
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of polysarcosine from air and moisture stable N-phenoxycarbonyl-N-methylglycine assisted by tertiary amine base†
Afroditi
Doriti
a,
Sarah M.
Brosnan
a,
Steffen M.
Weidner
b and
Helmut
Schlaad
*c
aMax Planck Institute of Colloids and Interfaces, Department of Colloid Chemistry, Research Campus Golm, 14424 Potsdam, Germany
bFederal Institute for Materials Research and Testing (BAM) – 1.3 Structure Analyses, Richard-Willstätter-Str. 11, 12489 Berlin, Germany
cUniversity of Potsdam, Institute of Chemistry, Karl-Liebknecht-Str. 24-25, 14476 Potsdam, Germany. E-mail: schlaad@uni-potsdam.de
First published on 8th April 2016
Abstract
Polysarcosine (Mn = 3650–20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, Đ ∼ 1.1) was synthesized from the air and moisture stable N-phenoxycarbonyl-N-methylglycine. Polymerization was achieved by in situ transformation of the urethane precursor into the corresponding N-methylglycine-N-carboxyanhydride, when in the presence of a non-nucleophilic tertiary amine base and a primary amine initiator.
000 g mol−1, Đ ∼ 1.1) was synthesized from the air and moisture stable N-phenoxycarbonyl-N-methylglycine. Polymerization was achieved by in situ transformation of the urethane precursor into the corresponding N-methylglycine-N-carboxyanhydride, when in the presence of a non-nucleophilic tertiary amine base and a primary amine initiator.
Poly(amino acids)s or polypeptides are considered as biocompatible, biodegradable, and can exhibit stimuli-responsive features,1–3 which makes them ideal candidates for advanced applications in targeted drug delivery,4–6 gene therapy,7 and tissue engineering.8 Synthetic polypeptide materials are most commonly prepared by ring-opening polymerization (ROP) of α-amino acid N-carboxyanhydrides (NCAs)9 or by polycondensation.10,11 The ROP of NCAs produces well-defined homo- and co-polypeptides with predictable molar mass and low dispersity; however, the wide spread adoption has been limited due in large part to the initial synthesis of the monomer. The production of the NCA monomers requires the use of toxic and harmful chemicals, i.e., phosgene and its derivatives, and also the purification and handling of the extremely moisture-sensitive NCA is tedious and difficult and subsequently quite costly.
To address these limitations, Endo and co-workers introduced a “greener” method to make polypeptides through the use of activated urethane derivatives of protected α-amino acids, which are in situ transformed into the corresponding NCA and polymerized when heated or in the presence of an initiator.12 The urethane derivative can be obtained from bis(aryl)carbonate instead of phosgene and is far more stable – even storable in air at room temperature – and easier to handle than the NCA monomer. Importantly, the polymerization occurs preferably via the NCA (not by polycondensation of the urethane precursor) in highly polar solvents, e.g., N,N-dimethylformamide (DMF) or dimethyl sulfoxide (DMSO), and is accelerated by the presence of amines.13,14 Although the mechanism of the reaction appears to be rather complex, it was successfully applied to the controlled synthesis of a variety of polypeptides.15–20
To date, this method has only been applied for the polymerization of activated urethane derivatives of α-amino acids, the only exception being N-(4-nitrophenoxycarbonyl)-L-proline).15 Polymerization of the secondary amine proline derivative implies that the in situ transformation into the NCA does not occur via an isocyanate intermediate,21 hence the method should be generally applicable to α-amino acid derivatives including N-alkylated amino acids. Poly(N-alkylglycine)s, known as polypeptoids,22 are an emerging class of polypeptides with similar beneficial and widely tunable properties, but have the added benefit of better solubility due to the lack of hydrogen bonding. Polypeptoids are typically prepared by primary amine-initiated ROP of their respective N-alkylglycine NCAs, which are even more susceptible to degradation with atmospheric moisture than α-amino acid NCAs.23
Herein, we report on the extension of the activated urethane derivative method to the simplest of the N-alkylglycines, i.e., N-methylglycine or sarcosine (Sar). The outline of the process is given in Scheme 1.
