
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid formation of regulated methacrylate networks yielding tough materials for lithography-based 3D printing†
Christian
Gorsche
ab,
Konstanze
Seidler
ab,
Patrick
Knaack
a,
Peter
Dorfinger
c,
Thomas
Koch
c,
Jürgen
Stampfl
bc,
Norbert
Moszner
bd and
Robert
Liska
*ab
aInstitute of Applied Synthetic Chemistry, Technische Universität Wien, Getreidemarkt 9/163 MC, 1060 Vienna, Austria. E-mail: robert.liska@tuwien.ac.at
bChristian-Doppler-Laboratory for Photopolymers in Digital and Restorative Dentistry, Getreidemarkt 9, 1060 Vienna, Austria
cInstitute of Materials Science and Technology, Technische Universität Wien, Getreidemarkt 9/308, 1060 Vienna, Austria
dIvoclar Vivadent AG, 9494 Schaan, Liechtenstein
First published on 22nd January 2016
Abstract
Multifunctional methacrylates are highly reactive monomers for radical photopolymerization, but yield brittle materials due to their inhomogeneous and highly crosslinked network architecture. Addition fragmentation chain transfer (AFCT) reagents serve as additives for the regulation of radical network formation and pave the way to photopolymer networks with high toughness. However, AFCT reagents (e.g. β-allyl sulfones) tend to have a negative influence on the reaction speed which limits them for lithography-based 3D fabrication. Vinyl sulfone esters are described as a new class of AFCT reagents for methacrylate-based photopolymerization without the drawback of retardation but good regulation of network architecture. The resulting materials show high network homogeneity, low shrinkage stress, and a significant increase in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond conversion and toughness. This promises great potential for vinyl sulfone esters as AFCT reagents in photopolymer applications. First 3D parts have been successfully fabricated via digital light processing.
C double bond conversion and toughness. This promises great potential for vinyl sulfone esters as AFCT reagents in photopolymer applications. First 3D parts have been successfully fabricated via digital light processing.
With growing research efforts and more importantly increasing interest of industry, additive manufacturing technologies1 are close to reaching the brink of breakthrough. A broad array of different technologies for various materials (e.g. metals,2,3 ceramics3,4 and polymers5–7) has already been established. The 3D structuring of polymeric materials can be achieved by almost any 3D printing technology, while metals and ceramics are mostly limited to specifically adjusted technologies. In general, extrusion- or melt-type techniques such as fused deposition modeling5 or selective laser sintering6 serve as good methods for the fabrication of thermoplastics with the drawback of low resolution, weak layer adhesion, and slow processing.
Taking advantage of photopolymerization, light-based technologies are developed (e.g. microstereolithography,7 digital light processing,7,8 lithography-based ceramic manufacturing9) that dramatically improve the resolution. Writing speeds and layer-to-layer interaction can also be significantly advanced.10 However, the accessible pallet of materials is limited to commercial acrylate-based monomers and their resulting polymer networks. Acrylate chemistry is suitable for light triggered 3D structuring due to the fast radical chain growth polymerization that forms stiff networks within seconds. The lack of control over this radical process yields materials which tend to be very brittle and this narrows the window of application. Consequently, attempts to improve the toughness of bulk photopolymers have been a focus in the literature.11 In general, methacrylates are the most popular type of monomer for lithography-based 3D printing as they are less cytotoxic and yield materials with higher heat resistance compared to acrylates. This also makes methacrylate-based photopolymers suitable materials for tissue engineering,12–15 dental application,16,17 nanoimprint lithography18,19 and applications in everyday life such as decorative and protective coatings.20,21 Crucial for the advancement of light-based 3D printing are new photopolymerizable formulations that form homogeneous networks via a radical step growth-like mechanism (similar to thiol–ene chemistry22) but without the drawbacks of low storage stability,23 strong odor,24 and retardation of the reaction. This could give access to 3D constructs with high toughness25 comparable to standard thermoplastics or ABS-type materials. In addition, polymerization induced shrinkage stress, which presents another problem for stereolithography,26 could also be lowered by the implementation of chain transfer.
Addition fragmentation chain transfer (AFCT) reagents (e.g. allyl sulfides27) have been mentioned as promising additives in photopolymerizable resins to regulate network formation, thus improving thermal and mechanical properties. Recently, β-allyl sulfones have been introduced as a potent class of AFCT reagents for the fabrication of tough crosslinked methacrylate-based networks.28,29 They have shown significant advantages over other AFCT reagents with various leaving30 or activating groups.31 The good coreactivity of β-allyl sulfones with methacrylates originates from a chain transfer constant (Ctr) of close to 1 (Ctr for thiols in methacrylate systems < 0.5).32,33 By definition Ctr equals the ratio of the rate constant for chain transfer (ktr) to the rate constant for chain propagation (kp; Ctr = ktr/kp). Furthermore, the regulation of methacrylate-based networks by β-allyl sulfones as AFCT reagents yields materials with tunable thermal and mechanical properties. During curing, the gelation can be moved to higher double bond conversion, which results in higher final conversion and reduced shrinkage stress. The cured materials exhibit significantly higher toughness while hardly having to sacrifice hardness or stiffness. However, the overall curing time for β-allyl sulfone containing resins is increased as the AFCT mechanism leads to some level of retardation, especially at higher concentrations. Focusing on lithography-based 3D printing, a crucial characteristic is the speed of the radical photopolymerization. Due to the layer-by-layer approach, the time needed for photocuring of a layer is multiplied for the amount of layers and can lead to a significant increase in production time.
