispersions at room temperature.
Organobase responsive properties of p(OEGMA28-block-PPMAx) copolymer nano-objects
While temperature is the most convenient trigger that can be employed for effecting order–order transitions in such soft matter nanoparticles it is also possible to employ alternative stimuli for inducing similar behaviour. Recently, Lovett et al.78 reported the aqueous RAFTDP-PISA synthesis of AB diblock copolymer nanoparticles composed of stabilizing poly(glycerol monomethacrylate) blocks with poly(2-hydroxypropyl methacrylate) as the core forming species utilizing a carboxylic acid functional trithiocarbonate as the RAFT chain transfer agent. The authors reported that the addition of NaOH to a worm nanoparticle phase induced an order–order transition to give spheres while a similar vesicle-to-worm morphology transition could also be induced. Such behaviour was fully reversible and rationalized in terms of the reversible ionization of the peripheral surface bound carboxylic acid functional groups.Since we employed CPADB as the RAFT CTA in our macro-CTA syntheses the final non-ionic p(OEGMA28-block-PPMAx) block copolymer nanoparticles also contained carboxylic acid groups at their periphery. Inspired by the work of Lovett et al.78 we examined the possibility of inducing similar morphology changes in a non-aqueous environment employing the organobase 1,8-diazabicycloundec-7-ene (DBU).
Utilizing the worm nanoparticles formed from p(OEGMA28-block-PPMA69) in methanol we examined the effect of adding increasing amounts of DBU on the nano-object morphology. The 2D TEM image in Fig. 3A shows the parent nanoparticles as a pure worm phase. The addition of 50 equivalents of DBU, based on –CO2H peripheral groups, induced a change from the worm nano-objects with end-to-end distances of multiple hundreds of nm to a mixture containing predominantly ‘oligomeric’ worm species with lengths of ca. 50–150 nm as well as a relatively small population of spheres, Fig. 3B. This change in morphology was also accompanied by a drop in the zeta potential from ca. −20 mV to −30 mV, as measured in methanol, indicating an increase in the anionic surface charge of the nano-objects upon the addition of DBU.
 | ||
| Fig. 3 High resolution TEM images of the nanoparticles formed by p(OEGMA28-block-PPMA69) in MeOH in (A) the absence of DBU; (B) the presence of 50 equivalents of DBU, and; (C) in the presence of 500 equivalents of DBU demonstrating the fundamental change in nano-object morphology with increasing [DBU]. | ||
In the case of adding 500 equivalents of DBU to the worm nanoparticles shown in 3A we observed a more pronounced impact on the nanoparticle morphology, Fig. 3C. The pure worm phase transforms into a near-pure spherical phase although there are still a number of worm nanoparticles remaining.
As noted above, according to Lovett et al.78 such base-induced transitions in non-ionic block copolymer nanoparticles are due to the ionization of the surface carboxylic acid functional groups with ionization leading to increased solvophilicity and a change in the relative volume fractions of the core and coronal blocks and hence the shape change. While such an effect can lead to a lowering of the packing parameter and account for the observed transition, the introduction of surface charge and therefore electrostatic repulsive interactions may also induce a change in aggregation number which could also be, partly, responsible for the morphological transition.
However, regardless of the exact cause for the order–order transition this represents, to the best of our knowledge, the first example in which an organobase has been employed in a non-aqueous environment to effect such a transition.
