
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled/living polymerization towards functional poly(p-phenylene vinylene) materials†
N.
Zaquen
a,
L.
Lutsen
bc,
D.
Vanderzande
bc and
T.
Junkers
*ab
aPolymer Reaction Design (PRD) Group, Institute for Materials Research (IMO), Hasselt University, Martelarenlaan 42, 3500 Hasselt, Belgium. E-mail: tanja.junkers@uhasselt.be
bIMEC associated laboratory IMOMEC, Wetenschapspark 1, 3590 Diepenbeek, Belgium
cDesign and Synthesis of Organic Semiconductors Group, Institute for Materials Research (IMO), Hasselt University, Martelarenlaan 42, 3500 Hasselt, Belgium
First published on 12th January 2016
Abstract
Poly(p-phenylene vinylene)s (PPVs) are an important class of highly fluorescent polymeric semiconductor materials. Despite their somewhat declining use in optoelectronic applications, PPV synthesis routes were in recent years significantly improved towards controlled/living polymerization. In this way, nowadays well-defined PPV structures that can be implemented in advanced polymer structures have become accessible, finding a potential application in new fields of research. This review summarizes the advances made and the types of polymers that have recently become available. Most notably, two polymerization approaches are compared, living polymerization towards well-defined PPVs via ring-opening metathesis polymerization (ROMP) and chain-transfer radical and anionic polymerization in the so-called sulfinyl precursor polymerization route.
1. Introduction
Since the discovery of polyacetylene as the first type of pi-conjugated polymer material in the 1970s,1 a broad variety of (semi)conducting polymer materials were developed.2–5 With plastic electronic applications such as organic light emitting diodes (OLEDs)6 and organic photovoltaics (OPVs),7–12 these materials found a secure position in innovative electronic applications and devices. Among the most common types of conjugated polymer materials, the class of poly(p-phenylene vinylene) (PPV) polymers play a special role.13,14 PPVs are robust polymers that are comparatively simple in their synthesis and well-reproducible in their physical characteristics (depending on the substituents on the 2 and 5 positions at the phenyl core). While initially interesting for their electroluminescent properties (the first OLED was made from PPV material),15,16 their use in optoelectronics has strongly declined over the past years due their somewhat lower performance in photovoltaic devices, compared to the use of e.g. pyrrole-based structures (Fig. 1).17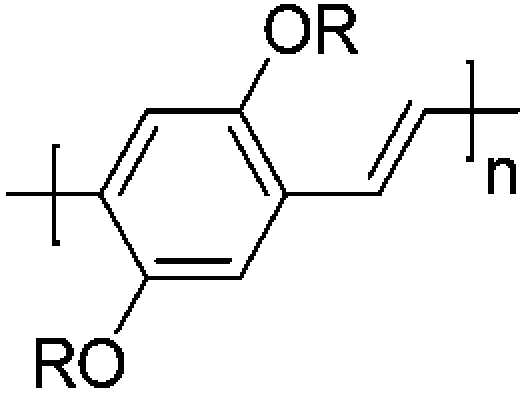 | ||
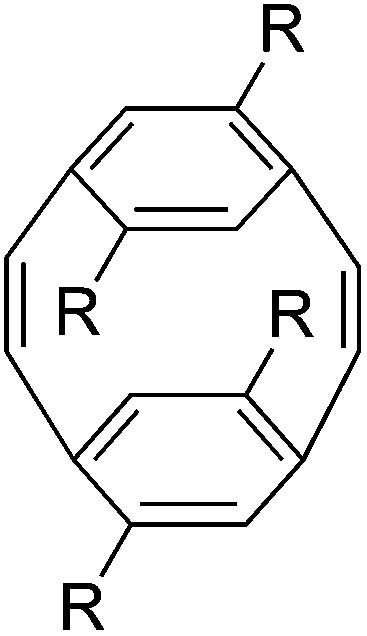
| Fig. 1 General structure of poly(p-phenylene vinylene)s (PPVs). | ||
However, the synthesis of PPVs has been well studied and a broad variety of synthesis techniques have been developed to date, making PPVs very valuable as work-horse materials.18 Next to step-growth procedures, which mostly provide oligo(phenylene vinylenes),19–22 most predominantly chain-growth polymerizations are used to obtain PPVs with moderate to high molecular weight, ranging between 4000 g mol−1 to 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1.23 Since the direct synthesis of PPV is often tedious, in many cases so-called precursor polymerization routes are employed,24,25 in which a precursor polymer is obtained from an initial polymerization, then converted into the conjugated polymer in a post-polymerization step. As mentioned above, all these synthesis methods are comparatively popular and straightforward in use; however, with only one notable exception (metathesis polymerization),26 reactions are fully uncontrolled. Uncontrolled means in this respect that no or only insufficient control over molecular weight is achieved, that end-groups of the polymers are not well defined and that dispersity is often high. As long as the main application of PPV was in optoelectronics, this was not an issue. For these applications, mechanical and optical properties (specifically the band gap) are of importance, thus ideally requiring good control over chain defects and microstructure but less over molecular weight and end-group functionalities.27 Functionality – when required – was usually introduced via attachment of side-chains at the 2 and 5 positions of the phenyl ring, which is also required to keep PPVs soluble and hence processable.28,29 Although controlled synthesis of PPVs for optoelectronic applications could be of interest from a fundamental point of view,30 the major focus in this review is placed on biomedical application-driven controlled synthesis of PPVs.