The radical AFCT mechanism of an ethyl ester activated β-allyl sulfone (EAS, Scheme 1a) causes retardation which can be attributed to two major factors. After fragmentation of the first addition product (intermediate radical, INT), the newly formed double bond can react with radicals and this results in a low reactive tertiary radical (T) that can delay the radical polymerization process. Aside from retardation, the reaction of the newly formed double bond adds another crosslink to the forming network and thus could contribute to a less uniform network. Moreover, the leaving sulfonyl radical (S) can also attack a new β-allyl sulfone, which would yield an intermediate radical (INT) that shows equal probability for fragmentation (kfrag) as for the back reaction (k−add) and in any case regenerates the original AFCT reagent EAS.
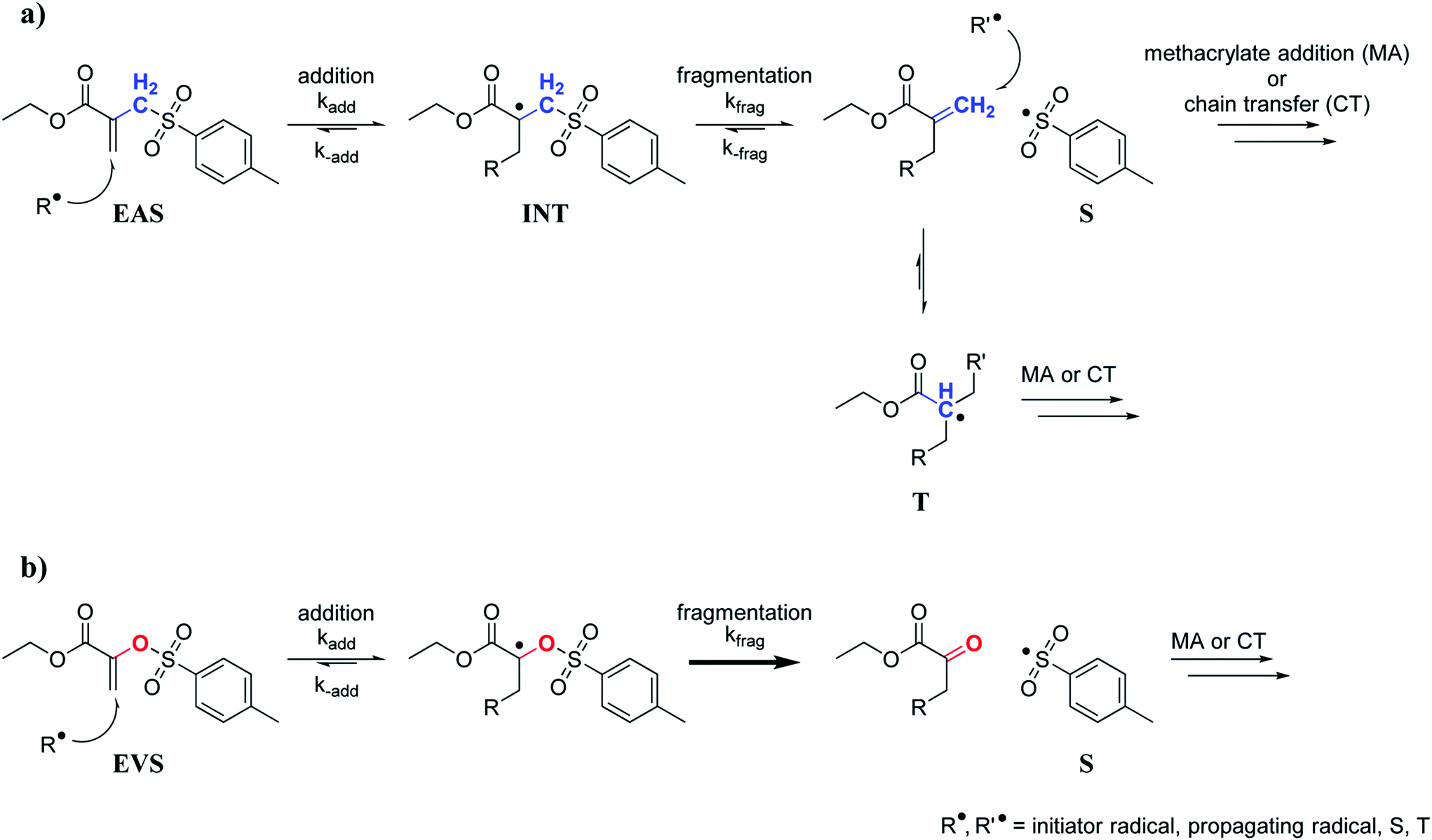 | ||
| Scheme 1 AFCT mechanism for an ethyl ester activated β-allyl sulfone (EAS, a) and an ethyl ester activated vinyl sulfone ester (EVS, b). | ||
Taking all of this into consideration, a new class of AFCT reagents with reduced retardation would be desired. This could then allow easy and fast 3D structuring of homogeneous photopolymer networks with high toughness. A number of different AFCT reagents have already been investigated.34,35 However, most reagents react analogously to β-allyl sulfones yielding a new reactive methacrylate-like double bond after fragmentation. Assuming the subsequent formation of a tertiary radical and the reversible addition of a sulfonyl radical to EAS are the major reasons for retardation, an AFCT reagent with an alternative radical mechanism needs to be developed. Vinyl ethers for instance are a class of AFCT reagents that form a non-reactive carbonyl group after fragmentation thus preventing the two described steps of retardation. Nevertheless, such compounds are known to show low coreactivity with methacrylates (Ctr significantly lower than 1)36,37 and are also prone to degrade hydrolytically making them unsuitable for many commercial applications. For an optimized AFCT reagent a double bond with similar reactivity as methacrylates would be needed, preferably with a Ctr close to 1, which could be achieved by ester activation.31 The key factor though seems to be the replacement of the methylene group of EAS with an O atom giving a vinyl sulfone ester (e.g. ethyl ester activated vinyl sulfone ester, EVS) as an AFCT reagent. The AFCT mechanism of EVS (Scheme 1b) might proceed similarly to EAS. However, due to the oxygen atom a carbonyl bond would be formed after the AFCT step, which would make this reaction step non-reversible and could drive the reaction equilibrium towards fragmentation. In addition, no tertiary radical would be formed yielding a faster polymerization.