Thermally responsive properties of p(OEGMA27-block-PPMAx) copolymer nano-objects: reversible, temperature-induced, order–disorder transitions in ethanol exploiting UCST-type behaviour
pOEGMA, and various functional copolymers containing this important building block, have documented UCST-type properties in various aliphatic alcohols although pOEGMA is known to be soluble in methanol above −10 °C.16 In an effort to exploit this interesting reversible phase behaviour and introduce a second thermal response (and the final, third smart characteristic) in p(OEGMA-PPMA) block copolymer nanoparticles prepared by RAFTDP-PISA we synthesized a small series of block copolymers at 20% w/w in ethanol with final compositions spanning the range p(OEGMA27-block-PPMA36) to p(OEGMA27-block-PPMA77), Table 1. Ethanol was chosen for this series of experiments given that pOEGMA has a documented TUCSTcp in this solvent of ca. 22.0 °C.16n) as measured by NMR spectroscopy and SEC, their dispersities (ĐM), observed upper critical solution temperature-type cloud points (TUCSTcp) and the nanoparticle morphology as observed by TEM at elevated temperature
| Compositiona | Solvent | PPMA% conv.a |
NMRna (SECn)b |
Đ
Mb |
Phase separation at RT |
T
UCSTcpc |
TEM @ 70 °C |
|---|---|---|---|---|---|---|---|
| a As determined by 1H NMR spectroscopy. b As measured by size exclusion chromatography in THF at a flow rate of 1.0 mL min−1. c As determined by turbidity measurements using a Crystal16. The cloud point is recorded at the temperature at which the transmittance dropped to 50%. | |||||||
| p(OEGMA27-b-PPMA36) | EtOH | 85 | 15700 (12900) |
1.30 | Y | 42 °C | S |
| p(OEGMA27-b-PPMA45) | EtOH | 74 | 17600 (13800) |
1.36 | Y | 63 °C | S + W |
| p(OEGMA27-b-PPMA56) | EtOH | 77 | 19800 (15800) |
1.38 | Y | 60 °C | S + W |
| p(OEGMA27-b-PPMA77) | EtOH | 90 | 24100 (16600) |
1.56 | Y | >70 °C | Aggregates |
This particular series of experiments were conducted with a pOEGMA macro-CTA with an n of 27 (NMRn = 8400; SECn = 7100; ĐM = 1.05) and was prepared via the homogeneous RAFT polymerization of the monomer with CPADB as the RAFT agent in acetonitrile. Subsequently, the pOEGMA27 macro-CTA was employed in the ethanolic RAFTDP-PISA synthesis of the p(OEGMA27-block-PPMAx) species listed in Table 1.
As anticipated the RAFTDP-PISA syntheses of these AB diblock copolymers, and their associated aggregates, yielded nano-objects that underwent an order–disorder transition with cooling. For example, in the case of the p(OEGMA27-block-PPMA36) species the RAFTDP-PISA synthesis proceeded smoothly but upon cooling a macroscopic phase separation was observed. Indeed, such behaviour was observed for all samples prepared in ethanol although the critical temperature at which phase separation occurred varied with the final p(OEGMA-block-PPMA) copolymer composition. Since this macroscopic phase separation is intimately tied to the UCST properties associated with the pOEGMA block we examined it more closely especially since such behaviour has not previously been reported in pOEGMA-based RAFTDP-PISA formulations.