000 g mol−1.23 Since the direct synthesis of PPV is often tedious, in many cases so-called precursor polymerization routes are employed,24,25 in which a precursor polymer is obtained from an initial polymerization, then converted into the conjugated polymer in a post-polymerization step. As mentioned above, all these synthesis methods are comparatively popular and straightforward in use; however, with only one notable exception (metathesis polymerization),26 reactions are fully uncontrolled. Uncontrolled means in this respect that no or only insufficient control over molecular weight is achieved, that end-groups of the polymers are not well defined and that dispersity is often high. As long as the main application of PPV was in optoelectronics, this was not an issue. For these applications, mechanical and optical properties (specifically the band gap) are of importance, thus ideally requiring good control over chain defects and microstructure but less over molecular weight and end-group functionalities.27 Functionality – when required – was usually introduced via attachment of side-chains at the 2 and 5 positions of the phenyl ring, which is also required to keep PPVs soluble and hence processable.28,29 Although controlled synthesis of PPVs for optoelectronic applications could be of interest from a fundamental point of view,30 the major focus in this review is placed on biomedical application-driven controlled synthesis of PPVs.
As PPVs and their derivatives have been mostly replaced in electronic applications, the question arises whether they can be used in other fields of application as well. An outstanding feature of PPVs is their inherent fluorescent properties,31–33 hence PPVs are excellent candidates for biomedical applications, where fluorescent properties are used broadly for imaging and in fact, a steep rise in the usage of conjugated polymers in bioimaging and theranostic applications has been seen in recent years.34–40 PPVs play a prominent role here due to their good performance with respect to fluorescence but also due to their high reliability in properties (good reproducibility of polymerizations, especially when using micro-reactor technology)41 and the relative purity in which they are obtained, in the absence of toxic catalysts and heavy metals. However, in order to employ these materials in biomedical research, their complexity and functionality must be inherently increased. To interact in – or to mimic – biological processes and environments, the ability to self-assemble is required, as is the potential to efficiently couple bioreceptors or other materials to the conjugated materials. In other words, controlled/living polymerization strategies have become a necessity for the further evolution of conjugated polymers. For various types of conjugated polymers, living polymerization strategies such as, for example, the Grignard metathesis polymerization (GRIM), were introduced,42 which gives access to complex block copolymer structures and which allows chain-end functionalization of polymer chains.
Here, we now focus on the strategies that can be used to control polymerizations leading to PPVs, namely on the ring-opening metathesis polymerization (ROMP) and the so-called anionic and radical precursor polymerization routes. This review is hence an update on a summary of (uncontrolled) PPV polymerization techniques that our group published a few years ago.18
