
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling molecular weight and polymer architecture during the Passerini three component step-growth polymerization†
S.
Oelmann
,
S. C.
Solleder
and
M. A. R.
Meier
*
Karlsruhe Institute of Technology (KIT), Institute of Organic Chemistry (IOC), Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany. E-mail: m.a.r.meier@kit.edu
First published on 20th January 2016
Abstract
A new approach to control the molecular weight and polymer architecture using the Passerini three-component step-growth polymerization is described. Starting from an AB-type monomer, linear homopolymers, diblock copolymers, as well as star-shaped polymers were synthesized in an efficient manner. By varying the ratio of the AB-type monomer and a suitable irreversible chain transfer agent (ICTA), different polymer architectures with specific molecular weights and high end-group fidelity were obtained.
In order to obtain better control over the material properties of polymers, it is essential to synthesize polymers with well-defined structures.1 Usually, living/controlled polymerization techniques, such as RAFT (reversible addition-fragmentation chain transfer) or ATRP (atom transfer radical polymerization), which are the most extensively studied controlled radical polymerization techniques, or anionic polymerization are used to prepare well-defined macromolecular architectures.2 However, well-defined polymers were also synthesized via polycondensation reactions by using monomers that selectively react with the polymer chain ends.3–5 For instance, our group synthesized defined macromolecular architectures using head-to-tail ADMET (acyclic diene metathesis) polymerization.6,7 This synthesis procedure was accomplished by taking advantage of the different reactivities of terminal double bonds and acrylates in olefin metathesis reactions.8
Recently, multicomponent reactions (MCRs) have been introduced to the field of polymer science as straightforward polymerization technique with a step-growth character.9 This type of reaction is performed with more than two starting materials in one-pot and leads to functional materials in a straightforward fashion. In polymer chemistry, isocyanide-based multicomponent reactions (IMCRs), such as the Passerini three-component reaction (Passerini-3CR), are often used.10,11 For instance, by using bifunctional components, a polymerization process is induced and substituted polyesters, polyamides, or poly(ester amide)s are obtained.12–14 Furthermore, with AB-type bifunctional monomers, it is possible to perform the reaction with only two components, which leads to an easier and more efficient polymerization process.15,16 It is also possible to synthesize multi-block copolymers with sequence-ordered sidegroups.17 Moreover, several applications of metal-catalyzed MCRs are described in polymer chemistry.18,19
This work focusses on the synthesis of well-defined polymer architectures employing the Passerini-3CR as polymerization technique. The approach relies on the use of a monocarboxylic acid as an irreversible chain transfer agent (ICTA) in combination with bifunctional monomer 3 and an isocyanide to achieve control over the molecular weight (Scheme 1).
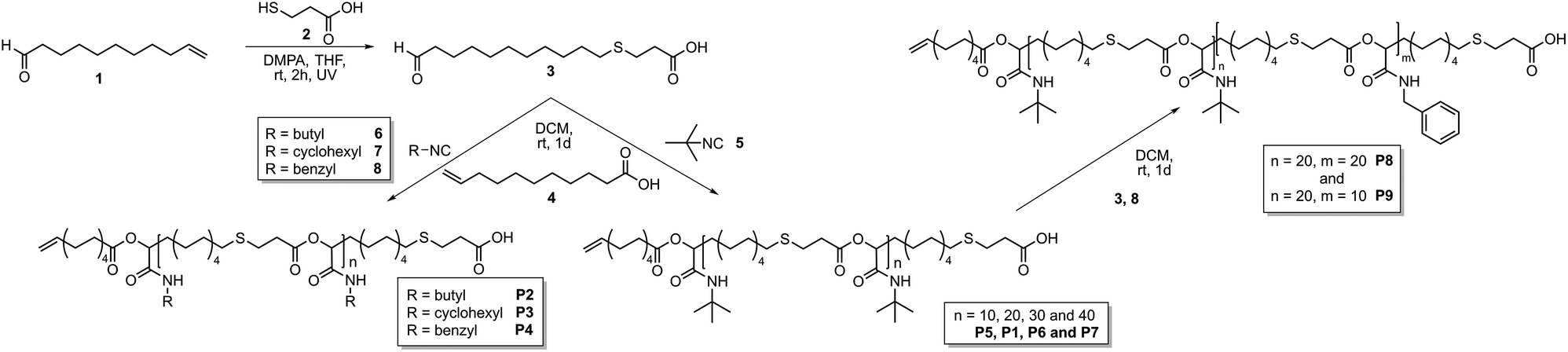 | ||
| Scheme 1 Synthesis of AB-type monomer 3, subsequent polymerization with different isocyanides 5–8 (left) and subsequent block copolymer formation (right). | ||