Utilizing turbidity measurements to determine the UCST-associated cloud-point (TUCSTcp) we tested all samples and examined their phase behaviour in heating and cooling cycles. As a representative example, Fig. 4 shows the heating and cooling curves obtained for the p(OEGMA27-block-PPMA36) copolymer in ethanol at 0.7 wt% at a heating/cooling rate of 0.2 °C min−1.
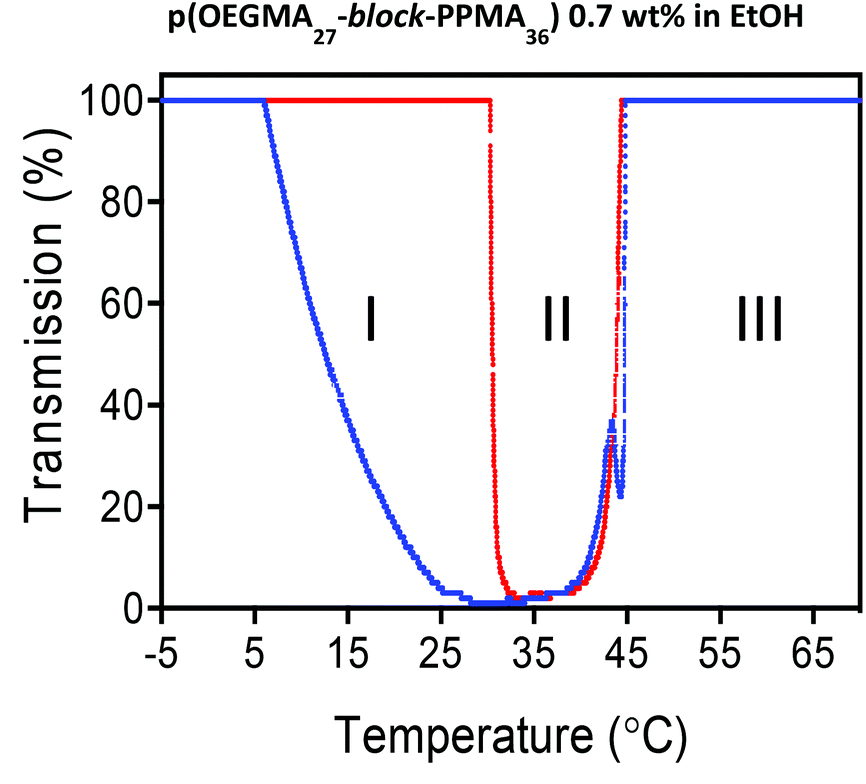 | ||
| Fig. 4 Heating (red) and cooling (blue) turbidity curves for the p(OEGMA27-block-PPMA36) copolymer, measured in EtOH, highlighting the presence of three distinct insolubility/dispersion regimes at 0.7 wt%. | ||
It is possible to identify three distinct regimes in the transmittance as a function of temperature. Consider first the heating profile (red data). At low temperature, from −5 to ca. 30 °C, we observed 100% transmittance (regime I). Under these conditions the block copolymer nanoparticulate species are in the form of a macroscopic precipitate. Just above this upper limit we observe a sharp change in the transmittance with it dropping to essentially zero. This corresponds to the onset of solvation of the pOEGMA block and the dispersion of the phase separated species although the nanoparticles are still in a largely ill-defined aggregated state (regime II). A further increase in the temperature results in another relatively sharp transition at ca. 42 °C with the transmittance returning to 100%. This transition corresponds to the full and efficient dispersion of spherical nanoparticles (as observed by TEM, vide infra) (regime III). The corresponding cooling curve mirrors, essentially perfectly, the heating curve in regimes III and II suggesting an initial rapid desolvation at the TUCSTcp (42 °C) resulting in the formation of ill-defined larger aggregates and observed clouding. This is followed, in regime I, by a more gradual transition over a range of ca. 20 °C ultimately resulting in full macroscopic precipitation. Notably, samples appeared clear in regime I (100% measured transmittance) because macroscopic aggregates precipitated below the optically observed region. Overall this represents a reversible disorder–order transitional process and corresponds to the second distinct thermal response exhibited by p(OEGMA-PPMA) block copolymers in the lower alcohols MeOH and EtOH.
The nature of these nanoparticles and aggregates was further analysed by DLS and final nanoparticle morphology at elevated temperature confirmed by TEM. For comparative purposes, Fig. 5A shows a replot of the heating curve for the p(OEGMA27-block-PPMA36) copolymer from 35 °C to 70 °C (left) with the corresponding hydrodynamic diameters as measured by DLS as a function of temperature with a TEM image of the final nanoparticle morphology at 70 °C shown inset (right).
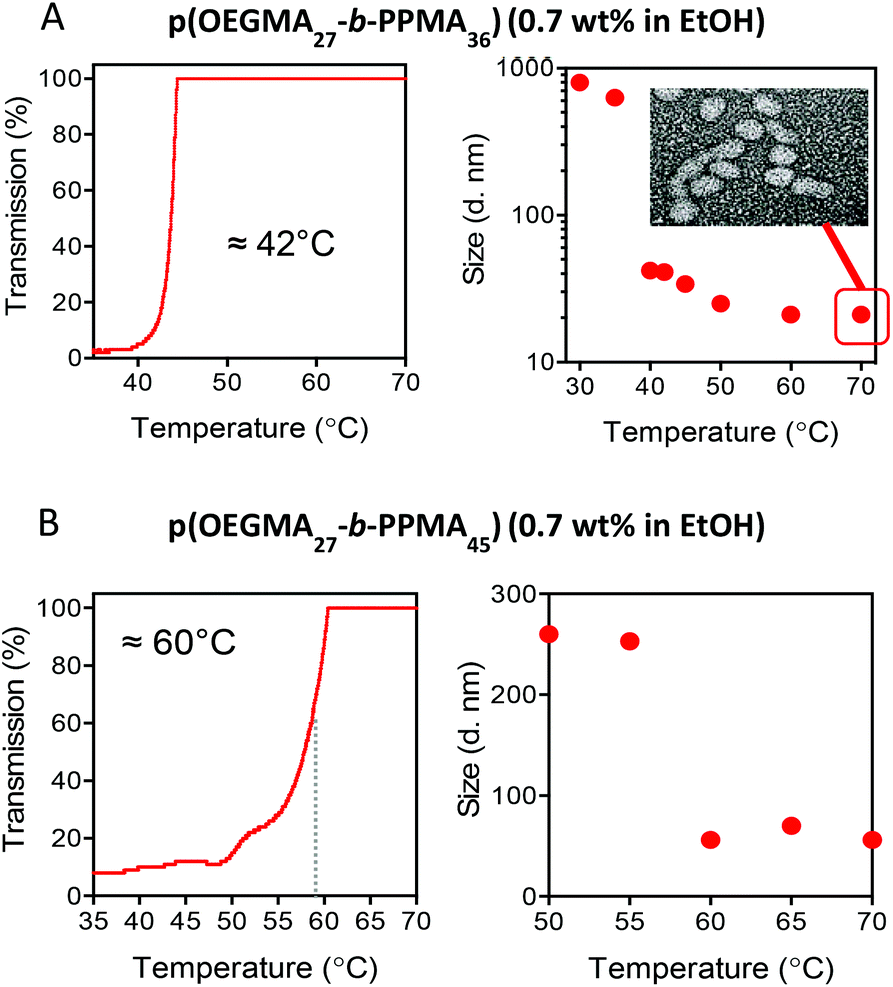 | ||
| Fig. 5 (A) Transmission vs. temperature plot for the heating cycle of the p(OEGMA27-block-PPMA36) copolymer nano-objects highlighting the TUCSTcp in EtOH at a concentration of 0.7 wt% (left) and the corresponding DLS measured change in hydrodynamic diameter as a function of increasing temperature with a TEM of the final nanoparticle morphology shown inset, and (B) the same plots for the p(OEGMA27-block-PPMA45) copolymer that exhibits a broader TUCSTcp transition. | ||