2. Controlled synthesis of PPVs
2.1 Ring-opening metathesis polymerization
Ring-opening metathesis polymerization (ROMP) is a chain-growth polymerization process where unsaturated cyclic olefins are converted into polymeric materials. The reaction is based on olefin metathesis, a process that found its origin in the 1950s.43–48 Here, a metal–carbene (alkylidene) complex undergoes reversible [2 + 2] cycloadditions with olefins, thereby completing metathesis via metallacyclobutane intermediates, as shown in Scheme 1.49,50 When employing ring-strained cyclic alkenes, olefin metathesis leads to a polymerization in which the cyclic monomers are converted into linear polymers with unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds being built into the main chain. ROMP is a widely used polymerization technique for the controlled synthesis of (complex) polymer architectures, giving access to a broad variety of interesting materials. A variety of catalysts as well as monomers are employed, with the Grubbs and Schrock catalyst as well as the norbornene monomer (NBE) and its derivatives being the most prominent ones.51,52
C bonds being built into the main chain. ROMP is a widely used polymerization technique for the controlled synthesis of (complex) polymer architectures, giving access to a broad variety of interesting materials. A variety of catalysts as well as monomers are employed, with the Grubbs and Schrock catalyst as well as the norbornene monomer (NBE) and its derivatives being the most prominent ones.51,52
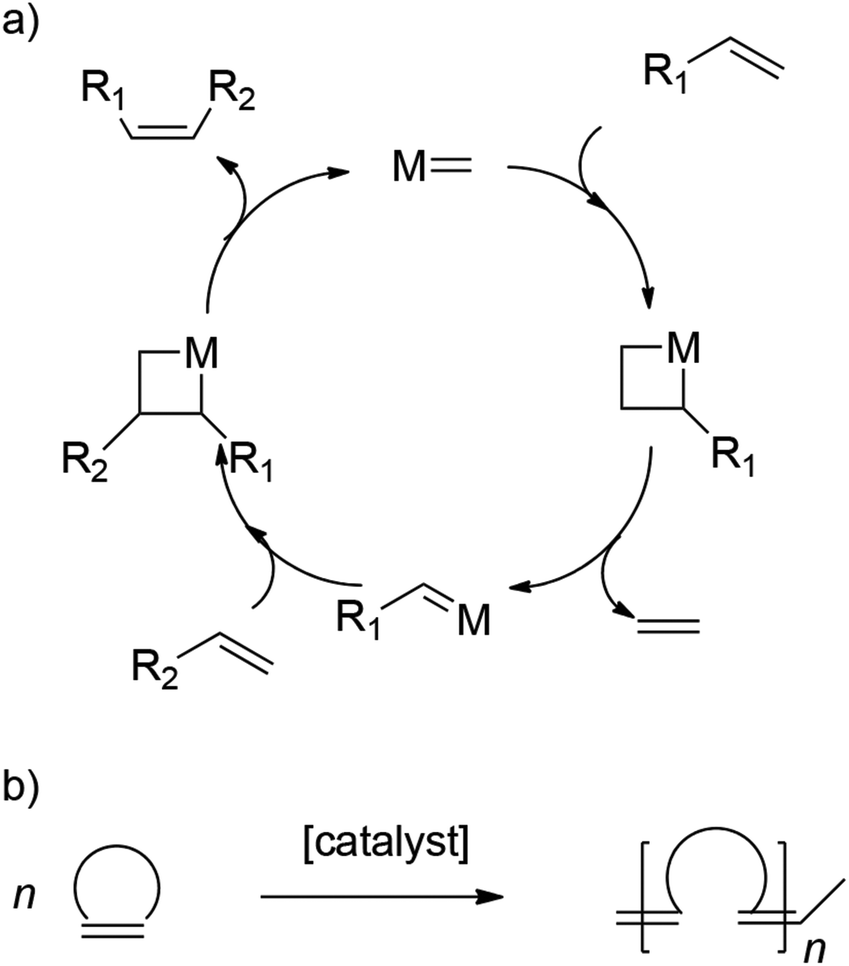 | ||
| Scheme 1 (a) Simplified olefin metathesis mechanism and (b) conversion of cyclic alkenes to polymers in ROMP. | ||
000 g mol−1 and a dispersity (Đ) of 1.23. Synthesis of this bicyclo[2.2.2]octadiene monomer (1) starts with a Diels–Alder reaction between the acetonide adduct of 3,5-cyclohexadiene-cis-1,2-diol and ethynyl p-tolyl sulfone. Reductive desulfonation of the anti cycloadduct, followed by acid-catalyzed hydrolysis yields a bicyclic diol, which can be converted into the desired biscarboxylate derivatives using Friedel Crafts acylation. Controlled polymerization of monomer 1 at room temperature using an olefin metathesis catalyst – [Mo(NAr)(C(H)CMe2Ph)–(OCMe2 (CF3))2] (Ar = 2,6-diisopropylphenyl) – is possible, yielding an intermediate with alternating cis and trans vinylene units (Fig. 2, structure 2). In the next step, thermolytic conversion enables a facile way to synthesize PPVs (3). 1H and 13C NMR spectra of the microstructure indicated that the polymer consists of the expected equal distribution of cis- and trans-vinylene units. The configuration of the metal complex inverts with each insertion of monomer, leading to an almost equal distribution of cis and trans vinylene units, with a small preference towards the trans distribution due to the preferred geometry of the methoxycarboxyl groups. Over the years, a variety of catalysts and monomers have been developed, by which control over microstructure became available. One example used similar reaction conditions and initiators as described above but a different type of monomer (see Table 1, structure 4), leading to a rare case of an almost pure cis adduct (98%). Under a flow of HCl(g) and a temperature of 190 °C, the cis-ROMP product is, however, conveniently converted into the preferred trans-PPV, as confirmed by UV spectroscopy (λmax = 424 nm) and the appearance of a strong absorption in the IR spectrum at 963 cm−1.58