The thus resulting carboxylic acid end-group subsequently allows the synthesis of block copolymers. Moreover, the use of a tricarboxylic acid as core unit should result in the formation of star-shaped homo- and copolymers. Thus, an AB-type monomer, containing a carboxylic acid and an aldehyde unit, was prepared (3, Scheme 1) by a thiol–ene reaction of 10-undecenal 1 and 3-mercaptopropionic acid 2.15 This type of AB-monomer was chosen for practical reasons discussed previously.15
The Passerini polymerization of monomer 3 was then performed in dichloromethane (DCM) at room temperature using four different isocyanides (5–8, Scheme 1) with a ratio of monomer 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :isocyanide 5–8:10-undecenoic acid 4 of 20:100:1. 10-Undecenoic acid 4 served as ICTA. Molecular weights of the obtained polymers were measured by SEC and additionally calculated from 1H NMR data. If this process proceeds as anticipated, monomer 3 as well as the formed oligomers of 3 and the respective isocyanide would add irreversibly to 4 (and later on growing polymer chains) until all monomer is consumed and the molecular weight would be predetermined by the ratio of 3:4, comparable to the monomer:initiatior ratio in controlled/living polymerizations. Indeed, for all investigated isocyanides 5–8, the expected molecular weights (formula S1†) corresponded well to the actual values determined by 1H NMR integration of the end group signal and the signals of the repeating units (Table S1†). SEC analysis revealed higher molecular weights than expected, which is due to the different hydrodynamic volumes compared to the linear PMMA calibration standards. The thermal properties of the polyesters were determined by DSC, which revealed an amorphous behaviour with glass transitions (Tg) ranging from −11 °C to 5 °C, depending on the isocyanide.
:isocyanide 5–8:10-undecenoic acid 4 of 20:100:1. 10-Undecenoic acid 4 served as ICTA. Molecular weights of the obtained polymers were measured by SEC and additionally calculated from 1H NMR data. If this process proceeds as anticipated, monomer 3 as well as the formed oligomers of 3 and the respective isocyanide would add irreversibly to 4 (and later on growing polymer chains) until all monomer is consumed and the molecular weight would be predetermined by the ratio of 3:4, comparable to the monomer:initiatior ratio in controlled/living polymerizations. Indeed, for all investigated isocyanides 5–8, the expected molecular weights (formula S1†) corresponded well to the actual values determined by 1H NMR integration of the end group signal and the signals of the repeating units (Table S1†). SEC analysis revealed higher molecular weights than expected, which is due to the different hydrodynamic volumes compared to the linear PMMA calibration standards. The thermal properties of the polyesters were determined by DSC, which revealed an amorphous behaviour with glass transitions (Tg) ranging from −11 °C to 5 °C, depending on the isocyanide.
Delighted by these results, the polymerization behaviour was further investigated by a step-wise addition of monomer 3 in portions of five equivalents. After the addition of each batch of fresh monomer, the reaction was allowed to proceed until no further change in molecular weight was observed by SEC (Fig. 1); longer times to reach a constant molecular weight were required after each addition (i.e. with higher molecular weight). Most importantly, we could clearly show by this experiment that the polymer chain grows until all monomer is consumed and that the polymerization can be continued, i.e. the chain extended, by simply adding additional amounts of monomer 3. Indeed, two hours after the addition of the first five equivalents of monomer (Fig. 1b), 1H NMR analysis of the crude reaction mixture showed full conversion of the aldehyde signal (Fig. S13†).