In order to investigate the assumptions made for the radical AFCT mechanism of EVS, the molecule was synthesized according to the literature.38 Then a reference formulation (A) of two commercial dimethacrylates (urethane dimethacrylate UDMA and 1,10-decanediol dimethacrylate D3MA) and formulations with added EAS (B) and EVS (C) have been studied. For formulations B and C 20 db% of the AFCT reagent were added, respectively (db% = double bond %, meaning that 20% of all double bonds in the resin formulation are AFCT-type double bonds). All resins were mixed with 1 wt% photoinitiator (Ivocerin39).
The photoreactivity of the three formulations A–C was tested via photo-DSC studies giving a first hint about coreactivity of the new AFCT reagent EVS with methacrylates. For all experiments filtered light (400–500 nm) was applied for 5 min and the DSC signal was recorded to give the photo-DSC plots (Fig. 1a). Taking a look at the photo-DSC plots, it can be easily seen how the pure dimethacrylate-based reference A shows the fastest reaction start with the time to reach the maximum of the heat evolution (tmax) at 4.6 s. The tmax value gives an idea about the gelation of the photopolymerization reaction and shows how both AFCT reagents, β-allyl sulfone EAS (B, tmax = 10.2 s) and vinyl sulfone ester EVS (C, tmax = 11.8 s), delay gelation. The EVS-based resin also slightly delays the photopolymerization at the beginning of the reaction and reaches tmax approximately at the same time as the EAS-based formulation. Nevertheless, a more significant value for the characterization of the speed of photopolymerization is the time when 95% of the total photopolymerization heat has been evolved (t95%). With the new EVS-based resin C there is retardation observed at the beginning stages of the radical reaction, which can be directly correlated to a delayed gelation. However, EVS causes a fast decline of the photopolymerization heat and reaches the end of the reaction significantly faster when compared to reference A (t95% = 65 s). With a t95% of 49 s, the EVS-based formulation shows faster photopolymerization compared to the rather slow EAS-based reaction which reaches t95% after 101 s (Fig. 1b). Here it needs to be stated, that the photo-DSC conversion solely represents methacrylate conversion, as AFCT consumption is almost energy neutral with equal amounts of bond formation and bond breakage.