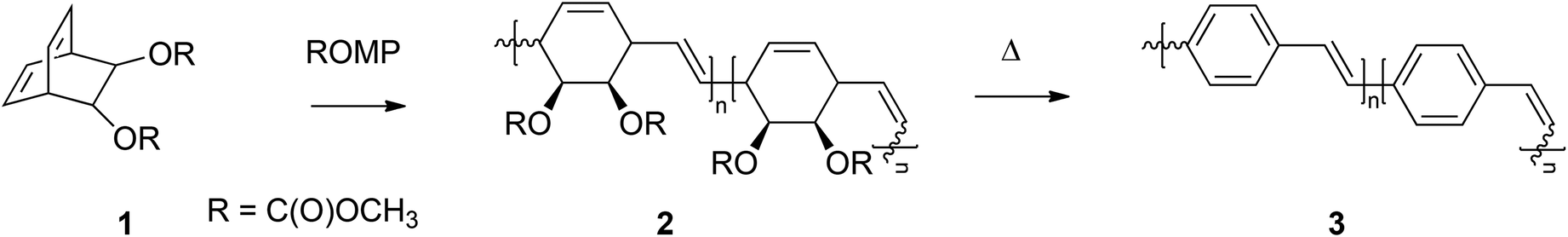 | ||
| Fig. 2 Precursor route using ROMP towards the synthesis of conjugated PPVs. | ||
| No. | Monomer structure | Catalyst | M appn/g mol−1 | Đ | Isomerism | Ref. |
|---|---|---|---|---|---|---|
| a All data given in here are associated with a certain error with respect to Mn, Mw and Đ values, as no precise Mark–Kuhn–Houwink–Sakurada (MKHS) parameters are available. | ||||||
| 1 |
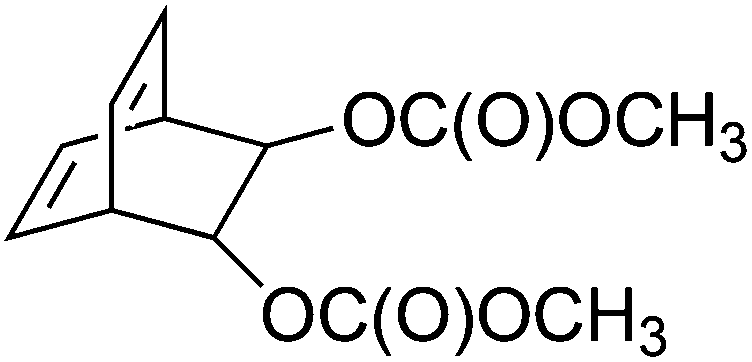
|
2nd generation Schrock catalyst | 46000 |
1.23 | Alternating cis and trans | 56 |
| 63000 |
1.34 | |||||
| 4 |
|
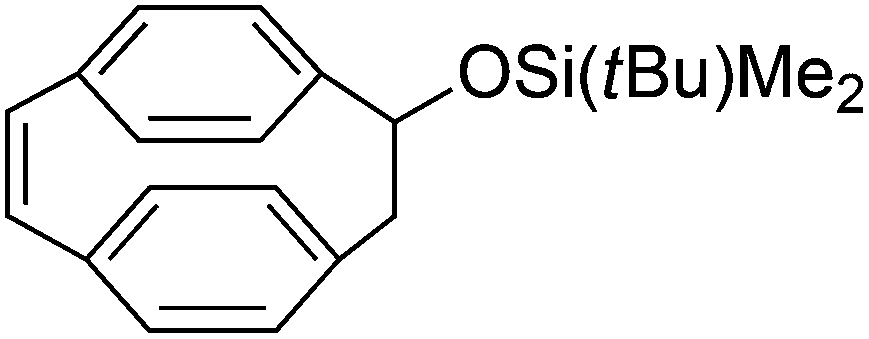
2nd generation Schrock catalyst | n.a | n.a | cis | 57 |
| 5 |
|
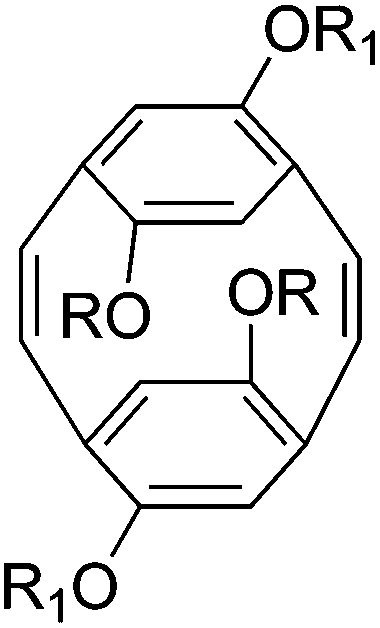
2nd generation ruthenium catalyst | 5000 | 1.22 | Alternating cis and trans | 58 |
| 25000 |
1.21 | |||||
| 6 |
|
3rd generation ruthenium catalyst | 10550 |
1.30 | Alternating cis and trans | 59 |
| 26210 |
1.32 | |||||
Generally it could be observed that the optical properties of PPVs strongly depend on the microstructure of the material. An alternating pattern in microstructures – similar built-in cis and trans adducts – will lead to higher electroluminescence efficiencies, as the folded cis-vinylene linkages reduce the interchain fluorescence-quenching reactions.59 As a result, the use of a 2nd generation Grubbs catalyst in combination with [2.2]paracyclophanedienes (5) leads to soluble PPV homopolymers of well-defined molecular weight and alternating microstructure. The living character was tested by the addition of a second batch of monomer, thereby increasing the molecular weight but maintaining similar low Đ values. Similar microstructures were obtained using microwave-assisted ROMP in combination with a 3rd generation Grubbs catalyst. The monomer, 4,12-di-2′-ethylhexyloxy-7,15-dimethoxy-[2.2]paracyclophane-1,9-diene (6), was synthesized using a two-step mechanism with a Wittig rearrangement, followed by a Hofmann elimination. Polymerization of monomer 6 for 1 h at 80 °C using microwave irradiation – compared to the 36 h needed to reach full conversion using conventional heating – indicated the benefits of microwave assisted polymerization. Molecular weights of up to 26000 g mol−1 in combination with a Đ of 1.32 underpinned the livingness of the polymerization. In the next step, pure trans-PPV could be obtained by prolonged irradiation of the product at 365 nm in THF, leading to maximum conversions of 95% trans product for reaction times of up to 36 h.60