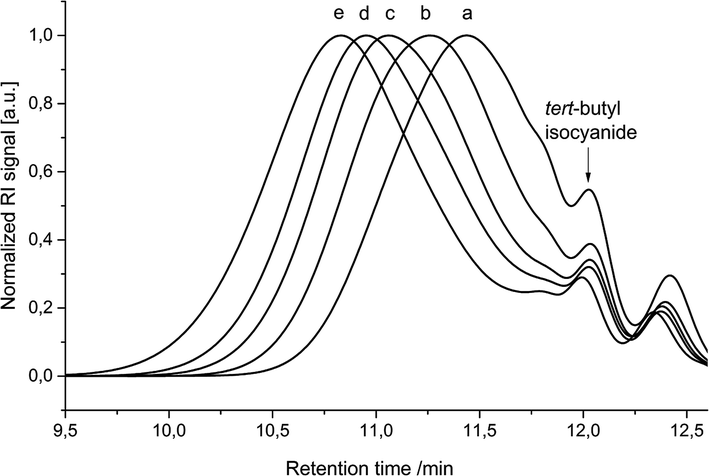 | ||
| Fig. 1 SEC chromatogram showing the growth of polyester P1 due to addition of 5 equivalents of monomer 3 after specific time periods measured from reaction mixture; (a = first (0 h), b = second (2 h), c = third (4.5 h), d = fourth addition (7.5 h), e = 24 h). | ||
To further demonstrate the control over the molecular weight using this method, different ratios of AB-type monomer 3 and ICTA 4 (10:1 to 40:1) were investigated (Fig. S1†). It was shown that the molecular weights of the resulting polymers calculated from 1H NMR data were in good agreement with the expected molecular weights (Table 1). Using a ratio of 3:4 of 40:1, a further addition of isocyanide 5 after 24 hours was necessary to obtain full conversion. A reason could be the increasing viscosity of the reaction mixture upon reaching higher molecular weights. By diluting the mixture with additional isocyanide 5, viscosity decreased while reactivity increased, leading to full conversion. Each linear homopolymer P1–P7 was characterized via mass spectroscopy (Fig. S14–S19†), showing the expected number of repeating units n as well as the expected theoretical masses, respectively. Unfortunately, proper ionization of P4 was not possible.
| Polymer | Ratio | M n calc. [g mol−1] | M n NMR [g mol−1] | M n SEC [g mol−1] | Đ |
|---|---|---|---|---|---|
| [3]:[4] |
M w/Mn | ||||
| P5 | 10:1 |
3760 | 4146 | 9100 | 1.60 |
| P1 | 20:1 |
7335 | 7377 | 12000 |
1.51 |
| P6 | 30:1 |
10911 |
10997 |
14200 |
1.40 |
| P7 | 40:1 |
14486 |
14515 |
15700 |
1.37 |
Starting from homopolymer P1, a chain extension by a second Passerini polymerization was performed with benzyl isocyanide 8 under the same reaction conditions yielding diblock copolymers P8 and P9 by the use of different ratios (monomer 3:homopolymer P1 20:1 (P8) and 10:1 (P9)). 1H NMR analysis of the resulting polymers P8 and P9 showed an excellent correlation of the double bond end group signals and the signals of the two different blocks. Symmetric molecular weight distribution, showing the presence of an unique block copolymer and full monomer conversion were observed by SEC analysis (Fig. S2†). The molecular weights calculated from 1H NMR data fit to the expected molecular weights of P8 (15300 g mol−1) and P9 (11300 g mol−1), respectively.