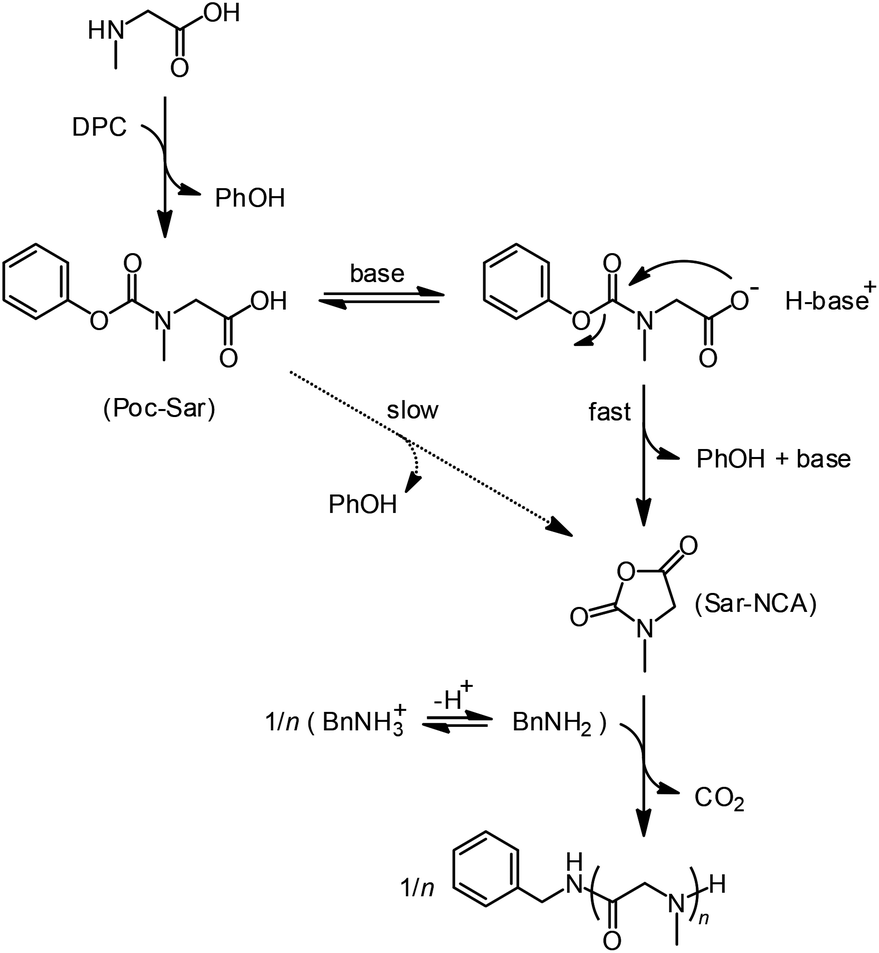 | ||
| Scheme 1 Synthesis of polysarcosine through in situ synthesis of N-methylglycine-NCA from an activated urethane precursor in the presence of tertiary amine base and primary amine initiator. | ||
N-Phenoxycarbonyl-sarcosine (Poc-Sar) was chosen as the activated urethane precursor because phenoxy is considered as a sufficiently good and so far most atom economic leaving group. Introducing electron withdrawing groups on the aromatic ring would increase the reactivity, but would decrease the atom efficiency and potentially make it less stable. Poc-Sar was prepared by the two-step reaction of sarcosine with tetrabutylammonium hydroxide and diphenyl carbonate (DCP)14 with a yield of 49%. The reaction could be simplified by reacting the sarcosine with KOH/LiCl and DCP to give the Poc-Sar with a similar yield of 52% (see ESI†).