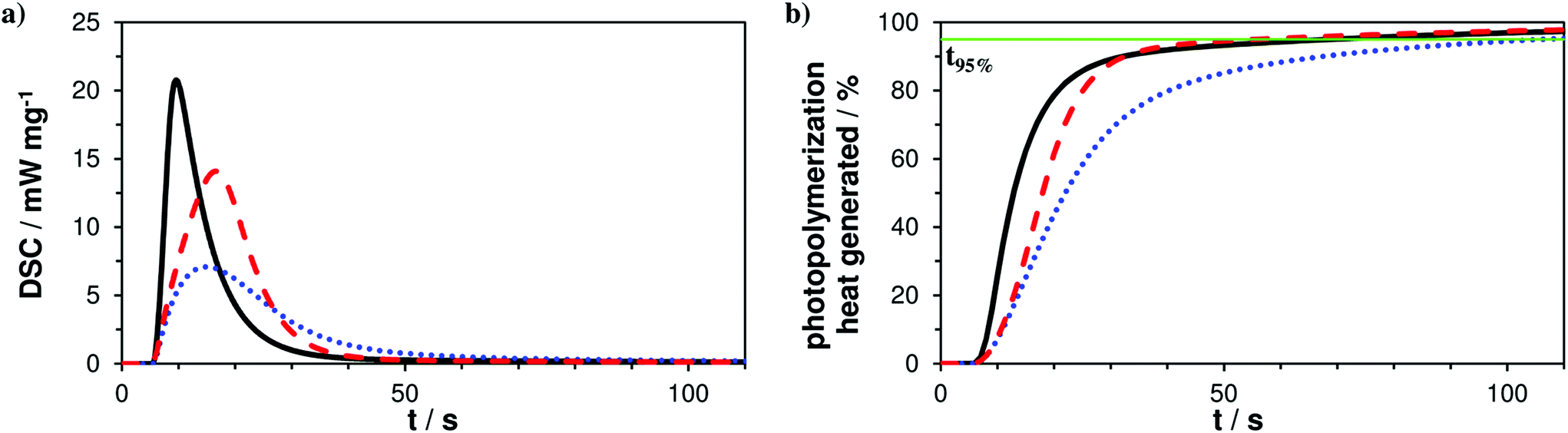 | ||
| Fig. 1 Photo-DSC plots (a) and % of photopolymerization heat generated (b) for reference A (—) and mixtures B (+ EAS, ⋯) and C (+ EVS, ---); irradiation starts after 5 s; line for 95% conversion (—, green). | ||
In order to evaluate the true double bond conversion (DBC) of the tested formulations A–C and also taking AFCT conversion into consideration, real time (RT)-FTIR-measurements were performed on 10 μm thin films that were irradiated with a light intensity of 15 mW cm−2 (5 min, 400–500 nm). FTIR spectra were recorded before and during irradiation and the double bond peak at ∼1638 cm−1 was integrated for the calculation of the DBC. The resulting final conversion values and DBC plots (Table S1,†Fig. 2) confirm that the EVS-based formulation C reacts very rapidly without any signs of retardation and even reaches significantly higher DBC (91%) compared to the reference formulation A (83%). With a final conversion of 76% the EAS-based formulation B performs worse than the reference. However, here it needs to be stated that this DBC plot has to be corrected which has been previously described.29 After the conversion of a β-allyl sulfone-based AFCT reagent (EAS), a new double bond is formed (Scheme 1) meaning that the double bond peak area in the IR spectra does not decrease. Therefore, this conversion step is not taken into consideration in the presented DBC plot. The β-allyl sulfone EAS reaches conversions comparable to the methacrylate during the reaction as Ctr is ∼1 and this would yield a 70–80% conversion of EAS that is not accounted for in the presented DBC plot. It can be assumed that the real DBC value that is reached during the photopolymerization is somewhere between 85–90% (Table S1†) and thus higher compared to reference formulation A.
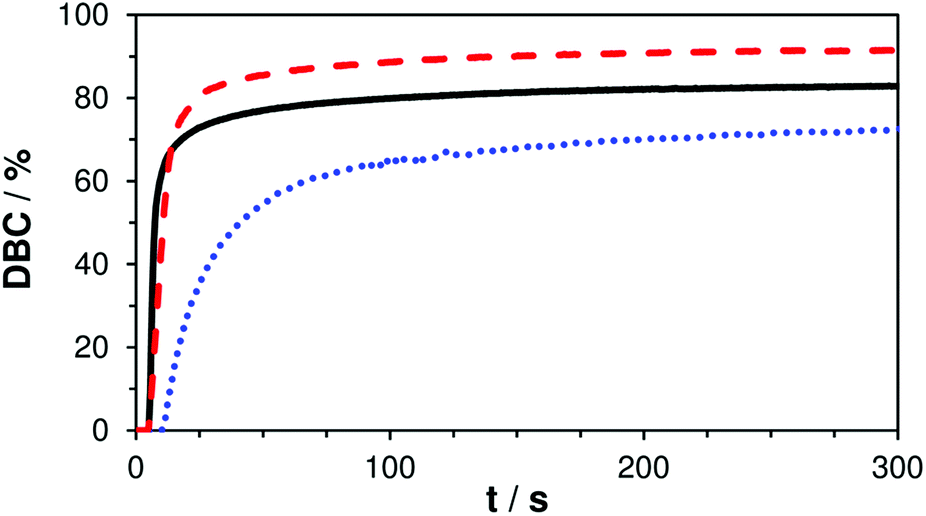 | ||
| Fig. 2 DBC plots of reference A (—) and mixtures B (+ EAS, ⋯) and C (+ EVS, ---); irradiation starts after 5 s. | ||
During photocuring shrinkage stress is created within the polymer networks, thus yielding materials with inferior mechanical properties. Fabricated polymer films tend to show warpage which is detrimental to a layer-based 3D-printing process. With photorheology studies a good measure for polymerization induced shrinkage stress can be evaluated by detecting the normal force of the measuring plate which gets pulled down by the sample that shrinks during curing. It was observed that both, formulations B (with EAS) and C (with EVS), lead to a noticeable reduction of shrinkage stress compared to the pure dimethacrylate reference resin A. For the EAS-based formulation B we observed a reduction in shrinkage stress of 33 ± 12% and for the EVS-based formulation C shrinkage stress was lowered by 12 ± 7%. It needs to be taken into consideration that the EVS-based formulation C yields a much higher double bond conversion (>8% compared to reference A) and therefore the reduction of shrinkage stress can be viewed as satisfactory.