Consider first the DLS data for the p(OEGMA27-block-PPMA36) copolymer, Fig. 5A right. At the onset of general dispersion, at around 30 °C, DLS indicated the presence of aggregates with an average hydrodynamic diameter, Dh, of ca. 800–900 nm. This is consistent with the presence of large ill-defined aggregates of nanoparticles associated with macroscopic phase separation. At the boundary between regimes II and III, at ca. 40 °C, we observed a dramatic drop in the measured Dh to a value of ca. 40.0 nm that continued to decrease with further heating to a final value of just above 20.0 nm at 70 °C. The TEM image shown inset confirms effective redispersion with nano-objects of a predominant spherical morphology clearly present with an average TEM size of ca. 30 nm which agrees reasonably well with the DLS data. To demonstrate that such UCST-type behaviour is uniform for such AB diblock nanoparticles in EtOH, Fig. 5B shows similar data for the p(OEGMA27-block-PPMA45) copolymer (full heating/cooling transmittance vs. temperature plots for the p(OEGMA27-block-PPMA45) and p(OEGMA27-block-PPMA56) copolymers are available in the ESI† along with the measured change in hydrodynamic diameter as a function of temperature for the p(OEGMA27-block-PPMA77) copolymer).
The transmission vs. temperature plot is plotted from the onset of regime II to the beginning of regime III. There are two clear differences between this plot and that in 5A for the p(OEGMA27-block-PPMA36) copolymer. Firstly, the TUCSTcp is approximately 20 °C higher and secondly, the measured transition is rather broad spanning a range of ca. 10 °C, compared to the sharp transition observed at 42 °C for the p(OEGMA27-block-PPMA36) species. The higher TUCSTcp appears to be due to the more solvophobic nature of p(OEGMA27-block-PPMA45) copolymer vs. the p(OEGMA27-block-PPMA36) species and suggests a rather significant influence of the solvophobic, phase separated block on the thermal behaviour of the coronal chains.81,82 Indeed, tuning phase transition temperatures via the introduction of more solvophobic (or solvophilic) comonomers is a well-established approach for controlling such temperature-dependent solubility behaviour. This is also likely the cause of the broader transition with the overall increased solvophobic nature of the nanoparticles resulting in a more gradual resolvation and finally effective redispersion.
The DLS data for the p(OEGMA27-block-PPMA45) block copolymer species is also consistent with the existence of fundamentally different physical regimes, Fig. 5B right. At the boundary between regimes II and III DLS indicated the presence of aggregates with an average Dh of ca. 275 nm – large but significantly smaller than those observed for the p(OEGMA27-block-PPMA36) copolymer. Increasing the temperature from 55 °C to 60 °C resulted in a significant drop in the measured Dh from ca. 250 nm to 50.3 nm. Again, this is entirely consistent with effective redispersion of the spherical nano-objects. The larger, DLS-measured, size for the nanoparticles formed by p(OEGMA27-block-PPMA45) copolymer compared to p(OEGMA27-block-PPMA36) is also consistent with an overall larger Xn of the former block copolymer versus the latter.