For polymerization of the Poc-Sar, we first had to induce the intramolecular condensation of the Poc-Sar into the corresponding Sar-NCA, which was monitored by 1H NMR spectroscopic analysis of aromatic protons of the released phenol (PhOH) co-product (Scheme 1). Initially, the reaction was conducted at ∼13 wt% (∼0.77 M) in DMSO-d6 solution at room temperature and 60 °C. After 1 day, no reaction was observed at room temperature and at 60 °C the conversion rose to only ∼5%. Evidently, the condensation of Poc-Sar requires elevated temperature, but still the reaction rate is very low even in a highly polar solvent like DMSO.
In order to accelerate the condensation of the Poc-Sar into the Sar-NCA, we considered to use a strong, non-nucleophilic tertiary amine base, such as triethylamine (TEA) (conjugated acid in DMSO: pKa 9.0) or diisopropylethylamine (DIPEA, Hünig's base) (pKa 8.5).24,25 The amine base would deprotonate the Poc-Sar and transform the carboxylic acid group into a more nucleophilic carboxylate. Ideally, the tertiary amine base does not initiate an uncontrolled polymerization of the Sar-NCA (which is a cyclic tertiary amide without acidic protons), which can only happen with an α-amino acid NCA (deprotonation of the NCA secondary amide and subsequent polymerization via the activated monomer mechanism).13,26 Also, the released PhO− should be readily protonated (PhOH, pKa 18.0 (DMSO))25 by Poc-Sar or by the protonated amine base, leading to a regeneration of the amine base (Scheme 1).
The intramolecular condensation of Poc-Sar in the presence of different amounts of TEA (0.2, 0.5, 1.0, and 2.0 equiv. with respect to Poc-Sar) in DMSO-d6 at 60 °C was monitored by 1H NMR spectroscopy; results are shown in Fig. 1. FT-IR analyses confirmed the transformation of Poc-Sar into Sar-NCA, by the two characteristic vibration bands (NCA, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch) at
O stretch) at ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 1775 and 1850 cm−1 (ESI†).26 As expected, the reaction was dramatically accelerated by the presence of TEA, and quantitative conversion of the Poc-Sar could be achieved within less than 8 hours (≥0.5 equiv. TEA) or 14 hours (0.2 equiv. TEA).
= 1775 and 1850 cm−1 (ESI†).26 As expected, the reaction was dramatically accelerated by the presence of TEA, and quantitative conversion of the Poc-Sar could be achieved within less than 8 hours (≥0.5 equiv. TEA) or 14 hours (0.2 equiv. TEA).
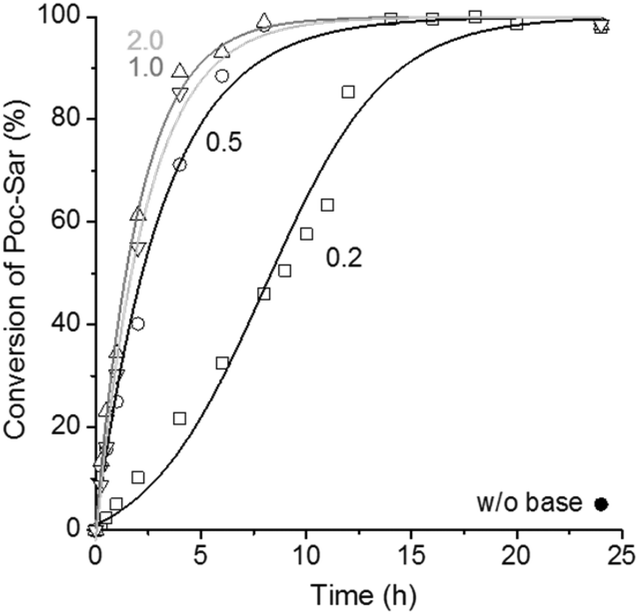 | ||
| Fig. 1 Conversion of Poc-Sar (by quantitative 1H NMR analysis of released PhOH; [Poc-Sar]0 = 0.77 M) in the absence and presence of tertiary amine base (TEA; 0.2, 0.5, 1.0, and 2.0 equiv.) in DMSO-d6 at 60 °C. Lines are to guide the eye. | ||
Next, the polymerization of Poc-Sar was performed using benzylamine (BnNH2) as the primary amine initiator in the presence of catalytic amounts of tertiary amine base, DIPEA (or TEA), i.e., 2 vol% in DMSO (0.16 equiv. with respect to Poc-Sar). The amount of base was reduced to 2 vol% because the tertiary amine, unlike the released PhOH, appeared to be a poor solvent for polysarcosine.