While photo-DSC, RT-FTIR, and photorheology experiments already showed a faster reaction, increased conversion, and reduced shrinkage stress of the EVS-based formulation C, there has been no proof of the actual activity towards radical chain growth regulation for the vinyl sulfone ester EVS.
Usually, the activity of AFCT reagents is characterized by their potential to regulate the final molecular weight of the polymer (NMR spectroscopy and GPC studies on linear polymers).34 However, in the case of highly crosslinked methacrylate-based networks GPC analysis cannot be performed due to insolubility. In general, with its uncontrolled chain growth mechanism radical polymerization based on multifunctional methacrylates yields inhomogeneous networks with very broad thermal phase transitions. A regulated, homogeneous polymer network could be characterized by a sharp thermal glass transition. Dynamic mechanical thermal analysis (DMTA) is a suitable tool for the characterization of thermal polymer phase transitions of polymer networks and thus can be used for the characterization of the network homogeneity. Polymer specimens of all three tested formulations have been fabricated using a silicon mold and curing with a Lumamat 100 light oven (400–580 nm, 20 min, 20 mW cm−2). DMTA analysis was performed in oscillation mode (1 Hz, 0.1%) from −100 to 200 °C with a heating rate of 2 °C min−1.
The obtained storage modulus (G′) and loss factor (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ) plots (Fig. 3) show very well how the introduced AFCT reagents in formulations B (EAS) and C (EVS) lead to significantly more homogeneous photopolymer networks. The very sharp thermal glass transitions with a full width at half maximum (fwhm) of ∼ 23 °C are very good indicators of that. The storage modulus at the rubbery state (G′r, Fig. 3a and Table S1†) is a measure for crosslink density and shows how the regulated networks B and C have a lower crosslink density compared to the methacrylate reference A. With its higher conversion, the EVS-based network C exhibits a higher G′r (6.6 MPa) compared to the EAS-based network B (4.5 MPa). Most importantly, the regulated networks do not show a significant softening effect at temperatures below glass transition, which can usually be expected for thiol–ene-based networks.29 The EAS-based network B shows with 837 MPa a slightly lower storage modulus at 20 °C (G′20) compared to the pure methacrylate-based reference A (G′20 = 987 MPa) while the EVS-based network C has, with 1060 MPa, an even higher G′20 value (Fig. 4). It is very promising that the EVS-based network C shows better mechanical properties compared to network B (EAS-based) and that the use of EVS as an AFCT reagent also significantly improves the reaction time.
δ) plots (Fig. 3) show very well how the introduced AFCT reagents in formulations B (EAS) and C (EVS) lead to significantly more homogeneous photopolymer networks. The very sharp thermal glass transitions with a full width at half maximum (fwhm) of ∼ 23 °C are very good indicators of that. The storage modulus at the rubbery state (G′r, Fig. 3a and Table S1†) is a measure for crosslink density and shows how the regulated networks B and C have a lower crosslink density compared to the methacrylate reference A. With its higher conversion, the EVS-based network C exhibits a higher G′r (6.6 MPa) compared to the EAS-based network B (4.5 MPa). Most importantly, the regulated networks do not show a significant softening effect at temperatures below glass transition, which can usually be expected for thiol–ene-based networks.29 The EAS-based network B shows with 837 MPa a slightly lower storage modulus at 20 °C (G′20) compared to the pure methacrylate-based reference A (G′20 = 987 MPa) while the EVS-based network C has, with 1060 MPa, an even higher G′20 value (Fig. 4). It is very promising that the EVS-based network C shows better mechanical properties compared to network B (EAS-based) and that the use of EVS as an AFCT reagent also significantly improves the reaction time.