While such thermal responsive behaviour may be considered detrimental it is important to note that from a practical viewpoint such UCST-type behaviour offers the potential for facile isolation and storage (in a dry state) of nanoparticles prepared by RAFTDP-PISA prior to redispersion and use in a targeted application in a manner similar to that reported by Kocik et al.83
Finally, to investigate more closely the molecular changes associated with the observed UCST-type behaviour we re-examined the temperature-dependent NMR experiments and performed a variable temperature experiment involving a heating-cooling cycle by first heating the p(OEGMA27-block-PPM36) sample in d6-ethanol from −5 °C to 70 °C with subsequent cooling back to 25 °C. Fig. 6A and B show the general structure of the block copolymer with key chemical components highlighted along with the acquired NMR spectra and a plot of the relative intensities of key signals as a function of temperature. The data in Fig. 6A (heating in d6-ethanol) shows many similarities to the data presented in Fig. 2 for a p(OEGMA28-block-PPMAx) copolymer heated from 25 to 63 °C in d4-methanol. At low temperature (−5 °C) all key resonances are absent with the exception of a signal associated with the pendent CH3 group of the pOEGMA side chains. The turbidity experiments suggested the presence of macroscopic precipitate under these conditions due to a UCST-type transition associated with the coronal pOEGMA chains. The NMR data suggests that even under these conditions of macroscopic phase separation the pOEGMA side chains are not fully desolvated but are clearly not sufficiently solvated to allow for effective dispersion. As the temperature is increased from −5 °C to 35 °C the intensity of the signal associated with the CH3 group also increases (although there is little evidence of any change in other key signals). This is consistent with the behaviour observed in regime I of the turbidity data – increasing solvation of the coronal pOEGMA chains results in an effect dispersion of the nanoparticles. At 45–55 °C we reach essentially full solvation of the coronal pOEGMA species and effective redispersion. This temperature range is consistent with the value of 42 °C identified in the turbidity experiments as the point at which full dispersion is accomplished. It is also during this range of 45–55 °C that signals associated with the inner pPPMA blocks begin to appear and, again, is entirely consistent with the data in Fig. 2 for similar AB diblock copolymers in d4-methanol. What is rather unique about this system however is the dual thermoresponsive nature of both the inner and outer blocks. Recall, the increased solvation of the inner pPPMA blocks can account for order–order transitions, such as worm-to-sphere morphology changes, while the decreased solvation of the outer pOEGMA block is responsible for an order–disorder transition. The reversibility, of this process is demonstrated by the cooling profile. Reducing the temperature from 70 to 25 °C results in a desolvation process for both the inner and outer chains. However, and as expected this is most dramatic for the inner pPPMA block.
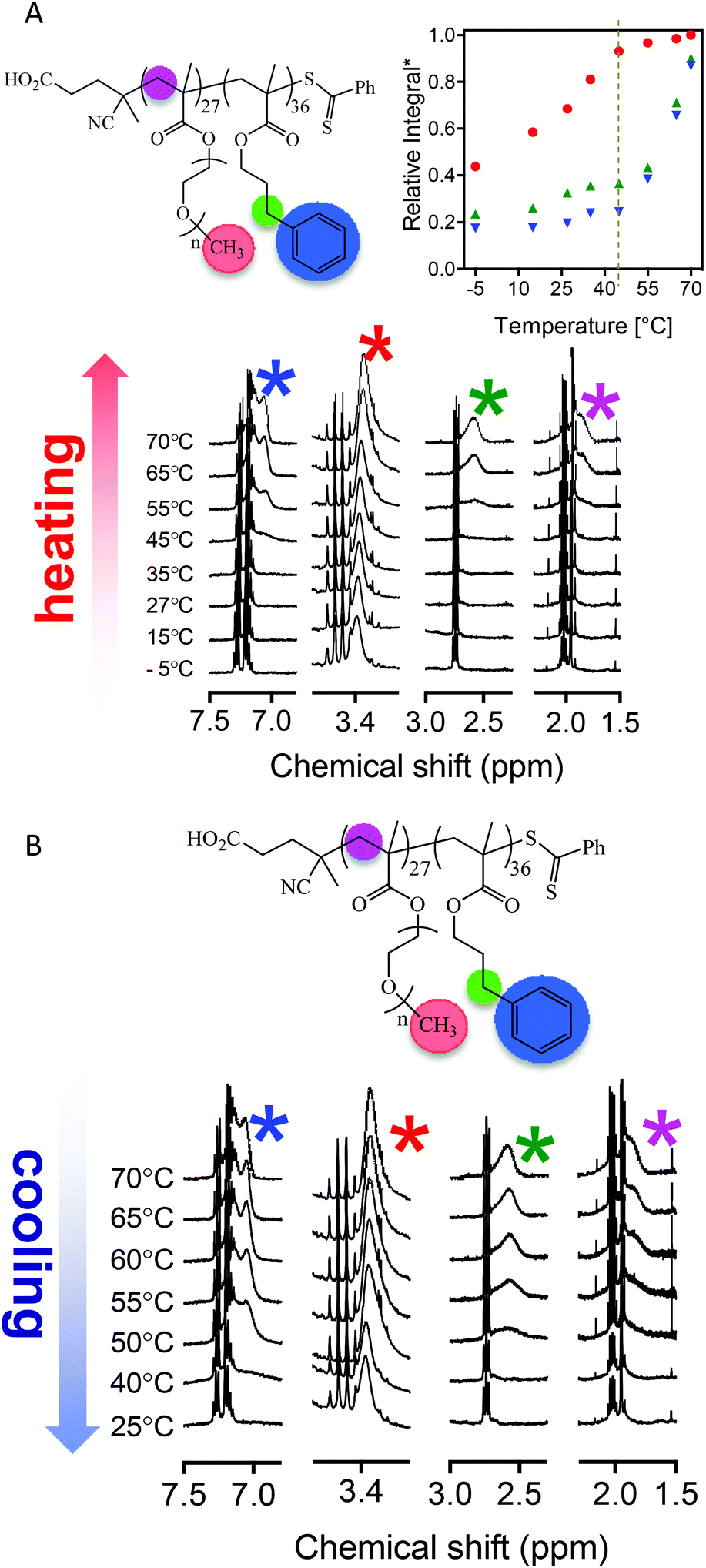 | ||