Until now, all PPV homopolymers that were synthesized by ROMP predominantly led to solubility issues with regard to analysis or processing of the materials. Tackling these solubility issues was mainly done by adjusting monomer structures and by the introduction of solubilizing side chains (Table 1, entry 4). However, another approach is to synthesize PPV-containing block copolymers as was first shown by Miao et al.58 Herein, PPVs were chain-extended with norbornadienes (NBE), leading to macromolecular structures containing 16% of PPV built into the chain, typical molecular weights of 26000 g mol−1 and dispersity values of 1.1 for the resulting copolymer. Lower molecular weight, soluble PPV-materials with negligible interference in the light absorption properties and mechanical flexibility of the material – due to the built-in NBE – were obtained. Similar research was performed by Bazan et al.61 where PPVs were chain-extended with 2,3-bis(trifluoromethyl)norbornadienes, leading to PPV-b-PNBE block copolymers (Fig. 3, left structure). In this way, soluble PPV20-b-PNBE200 was synthesized, for which unfortunately no information about the photophysical properties of the block copolymer was reported. Lately, Porz et al.62 also increased the solubility of PPV chains by block copolymerization with NBE (Fig. 3, right structure). Employment of a double ROMP approach led first to the polymerization of NBE monomer, after which chain extension with PPVs led to the desired PPV-b-PNBE block copolymers. GPC results for the block copolymer indicated molecular weights of 28500 g mol−1 with a Đ of 1.8. Incorporation of roughly 1–5 PPV units was achieved in this work, which corresponds to a maximum of 2000 g mol−1 for the PPV unit. A comparatively higher polydispersity of 1.8 was associated with undesired cyclization reactions. UV-Vis and fluorescence analysis showed a redshift in the absorption as well as emission spectrum after copolymerization of the PPV with NBE. Furthermore, analysis of the quantum yield revealed a significant decrease (0.12 instead of 0.68), clearly showing the negative effect of copolymerization on the photophysical properties of PPVs. Although significant changes in the photo-optical properties with respect to the PPV materials was obtained, chain extension of PPV on NBE did lead to the desired solubility for the PPV materials. Turner et al.63 demonstrated that it is possible to synthesize PPV homopolymers containing an α-bromoester via ROMP. Reactivation of the bromine end-group using atom transfer radical polymerization (ATRP) reaction conditions in combination with methyl methacrylate (MMA) as monomer, led to the well-defined synthesis of PPV-b-PMMA copolymers.64 A variety of PPV homopolymers with a Mn of 22800 g mol−1 and a Đ of 1.44 was successfully chain-extended with MMA, to yield apparent Mn values of 54500 g mol−1 and a somewhat lower Đ of 1.32. In addition, results for absorbance, emission and quantum yield indicate no significant change in the PPV characteristics upon di block copolymer formation. Hence, the PPV-conjugated backbone is not affected by the ATRP reaction conditions.
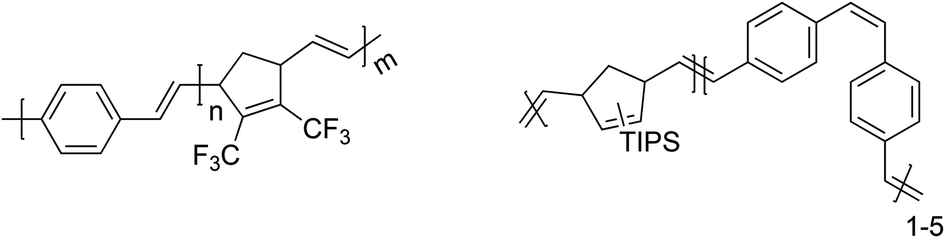 | ||
| Fig. 3 Chemical structures of PPV block copolymers with NBE-type monomers. | ||
Thus, as shown, ROMP can be efficiently used to access PPVs with all the features of a living polymerization. Block copolymerizations are, at least to date, limited to block extensions via ROMP – hence limiting the choice of available materials. At the same time, accessibility of the cyclic monomers is relatively low, which probably explains why the ROMP route is – compared to other synthesis pathways – not studied in high detail. Despite these disadvantages, the technique has high potential.