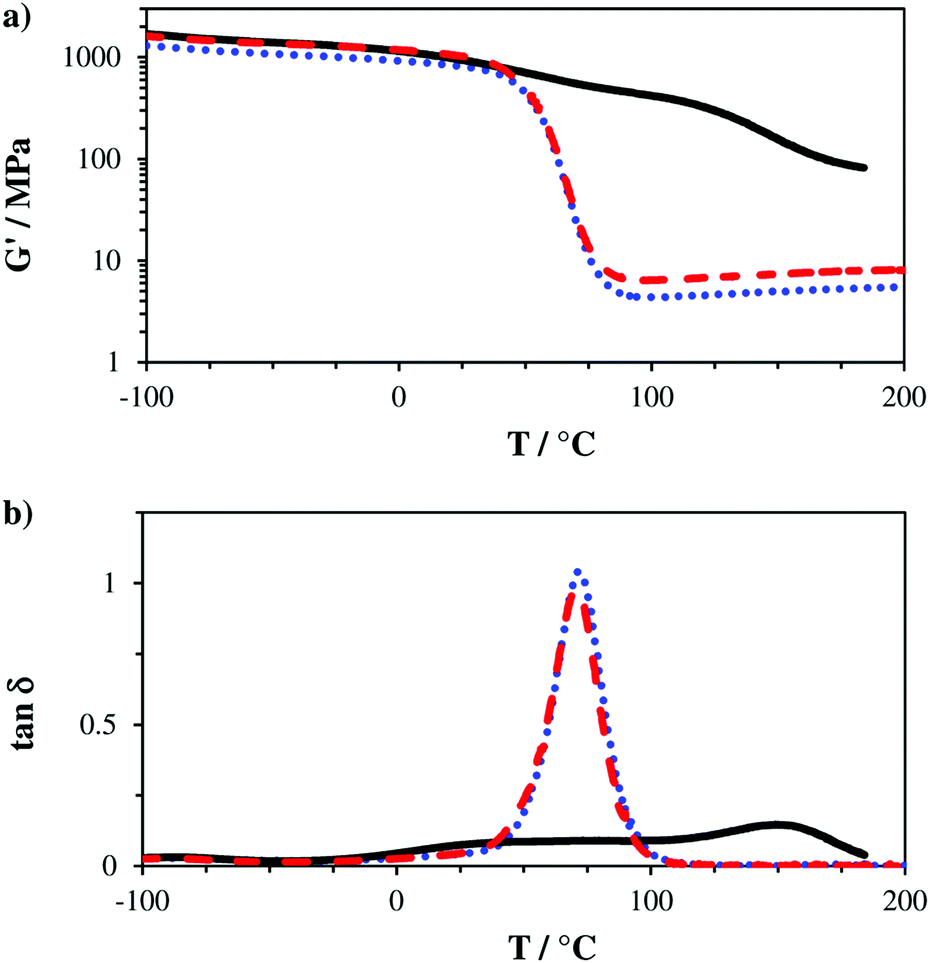 | ||
| Fig. 3 Storage modulus (G′, a) and loss factor (tanδ, b) plots of reference A (—) and mixtures B (+ EAS, ⋯) and C (+ EVS, ---). | ||
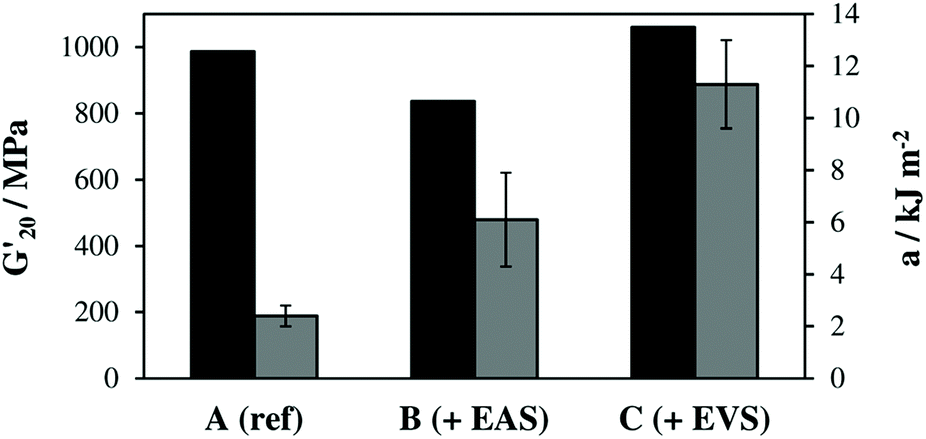 | ||
| Fig. 4 Storage modulus at 20 °C (G′20, black) and Dynstat impact resistance (a, grey). | ||
The brittleness of polymerized (multi)methacrylate-based networks can be attributed to their rather inhomogeneous highly crosslinked network architecture, which results from the fast radical curing process. By regulating this reaction, more homogeneous networks were achieved and Dynstat impact tests were performed to establish a measure for toughness (Fig. 4). The reference formulation A (pure dimethacrylate mixture) exhibited the poorest impact resistance of ∼2.4 kJ m−2. Both AFCT reagents yield more regulated network architectures and thus increase toughness by a factor of ∼3 (for EAS-based networks, B) and ∼5 (for EVS-based networks, C). The significantly increased toughness for the EVS-based polymers can additionally be attributed to fewer possible side reactions during radical polymerization compared to EAS-based materials (Scheme 1).
3D fabrication via digital light processing (DLP, Fig. 5a) of formulations containing approximately 20 db% β-allyl sulfone EAS have not been successful. The slow reaction progress makes fast curing and a good layer to layer adhesion challenging. Concomitantly with the increased processing time, over-exposure of every single layer results in bad resolution due to light scattering. Thus, also layer separation during the 3D printing process can occur due to improper curing of the individual layers. With the AFCT reagent EVS instead of EAS the formation of regulated dimethacrylate networks proceeds much faster. Due to this dramatically improved curing speed of formulations with EVS as the AFCT reagent (formulation C), first 3D parts of tough, regulated methacrylate-based materials were successfully fabricated (Fig. 5b).