| Fig. 6 1H NMR spectra of p(OEGMA27-block-PPMA36) recorded in d6-ethanol; (A) heating from −5 °C to 70 °C (inset: relative integral as a function of temperature. The relative integral values are normalized utilizing the absolute integral value of the vinyl proton peaks from residue PPMA monomer based on the assumption that the pOEGMA block is fully solvated at 70 °C), and (B) cooling from 70 °C to 25 °C. | ||
Conclusions
Herein we have described the triply-responsive nature of soft matter nanoparticles based on AB diblock copolymers of poly[oligo(ethylene glycol) methyl ether methacrylate-block-3-phenylpropyl methacrylate]. We have shown that these nanoparticles can exhibit two distinct, fully reversible, thermal responses – a heating induced worm-to-sphere morphological (order–order) transition in methanol and an order–disorder transition based on upper critical solution temperature-based behaviour associated with the poly[oligo(ethylene glycol) methyl ether methacrylate] coronal chains when the nano-objects are dispersed in ethanol. Additionally, given the presence of surface carboxylic acid functional groups (as a result of the RAFT syntheses utilizing an acid-containing chain transfer agent) we have, for the first time, demonstrated an organobase-mediated worm-to-sphere order–order transition. This latter response is complementary to base-mediated transitions in aqueous media reported previously.Acknowledgements
ABL thanks the Australian Research Council (ARC) for funding via a Future Fellowship (FT110100046). Curtin University is acknowledged for funds for support of YP. The authors acknowledge the facilities and the scientific and technical assistance of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy, Characterisation & Analysis, The University of Western Australia, a facility funded by the University, State and Commonwealth Governments.Notes and references
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- J. Rodriguez Hernandez, F. Checot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef CAS.
- S. J. Holder and N. A. J. M. Sommerdijk, Polym. Chem., 2011, 2, 1018–1028 RSC.
- T. Gadt, N. S. Ieong, G. Cambridge, M. A. Winnik and I. Manners, Nat. Mater., 2009, 8, 144–150 CrossRef PubMed.
- Y. Yu and A. Eisenberg, J. Am. Chem. Soc., 1997, 119, 8383–8384 CrossRef CAS.
- G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS.
- M. Moffitt, K. Khougaz and A. Eisenberg, Acc. Chem. Res., 1996, 29, 95–102 CrossRef CAS.
- A. B. Lowe, M. Torres and R. Wang, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5864–5871 CrossRef CAS.
- P. J. Roth, T. P. Davis and A. B. Lowe, Macromolecules, 2012, 45, 3221–3230 CrossRef CAS.
- J. Y. Quek, P. J. Roth, R. A. Evans, T. P. Davis and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 394–404 CrossRef CAS.
- B. Yu, A. B. Lowe and K. Ishihara, Biomacromolecules, 2009, 10, 950–958 CrossRef CAS PubMed.
- V. Butun, F. F. Taktak and C. Tuncer, Macromol. Chem. Phys., 2011, 212, 1115–1128 CrossRef.
- N. A. Plate, T. L. Lebedeva and L. I. Valuev, Polym. J., 1999, 31, 21–27 CrossRef CAS.
- H. Feil, Y. H. Bae, J. DFeijen and S. W. Kim, Macromolecules, 1993, 26, 2496–2500 CrossRef CAS.
- J. Seuring and S. Agarwal, Macromol. Rapid Commun., 2012, 33, 1898–1920 CrossRef CAS PubMed.