2.2 Radical and anionic polymerization
As already mentioned above, PPV materials are accessible via step growth (direct routes) or chain growth (indirect or precursor route) mechanisms. Extensive work on the so-called ‘direct’ routes – all step growth and hence inherently non-living – has been performed over the last decades, employing Wittig,65–67 Horner,64,68 McMurry,69,70 Knoevenagel71,72 and Siegrist73 polycondensation reactions or the palladium-catalyzed Heck,74,75 Stille76 and Suzuki77 coupling reactions.To overcome the inherent drawbacks of step-growth polymerization (limited molecular weight, necessity for high functional conversions), the so called ‘indirect’ or ‘precursor’ quinodimethane routes were introduced (see Scheme 2), resulting in high-molecular-weight polymers, which were relatively easy and cost effective to synthesize.23 Precursor routes follow a chain growth mechanism, which in principle could be employed to ultimately achieve living polymerization reaction conditions. Polymerization proceeds starting from a premonomer, which upon the addition of a base forms an actively polymerizing p-quinodimethane monomer species.78 The in situ-formed monomer spontaneously polymerizes via biradical formation, which then follows for most parts a classical free-radical polymerization (FRP) pathway, leading to precursor polymers (see Scheme 2). In a second step, thermal elimination of the precursor polymer yields the formation of the desired conjugated polymer.79,80 Different ‘precursor’ routes were established, depending on the choice of leaving group (L) and polarizer (P) attached to the premonomer (Scheme 2). The L group is eliminated from the premonomer and the P group from the prepolymer. Symmetric monomers were employed in the Gilch,81 Wessling,82–84 Xanthate28,85 and dithiocarbamate86 (DTC) routes, whereas the sulfinyl87–90 route starts from an asymmetric premonomer. In this way, the polymerization and elimination processes are completely decoupled, allowing full analysis and improved control over the reaction. However, care has to be taken, as complete decoupling of these processes does require carefully selected reaction conditions. In addition, low defect levels as a result of good microstructural control – mainly head-to-tail attachment during the polymerization – were obtained when employing the sulfinyl route, leading to polymers with superior performances with regard to optoelectronic properties.91,92