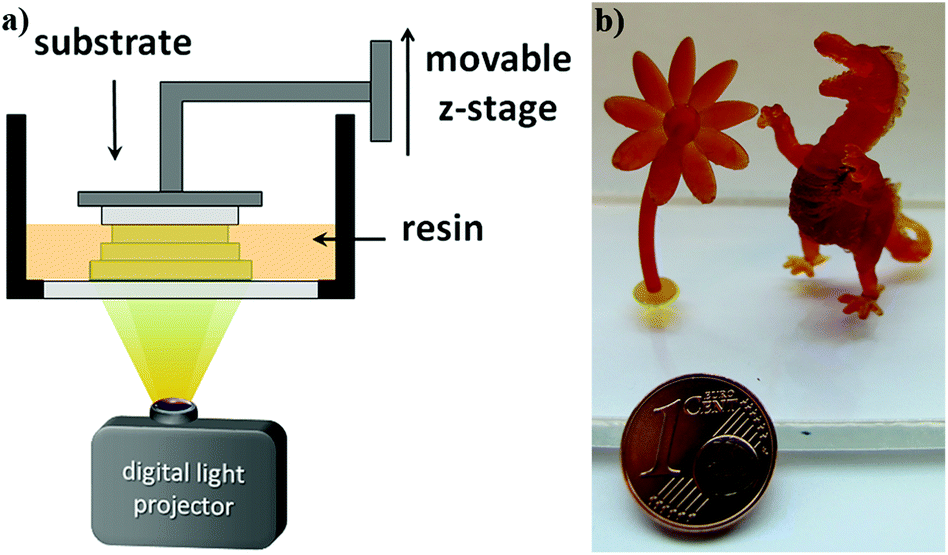 | ||
| Fig. 5 DLP set up (a) and 3D structures fabricated with formulation C (20 db% EVS, b). | ||
Conclusions
In conclusion, it can be stated that the previously published β-allyl sulfones serve as very potent AFCT reagents for methacrylate-based photopolymerization paving the way for materials with high toughness. Nevertheless, delayed curing of such resin formulations is unfavorable for applications where rapid photopolymerization is indispensable (e.g. lithography-based 3D structuring). The use of ester activated vinyl sulfone esters (EVS) in methacrylate-based formulations has been shown to significantly improve this drawback of AFCT reagents in 3D structuring. With the formation of a non-reactive carbonyl bond instead of a new double bond these potent AFCT reagents enable network regulation without retardation of the radical polymerization process. First examples of 3D fabricated constructs have been successfully presented. Nevertheless, during 3D printing a delayed gelation was noticed but is expected to happen at higher conversion (compared to the pure dimethacrylate reference formulation) yielding photopolymer networks with reduced shrinkage stress and increased double bond conversion. The established EVS will be subjected to further exploration as it promises great potential for applications where tough materials are desired (e.g. biomedical applications, coatings, nanoimprint lithography, 3D structuring). There is also a need for investigating the AFCT mechanism in greater detail to gain more understanding over the single reaction steps like addition, back reaction, fragmentation and reinitiation. Moreover, networks with multifunctional EVS-based AFCT reagents could be studied to develop a method for tuning parameters such as network density, migration of monomers, mechanical properties, and viscosity.Acknowledgements
We would like to thank the Christian Doppler Research Association and the company Ivoclar Vivadent AG (Liechtenstein) for their financial support. The authors also thank the Austrian Science fund (FWF) for funding (project P27059).References
- T. Wohlers , Wohlers Report 2015: 3D printing and additive manufacturing state of industry.
- L. E. Murr, E. Martinez, K. N. Amato, S. M. Gaytan, J. Hernandez, D. A. Ramirez, P. W. Shindo, F. Medina and R. B. Wicker, J. Mater. Res. Technol., 2012, 1, 42–54 CrossRef CAS.
- D. L. Bourell, H. L. Marcus, J. W. Barlow and J. J. Beaman, Int. J. Powder Metall., 1992, 28, 369–381 CAS.
- A. Zocca, P. Colombo, C. M. Gomes and J. Guenster, J. Am. Ceram. Soc., 2015, 98, 1983–2001 CrossRef CAS.
- L. Novakova-Marcincinova and J. Novak-Marcincin, Appl. Mech. Mater., 2013, 309, 133–140 CrossRef.
- R. D. Goodridge, C. J. Tuck and R. J. M. Hague, Prog. Mater. Sci., 2012, 57, 229–267 CrossRef CAS.
- R. Liska, M. Schuster, R. Infuehr, C. Turecek, C. Fritscher, B. Seidl, V. Schmidt, L. Kuna, A. Haase, F. Varga, H. Lichtenegger and J. Stampfl, J. Coat. Technol. Res., 2007, 4, 505–510 CrossRef CAS.
- P. Tesavibul, R. Felzmann, S. Gruber, R. Liska, I. Thompson, A. R. Boccaccini and J. Stampfl, Mater. Lett., 2012, 74, 81–84 CrossRef CAS.
- M. Schwentenwein and J. Homa, Int. J. Appl. Ceram. Technol., 2015, 12, 1–7 CrossRef CAS.