To extend this study, ICTA 4 was replaced by a trifunctional core unit 9, which should result in the formation of a star-shaped homopolymer and subsequently a star-shaped copolymer (Scheme 2). The Passerini polymerization was performed with tert-butyl isocyanide 5 and AB-type monomer 3 with a ratio of monomer 3:isocyanide 5:trimesic acid 9 of 60:300:1.
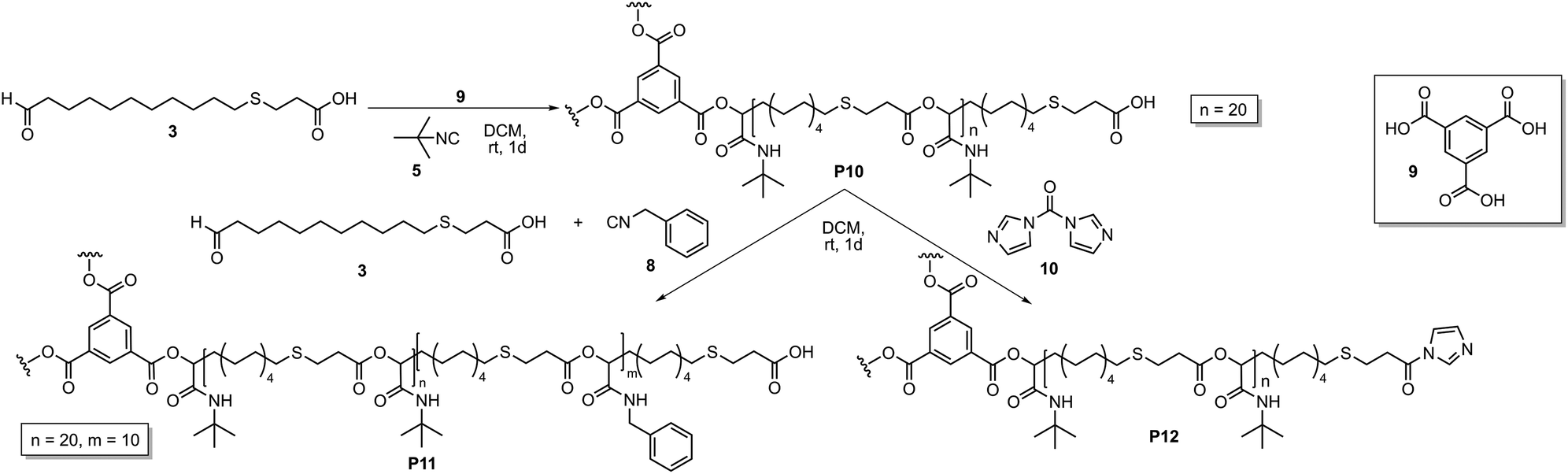 | ||
| Scheme 2 Synthesis of star-shaped homopolymer P10, subsequent copolymerization with benzyl isocyanide 8 (left) and activation of the end group with CDI 10 (right). | ||
Subsequent SEC analysis revealed this time lower molecular weights than the theoretical calculation, which is typical for star-shaped polymers (Fig. S3†). 1H NMR analysis of star-shaped homopolymer P10 indicated complete conversion of the ICTA by a slight shift of the aromatic protons a of the core unit compared to the protons of trimesic acid 9 (Fig. 2, top). With a ratio of 60:1 (AB-type monomer 3:core 9), full monomer conversion was detected by 1H NMR. The ratio of the protons a of the core, to the protons b and c of the arms, confirmed this assumption. This star-shaped polyester P10 was chain-extended by a further Passerini reaction with benzyl isocyanide 8 under the same reaction conditions, leading to star-shaped block-copolymer P11 (Fig. 2, middle). While the first block still exhibits 20 monomer units, the second block was synthesized with a ratio of 10:1 (AB-type monomer 3:homopolymer P10). This leads to an overall ratio of the two blocks of 20:10, which was confirmed by the integrated signal of protons b–c and d–g.
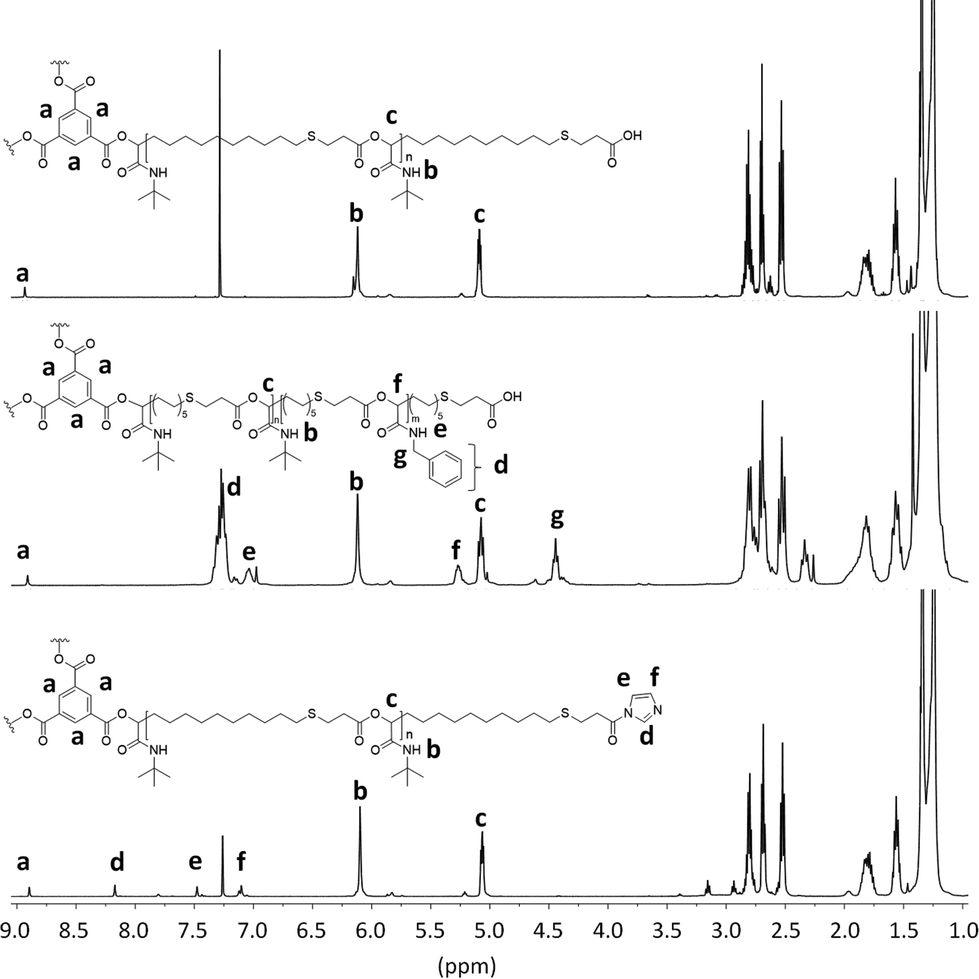 | ||
| Fig. 2 1H NMR spectra of star-shaped homopolymer P10, star shaped copolymer P11 and esterified star-shaped homopolymer P12. | ||
In order to investigate the reactivity and end-group fidelity of the carboxylic acid end groups, P10 was activated with carbonyldiimidazole 10 (CDI). The reaction was carried out in DCM at room temperature for one day to obtain polyester P12. The modification of the end group with CDI was verified by 1H NMR analysis (Fig. 2, bottom). The ratio of the protons e, d and f to protons a of the core unit indicate full conversion of the end groups. This proves that the carboxylic acid chain ends are still reactive and further modifications can be carried out.