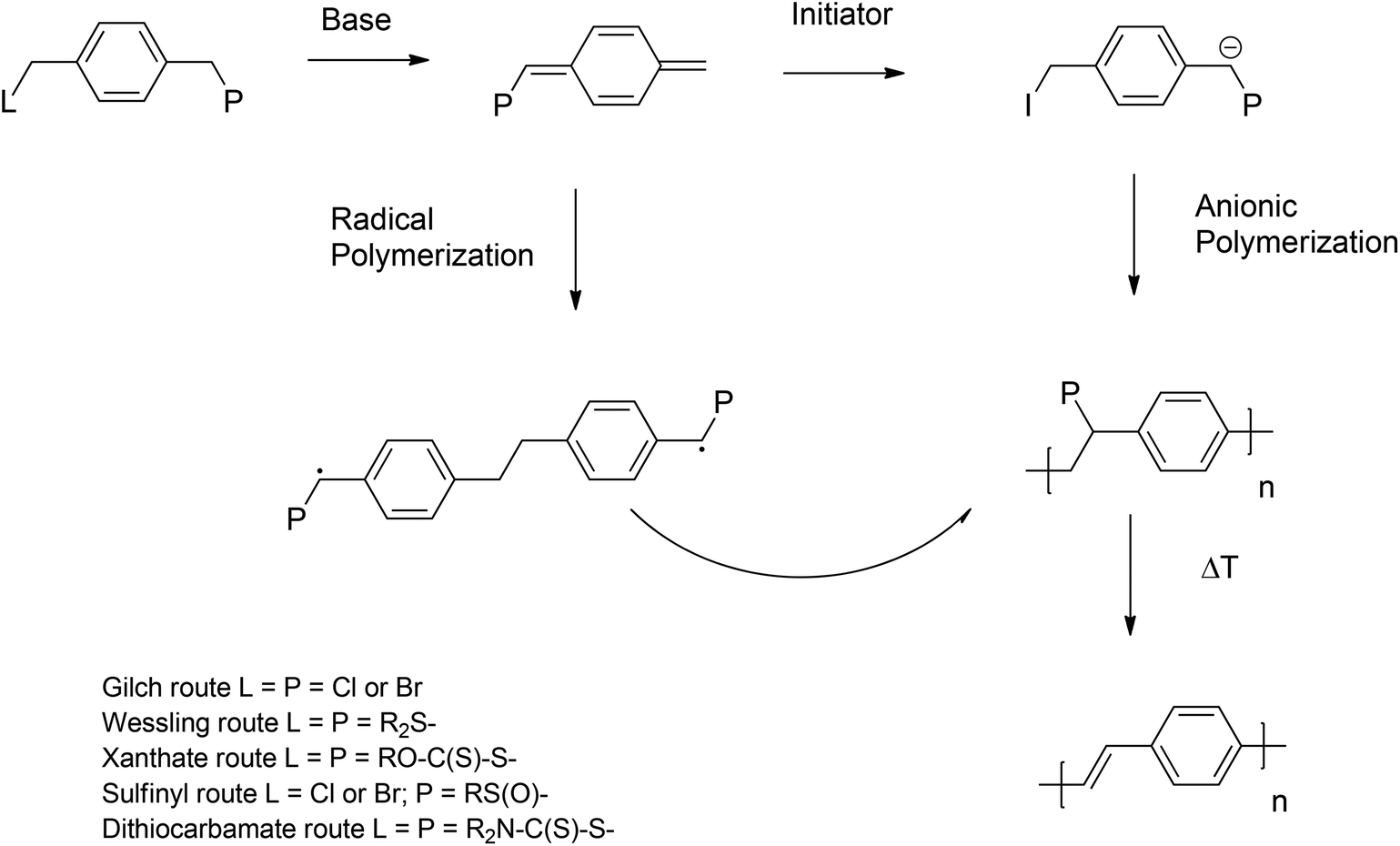 | ||
| Scheme 2 General scheme for the synthesis of PPVs using quinodimethane ‘precursor’ routes. | ||
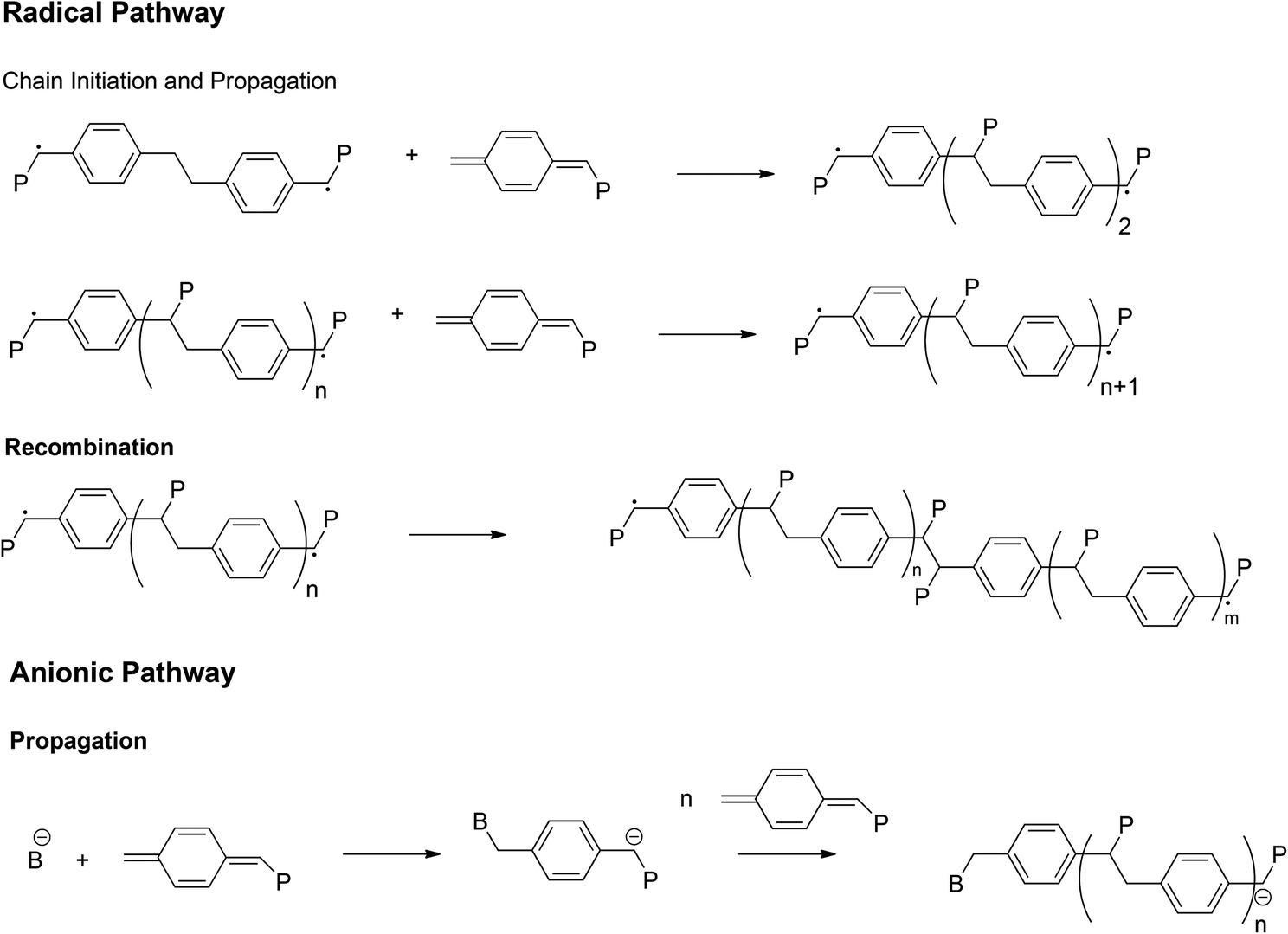 | ||
| Fig. 4 The most important reactions in the mechanisms of p-quinodimethane polymerization in the radical or anionic pathway.40 | ||
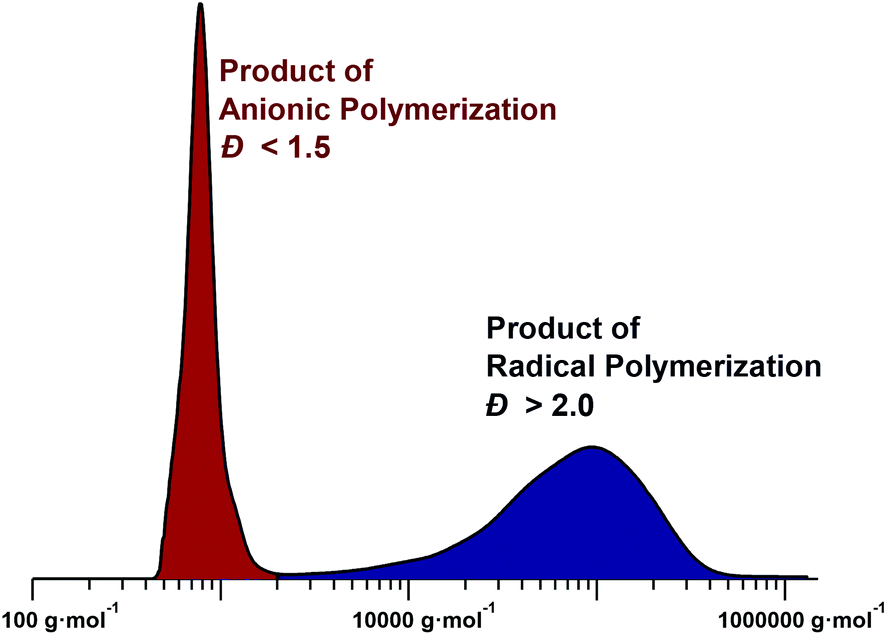 | ||
| Fig. 5 Typical GPC chromatogram for the polymerization of a sulfinyl premonomer in NMP (right peak product of radical polymerization, left peak product of anionic polymerization). See ESI† for experimental details. | ||
Initially, the low molecular weight material was considered simply as a side product and was removed from the desired high-molecular-weight material by selective precipitation. Only later, investigations into the polymerization mechanism of the sulfinyl route led to the discovery that the type of solvent and base used during the polymerization can suppress either the radical or anionic pathway, leading to monomodal distributions in the product mixtures.95 Polymerizations in sec-butanol, using NatBuO as base, led to a purely radical polymerization mechanism with high-molecular-weight polymers (Mn > 50000 g mol−1). Under these conditions, anionic polymerizations cannot proceed due to the protic solvent. The polymerization showed an increase in yield with increasing temperature, which can be interpreted as a direct result of an increasing initiation and propagation rate, compared to the termination rate.18 At the same time, a decrease in molecular weight was also observed, which can again be linked to higher initiation rates. In addition, the effect of the monomer concentration on the polymerization was investigated. The results clearly indicated no significant difference in molecular weight upon increasing the monomer concentration, verifying the self-initiation character of the reaction. Addition of a second batch of monomer during the polymerization revealed no increase in molecular weight, confirming that the reaction is effectively non-living. The radical nature of the polymerization was ultimately proven by the addition of a radical scavenger, 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO). In the presence of TEMPO, no polymerization occurred at all when aprotic solvents were used.94–97