- P. J. Roth, F. D. Jochum and P. Theato, Soft Matter, 2011, 7, 2484–2492 RSC.
- P. J. Roth, T. P. Davis and A. B. Lowe, Macromolecules, 2012, 45, 3221–3230 CrossRef CAS.
- P. J. Roth, T. P. Davis and A. B. Lowe, Polym. Chem., 2012, 3, 2228–2235 RSC.
- Q. Zhang and R. Hoogenboom, Prog. Polym. Sci., 2015, 48, 122–142 CrossRef CAS.
- G. Vancoillie, D. Frank and R. Hoogenboom, Prog. Polym. Sci., 2014, 39, 1074–1095 CrossRef CAS.
- S. Abbas, Z. Li, H. Hassan and T. P. Lodge, Macromolecules, 2007, 40, 4048–4052 CrossRef CAS.
- I. LaRue, M. Adam, M. Pitsikalis, N. Hadjichristdis, M. Rubenstein and S. S. Sheiko, Macromolecules, 2006, 39, 309–314 CrossRef CAS.
- A. O. Moughton and R. K. O'Reilly, Chem. Commun., 2010, 46, 1091–1093 RSC.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- Y. T. Li and S. P. Armes, Angew. Chem., Int. Ed., 2010, 49, 4042–4046 CrossRef CAS PubMed.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- S. Sugihara, S. P. Armes, A. Blanazs and A. L. Lewis, Soft Matter, 2011, 7, 10787–10793 RSC.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, J. Am. Chem. Soc., 2011, 133, 15707–15713 CrossRef CAS PubMed.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- P. Chambon, A. Blanazs, G. Battaglia and S. P. Armes, Macromolecules, 2012, 45, 5081–5090 CrossRef CAS.
- M. Semsarilar, E. R. Jones, A. Blanazs and S. P. Armes, Adv. Mater., 2012, 24, 3378–3382 CrossRef CAS PubMed.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- D. Zehm, L. P. D. Ratcliffe and S. P. Armes, Macromolecules, 2013, 46, 128–139 CrossRef CAS.
- M. J. Derry, L. A. Fielding and S. P. Armes, Polym. Chem., 2015, 6, 3054–3062 RSC.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Macromolecules, 2014, 47, 1664–1671 CrossRef CAS.
- G. Zheng and C. Pan, Macromolecules, 2006, 39, 95–102 CrossRef CAS.
- W. Cai, W. Wan, C. Hong, C. Huang and C. Pan, Soft Matter, 2010, 6, 5554–5561 RSC.
- C.-Q. Huang and C.-Y. Pan, Polymer, 2010, 51, 5115–5121 CrossRef CAS.
- W.-M. Wan and C.-Y. Pan, Polym. Chem., 2010, 1, 1475–1484 RSC.
- W.-M. Wan and C.-Y. Pan, Macromolecules, 2010, 43, 2672–2675 CrossRef CAS.
- W.-M. Wan, X.-L. Sun and C.-Y. Pan, Macromol. Rapid Commun., 2010, 31, 399–404 CrossRef CAS PubMed.
- W.-D. He, X.-L. Sun, W.-M. Wan and C.-Y. Pan, Macromolecules, 2011, 44, 3358–3365 CrossRef CAS.
- X. Wang, J. Xu, Y. Zhang and W. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2452–2462 CrossRef CAS.
- M. Dan, F. Huo, X. Zhang, X. Wang and W. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1573–1584 CrossRef CAS.
- F. Huo, X. Wang, Y. Zhang, X. Zhang, J. Xu and W. Zhang, Macromol. Chem. Phys., 2013, 214, 902–911 CrossRef CAS.
- X. Xiao, S. He, M. Dan, Y. Su, F. Huo and W. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3177–3190 CrossRef CAS.
- M. Dan, F. Huo, X. Xiao, Y. Su and W. Zhang, Macromolecules, 2014, 47, 1360–1370 CrossRef CAS.
- C. Gao, Q. Li, Y. Cui, F. Huo, S. Li, Y. Su and W. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2155–2165 CrossRef CAS.
- C. Gao, S. Li, Q. Li, P. Shi, S. A. Shah and W. Zhang, Polym. Chem., 2014, 5, 6957–6966 RSC.
- X. He, Q. Li, P. Shi, Y. Cui, S. Li and W. Zhang, Polym. Chem., 2014, 5, 7090–7099 RSC.
- Q. Li, C. Gao, S. Li, F. Huo and W. Zhang, Polym. Chem., 2014, 5, 2961–2972 RSC.