In summary, a new approach for the synthesis of polymers with defined macromolecular architecture by a non-classic step-growth polymerization technique was demonstrated. An AB-type monomer containing an aldehyde and carboxylic acid moiety was synthesized and polymerized in a Passerini reaction with four different isocyanides and an ICTA. With this simple polyaddition process, α-amide substituted polyesters with controlled molecular weights were synthesized. Furthermore, diblock copolymers, star-shaped homo- and copolymers could be synthesized in this way. Analysis by NMR as well as GPC-ESI-MS clearly confirmed the structures of the investigated polymers and strongly indicated the absence of cyclizations or other side-reactions. By modifying the carboxylic acid end group, further reactions can be performed on the polymer to tune the properties of these materials. The described strategy offers a variety of straightforward new possibilities for the design of defined polymer architectures.
Acknowledgements
We thank Prof. Barner-Kowollik and his group for access to the GPC-ESI-MS equipment. We are grateful to the Deutsche Forschungsgemeinschaft (German Research Council, DFG) in the context of §91b application under the auspices of Prof. C. Barner-Kowollik for funding the orbitrap mass spectrometer.Notes and references
- J. M. J. Fréchet, Science, 1994, 263, 1710 Search PubMed.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93 CrossRef CAS.
- T. Yokozawa and A. Yokoyama, Prog. Polym. Sci., 2007, 32, 147 CrossRef CAS.
- T. Yokozawa and A. Yokoyama, Chem. Rec., 2005, 5, 547 CrossRef PubMed.
- A. Sandeaua, S. Mazieres and M. Destarac, Polymer, 2012, 53, 5601 CrossRef.
- L. Montero de Espinosa and M. A. R. Meier, Chem. Commun., 2011, 47, 1908 RSC.
- A. Sehlinger, L. Montero de Espinosa and M. A. R. Meier, Macromol. Chem. Phys., 2013, 214, 2821 CrossRef CAS.
- M. Unverferth and M. A. R. Meier, Polymer, 2014, 55, 5571 CrossRef CAS.
- O. Kreye, T. Tóth and M. A. R. Meier, J. Am. Chem. Soc., 2011, 133, 1790 CrossRef CAS PubMed.
- J. G. Rudick, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3985 CrossRef CAS.
- M. Passerini, Gazz. Chem. Ital., 1921, 51, 126 CAS.
- A. Sehlinger and M. A. R. Meier, Adv. Polym. Sci., 2015, 269, 61 CrossRef.
- X.-X. Deng, L. Li, Z.-L. Li, A. Lv, F.-S. Du and Z.-C. Li, ACS Macro. Lett., 2012, 1, 1300 CrossRef CAS.
- Y.-Z. Wang, X.-X. Deng, L. Li, Z.-L. Li, F.-S. Du and Z.-C. Li, Polym. Chem., 2013, 3, 444 RSC.
- A. Sehlinger, R. Schneider and M. A. R. Meier, Eur. Polym. J., 2014, 50, 150 CrossRef CAS.
- L.-J. Zhang, X.-X. Deng, F.-S. Du and Z.-C. Li, Macromolecules, 2013, 46, 9554 CrossRef CAS.
- A. Lv, X.-X. Deng, L. Li, Z.-L. Li, Y.-Z. Wang, F.-S. Du and Z.-C. Li, Polym. Chem., 2013, 13, 3659 RSC.
- C. Y. K. Chan, N.-W. Tseng, J. W. Y. Lam, J. Liu, R. T. K. Kwok and B. Z. Tang, Macromolecules, 2013, 9, 3246 CrossRef.
- R. Kakuchi and P. Theato, ACS Macro. Lett., 2013, 5, 419 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR. See DOI: 10.1039/c5py02030a |
| This journal is © The Royal Society of Chemistry 2016 |