For the first polymerization attempt (entry 1 in Table 1), Poc-Sar (1 equiv.) was dissolved in DMSO ([Poc-Sar]0 = 0.77 M) and then the BnNH2 (0.02 equiv.) and DIPEA (0.16 equiv.) were added at room temperature. The mixture was then heated to 60 °C and stirred for 24 h. The product was precipitated in acetone, dried in vacuum (isolated yield: 75%), and analyzed by 1H NMR spectroscopy (Mn = 3650 g mol−1; end group analysis), size exclusion chromatography (SEC) (Mappn = 4580 g mol−1, Đapp = 1.11), and MALDI-TOF mass spectrometry (Mp = 3200 g mol−1, Fig. 2). 1H NMR and MALDI-TOF MS identified the product as polysarcosine (mass of repeat unit, m(Sar)–m(H2O) = (89.1–18.0) Da = 71.1 Da) carrying α-BnNH and ω-H end groups (Fig. 2). Evidently, the polymerization was initiated by the BnNH2 (and not by DIPEA or PhOH27) and proceeded via the in situ formed Sar-NCA (and not via the Poc-Sar through polycondensation) as depicted in Scheme 1. Furthermore, the number-average molar mass of the polysarcosine was very close to the calculated molar mass, and the molar mass distribution was monomodal and narrow (Table 1).
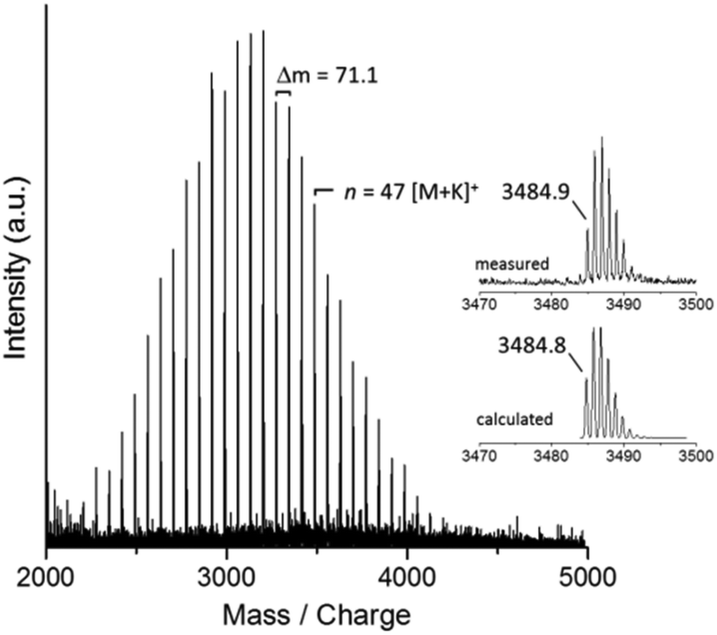 | ||
| Fig. 2 MALDI-TOF mass spectrum of polysarcosine (K+ adducts) as prepared by Poc-Sar/BnNH2/DIPEA/DMSO at 60 °C (entry 1 in Table 1). | ||
| Entry | Base | n |
M
n
calb (g mol−1) |
Conv.c (%) |
M
nd (g mol−1) |
Đ
appe |
|---|---|---|---|---|---|---|
| a n = [Poc-Sar]0/[BnNH2]0, calculated average degree of polymerization. b Calculated average molar mass, Mcaln = (n × 71.1 + 107.2) g mol−1. c Conversion of Poc-Sar, by 1H NMR. d Number-average molar mass, by 1H NMR (600 MHz, D2O) end group analysis. e Apparent dispersity index, by SEC (eluent: 0.058 M LiBr in N-methyl-2-pyrrolidone (NMP), 60 °C, polystyrene calibration). f See ESI. | ||||||
| 1 | DIPEA | 50 | 3660 | >99 | 3650 | 1.11 |
| 2 | TEA | 68 | 4940 | >99 | 4440 | 1.11 |
| 3 | DIPEA | 96 | 6930 | >99 | 7780 | 1.10 |
| 4 | DIPEA | 194 | 13900 |
>99 | 16500 |
1.08 |
| 5 | DIPEA | 307 | 21940 |
>99 | 20000 |
(1.2)f |
It is important to note that initially the BnNH2 should be protonated (BnNH3+), by reaction with the carboxylic acid group of Poc-Sar ([COOH]0 > [amine]0), thus is non-nucleophilic. Transformation of the Poc-Sar into the Sar-NCA, aided by the tertiary amine base, decreases the carboxylic acid content and causes a continuous shift of the primary ammonium–amine equilibrium (Scheme 1) toward BnNH2, which then