- S. Li, X. He, Q. Li, P. Shi and W. Zhang, ACS Macro Lett., 2014, 3, 916–921 CrossRef CAS.
- Y. Pei, N. C. Dharsana, J. A. van Hensbergen, R. P. Burford, P. J. Roth and A. B. Lowe, Soft Matter, 2014, 10, 5787–5796 RSC.
- Y. Pei and A. B. Lowe, Polym. Chem., 2014, 5, 2342–2351 RSC.
- Y. Pei, L. Thurairajah, O. R. Sugita and A. B. Lowe, Macromolecules, 2014, 48, 236–244 CrossRef.
- Y. Pei, N. C. Dharsana and A. B. Lowe, Aust. J. Chem., 2015, 68, 939–945 CrossRef CAS.
- Y. Pei, J.-M. Noy, P. J. Roth and A. B. Lowe, Polym. Chem., 2015, 6, 1928–1931 RSC.
- Y. Pei, J.-M. Noy, P. J. Roth and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2326–2335 CrossRef CAS.
- Y. Pei, O. Sugita, L. Thurairajah and A. B. Lowe, RSC Adv., 2015, 5, 17636–17646 RSC.
- M. Zong, K. J. Thurecht and S. M. Howdle, Chem. Commun., 2008, 5942–5944 RSC.
- J. Jennings, M. Beija, A. P. Richez, S. D. Cooper, P. E. Mignot, K. J. Thurecht, K. S. Jack and S. M. Howdle, J. Am. Chem. Soc., 2012, 134, 4772–4781 CrossRef CAS PubMed.
- J. Jennings, M. Beija, J. T. Kennon, H. Willcock, R. K. O'Reilly, S. Rimmer and S. M. Howdle, Macromolecules, 2013, 46, 6843–6851 CrossRef CAS.
- Q. Zhang and S. Zhu, ACS Macro Lett., 2015, 4, 755–758 CrossRef CAS.
- G. Liu, Q. Qiu, W. Shen and Z. An, Macromolecules, 2011, 44, 5237–5245 CrossRef CAS.
- W. Shen, Y. Chang, G. Liu, H. Wang, A. Cao and Z. An, Macromolecules, 2011, 44, 2524–2530 CrossRef CAS.
- G. Liu, Q. Qiu and Z. An, Polym. Chem., 2012, 3, 504–513 RSC.
- X. Shi, M. Miao and Z. An, Polym. Chem., 2013, 4, 1950–1959 RSC.
- Y. Xu, Y. Li, X. Cao, Q. Chen and Z. An, Polym. Chem., 2014, 5, 6244–6255 RSC.
- K. Ma, Y. Xu and Z. An, Macromol. Rapid Commun., 2015, 36, 566–570 CrossRef CAS PubMed.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS PubMed.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2013, 4, 873–881 RSC.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, Soft Matter, 2012, 8, 7753–7767 RSC.
- N. J. Warren and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Doudlas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
- V. J. Cunningham, L. P. D. Ratcliffe, A. Blanazs, N. J. Warren, A. J. Smith, O. O. Mykhaylyk and S. P. Armes, Polym. Chem., 2014, 5, 6307–6317 RSC.
- L. A. Fielding, J. A. Lane, M. J. Derry, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 5790–5798 CrossRef CAS PubMed.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
- J. R. Lovett, N. J. Warren, L. P. D. Ratcliffe, M. K. Kocik and S. P. Armes, Angew. Chem., Int. Ed., 2014, 53, 1279–1283 CrossRef PubMed.
- S. H. Thang, Y. K. Chong, R. T. A. Mayadunne, G. Moad and E. Rizzardo, Tetrahedron Lett., 1999, 40, 2435–2438 CrossRef CAS.
- Y. Pei, K. Jarrett, L. G. Garces, M. Saunders, J. P. Croue, P. J. Roth, C. E. Buckley and A. B. Lowe, RSC Adv., 2016, 6, 28130–28139 RSC.
- Y. Zhu, J.-M. Noy, A. B. Lowe and P. J. Roth, Polym. Chem., 2015, 6, 5705–5718 RSC.
- Y. Zhu, J. Y. Quek, A. B. Lowe and P. J. Roth, Macromolecules, 2013, 46, 6475–6484 CrossRef CAS.
- M. K. Kocik, O. O. Mykhaylyk and S. P. Armes, Soft Matter, 2014, 10, 3984–3992 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py00254d |
| ‡ Current address: Department of Chemistry, Faculty of Engineering and Physical Sciences, University of Surrey, Guildford, GU2 7XH, UK. |
| This journal is © The Royal Society of Chemistry 2016 |