- J. R. Tumbleston, D. Shirvanyants, N. Ermoshkin, R. Janusziewicz, A. R. Johnson, D. Kelly, K. Chen, R. Pinschmidt, J. P. Rolland, A. Ermoshkin, E. T. Samulski and J. M. DeSimone, Science, 2015, 347, 1349–1352 CrossRef CAS PubMed.
- S. C. Ligon-Auer, M. Schwentenwein, C. Gorsche, J. Stampfl and R. Liska, Polym. Chem., 2016, 7, 257–286 RSC.
- S. Tielens, H. Declercq, T. Gorski, E. Lippens, E. Schacht and M. Cornelissen, Biomacromolecules, 2007, 8, 825–832 CrossRef CAS PubMed.
- A. Tiwari, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, Acta Biomater., 2009, 5, 3441–3452 CrossRef CAS PubMed.
- H. Wang, Y. Feng, B. An, W. Zhang, M. Sun, Z. Fang, W. Yuan and M. Khan, J. Mater. Sci.: Mater. Med., 2012, 23, 1499–1510 CrossRef CAS PubMed.
- Y. Zuo, X. Liu, D. Wei, J. Sun, W. Xiao, H. Zhao, L. Guo, Q. Wei, H. Fan and X. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 10386–10394 CAS.
- N. Moszner and U. Salz, Macromol. Mater. Eng., 2007, 292, 245–271 CrossRef CAS.
- N. Moszner and T. Hirt, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4369–4402 CrossRef CAS.
- Y. Hirai, T. Yoshikawa, N. Takagi, S. Yoshida and K. Yamamoto, J. Photopolym. Sci. Technol., 2003, 16, 615–620 CrossRef CAS.
- D. Matsukawa, H. Wakayama, K. Mitsukura, H. Okamura, Y. Hirai and M. Shirai, J. Mater. Chem., 2009, 19, 4085–4087 RSC.
- J. E. Fleischer, Mod. Paint Coat., 2001, 91, 21–22 CAS.
- Y. Abe, DIC Tech. Rev., 2005, 11, 1–20 CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- P. Esfandiari, S. C. Ligon, J. J. Lagref, R. Frantz, Z. Cherkaoui and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4261–4266 CrossRef CAS.
- N. Moszner, P. Burtscher, U. K. Fischer, H. Ritter, M. Tabatabai and A. Utterodt, Dental materials on the basis of low-odor thiols, WO2015124797A1, 2015 Search PubMed.
- A. Mautner, X. Qin, H. Wutzel, S. C. Ligon, B. Kapeller, D. Moser, G. Russmueller, J. Stampfl and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 203–212 CrossRef CAS.
- D. Karalekas and A. Aggelopoulos, J. Mater. Process. Technol, 2003, 136, 146–150 CrossRef CAS.
- H. Y. Park, C. J. Kloxin, A. S. Abuelyaman, J. D. Oxman and C. N. Bowman, Macromolecules, 2012, 45, 5640–5646 CrossRef CAS PubMed.
- C. Gorsche, M. Griesser, G. Gescheidt, N. Moszner and R. Liska, Macromolecules, 2014, 47, 7327–7336 CrossRef CAS.
- C. Gorsche, T. Koch, N. Moszner and R. Liska, Polym. Chem., 2015, 6, 2038–2047 RSC.
- S. C. Ligon, K. Seidler, C. Gorsche, M. Griesser, N. Moszner and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 394–406 CrossRef CAS.
- P. Gauss, S. C. Ligon, M. Griesser, C. Gorsche, H. Svajdlenkova, T. Koch, N. Moszner and R. Liska, J. Polym. Sci., Part A: Polym. Chem., 2015 Search PubMed , ahead of print.
- T. Sato, M. Seno, M. Kobayashi, T. Kohno and H. Tanaka, Eur. Polym. J., 1995, 31, 29–34 CrossRef CAS.
- L. Lecamp, F. Houllier, B. Youssef and C. Bunel, Polymer, 2001, 42, 2727–2736 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- Y. Yagci and I. Reetz, React. Funct. Polym., 1999, 42, 255–264 CrossRef CAS.
- G. F. Meijs and E. Rizzardo, Makromol. Chem., 1990, 191, 1545–1553 CrossRef CAS.
- E. Rizzardo, G. F. Meijs and S. H. Thang, Free radical chain transfer polymerization processes, US5932675A, 1999 Search PubMed.
- D. H. R. Barton, C.-Y. Chern and J. C. Jaszberenyi, Tetrahedron, 1995, 51, 1867–1886 CrossRef CAS.
- N. Moszner, F. Zeuner, I. Lamparth and U. K. Fischer, Macromol. Mater. Eng., 2009, 294, 877–886 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, synthesis of EAS and EVS; experimental description for photo-DSC, RT-FTIR, and photorheology measurements, DMTA, Dynstat impact tests, and digital light processing; Table S1 with a summary of numerical results. See DOI: 10.1039/c5py02009c |
| This journal is © The Royal Society of Chemistry 2016 |