can attack the Sar-NCA and initiate the polymerization.26,28,29 Since the propagating chain end is a secondary amine, the polymerization rate should also be affected by the ammonium–amine equilibrium (actually, the rate should increase during polymerization). However, as long as the ammonium–amine exchange is faster than monomer addition, the resulting polysarcosine should still have a narrow molar mass distribution.30,31
A series of polysarcosines with different chain lengths (n = 50–307; Mcaln = 3660–21940 g mol−1) were then prepared from Poc-Sar ([Poc-Sar]0 = 0.77 M) in 2 vol% DIPEA (or TEA) in DMSO; results are summarized in Table 1 (entries 1–5). Always full conversion of Poc-Sar was achieved within 24 h (1H NMR), and the products did not contain residual Sar-NCA (FT-IR) (yields: n/d, except 75% for entry 1). The number-average molar masses (Mn; 1H NMR) of the polysarcosines were close to the calculated values (usually within ±10% error) and the molar mass distributions were narrow (Đapp ∼ 1.1; SEC) (see ESI†), indicating that the polymerization proceeded in a well-controlled manner.
In summary, we demonstrated the successful preparation of polysarcosine (Mn = 3650–20000 g mol−1, Đ ∼ 1.1) from the stable Poc-Sar precursor, which was in situ transformed into Sar-NCA monomer, using primary amine as the initiator in a 2 vol% solution of a tertiary amine base (DIPEA or TEA) in DMSO at 60 °C. The intramolecular condensation of Poc-Sar and subsequent polymerization of the Sar-NCA are accelerated by the non-nucleophilic tertiary amine base, due to the deprotonation (activation) of the Poc-Sar and shifting of the ammonium–amine equilibrium toward the amine.
Current work is devoted to a more detailed mechanistic and kinetic analysis of the process, which actually is a two-step cascade reaction involving acid–base equilibria, and application to other stable urethane precursors (variation of the N-alkyl chain and leaving group) and the synthesis of (block) copolypeptoids.
Boonya Thongrom, Sascha Prentzel, Angela Krtitschka (UP), and Marlies Gräwert (MPI-KG) are thanked for their valuable contributions to this work. Financial support was given by the Max Planck Society and the University of Potsdam. S. M. B acknowledges support by the National Science Foundation Postdoctoral Research Fellowship in Biology under Grant No. DBI-1308104.
Notes and references
- T. J. Deming, Prog. Polym. Sci., 2007, 32, 858–875 CrossRef CAS
.
- G. J. M. Habraken, A. Heise and P. D. Thornton, Macromol. Rapid Commun., 2012, 33, 272–286 CrossRef CAS PubMed
- J. Huang and A. Heise, Chem. Soc. Rev., 2013, 42, 7373–7390 RSC
- A. Carlsen and S. Lecommandoux, Curr. Opin. Colloid Interface Sci., 2009, 14, 329–339 CrossRef CAS
- I. W. Hamley, Soft Matter, 2011, 7, 4122–4138 RSC
- H. Cabral and K. Kataoka, J. Controlled Release, 2014, 190, 465–476 CrossRef CAS PubMed
- L. Yin, H. Tang, K. H. Kim, N. Zheng, Z. Song, N. P. Gabrielson, H. Lu and J. Cheng, Angew. Chem., Int. Ed., 2013, 52, 9182–9186 CrossRef CAS PubMed
- D. L. Nettles, A. Chilkoti and L. A. Setton, Adv. Drug Delivery Rev., 2010, 62, 1479–1485 CrossRef CAS PubMed
- H. R. Kricheldorf, Angew. Chem., Int. Ed., 2006, 45, 5752–5784 CrossRef CAS PubMed
- D. Chow, M. L. Nunalee, D. W. Lim, A. J. Simnick and A. Chilkoti, Mater. Sci. Eng., R, 2008, 62, 125–155 CrossRef PubMed
- M. Rodriguez-Garcia, A. J. Surman, G. J. T. Cooper, I. Suarez-Marina, Z. Hosni, M. P. Lee and L. Cronin, Nat. Commun., 2015, 6, 8385 CrossRef CAS PubMed
- Y. Fujita, K. Koga, H.-K. Kim, X.-S. Wang, A. Sudo, H. Nishida and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5365–5370 CrossRef CAS
- Y. Kamei, A. Sudo and T. Endo, Macromolecules, 2008, 41, 7913–7919 CrossRef CAS
- S. Yamada, K. Koga and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2527–2532 CrossRef CAS
- Y. Kamei, A. Sudo, H. Nishida, K. Kikukawa and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2525–2535 CrossRef CAS
- S. Yamada, K. Koga, A. Sudo, M. Goto and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3726–3731 CrossRef CAS
- S. Yamada, S. Atsushi, M. Goto and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2013, 53, 4565–4571 Search PubMed
- S. Yamada, A. Sudo, M. Goto and T. Endo, RSC Adv., 2014, 4, 29890–29896 RSC
- S. Wong and Y. J. Kwon, J. Polym., Sci. Part A: Polym. Chem., 2015, 53, 280–286 CrossRef CAS
- Z. Yang, Z. Mao and J. Ling, Polym. Chem., 2016, 7, 519–522 RSC
- K. W. Ehler and L. E. Orgel, Biochim. Biophys. Acta, 1976, 434, 233–243 CrossRef CAS
- N. Gangloff, J. Ulbricht, T. Lorson, H. Schlaad and R. Luxenhofer, Chem. Rev., 2016, 116, 1753–1802 CrossRef CAS PubMed
- F. Wessely, K. Riedl and H. Tuppy, Monatsh. Chem., 1950, 81, 861–872 CrossRef CAS
- S. D. Lepore, A. Khoram, D. C. Bromfield, P. Cohn, V. Jairaj and M. A. Silvestri, J. Org. Chem., 2005, 70, 7443–7446 CrossRef CAS PubMed
- http://www.chem.wisc.edu/areas/reich/pkatable/ .
- C. D. Vacogne and H. Schlaad, Chem. Commun., 2015, 51, 15645–15648 RSC
- B. A. Chan, S. Xuan, M. Horton and D. Zhang, Macromolecules, 2016, 49, 2002–2012 CrossRef CAS
- C. Fetsch, A. Grossmann, L. Holz, J. F. Nawroth and R. Luxenhofer, Macromolecules, 2011, 44, 6746–6758 CrossRef CAS
- N. Gangloff, C. Fetsch and R. Luxenhofer, Macromol. Rapid Commun., 2013, 34, 997–1001 CrossRef CAS PubMed
- I. Dimitrov and H. Schlaad, Chem. Commun., 2003, 2944–2945 RSC
- J.-F. Lutz, D. Schütt and S. Kubowicz, Macromol. Rapid Commun., 2005, 26, 23–28 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Analytical instrumentation and methods, chemicals, experimental procedures, and additional analytical data (1H/13C NMR, FT-IR, MALDI-TOF MS, and SEC). See DOI: 10.1039/c6py00221h |
| This journal is © The Royal Society of Chemistry 2016 |