In contrast, a purely anionic polymerization pathway is followed if the reaction conditions are adjusted carefully. In principle, in order to achieve anionic polymerization conditions, radical formation must be supressed and a base or anionic initiator must be used that is able to effectively start chain growth at a balanced rate (too-fast initiation will favour oligomer formation rather than polymerization as the base is necessarily present in molar excess to facilitate premonomer elimination). A variety of bases – NatBuO, lithium diisopropyl amide (LDA) and lithium bis(trimethylsilyl) amide (LiHMDS) – were tested in aprotic solvents, leading to the sterically hindered LiHMDS as the most suitable base. The latter showed a reduction in the rate of initiation – due to steric hindrance – leading to PPVs of significant molecular weight. THF was chosen as the optimal solvent as it is able to stabilize the anionic chain ends sufficiently and under these conditions, THF in combination with LiHMDS, an exclusively anionic pathway for the synthesis of PPVs via the sulfinyl route could be achieved. Verification of the anionic character was tested by adding TEMPO, which in this case did not quench the polymerization; see Table 2 for a collation of quench tests for the various polymerization pathways. Why no radical polymerization occurs in THF with LiHMDS is not fully clear; it can, however, be speculated that the anionic polymerization is simply faster than biradical formation from monomer self-initiation. While the radical route proceeds typically within several minutes, anionic polymerizations reach full monomer conversion on a timescale of seconds, often during mixing of the components.
94
| Base | Solvent | Additive | M appw/g mol−1 | Đ | Yield % |
|---|---|---|---|---|---|
| a Reactions are performed at room temperature with [M]I = 0.05 M. b Molecular weights shown are for non-eliminated prepolymers. | |||||
| NatBuO | sec-BuOH | None | 208400 |
4.0 | 52 |
| NatBuO | sec-BuOH | TEMPO | 9800 | 1.4 | <1 |
| NatBuO | THF | None | 1324100 |
6.9 | 79 |
| NatBuO | THF | TEMPO | 111800 |
2.7 | 21 |
| LiHMDS | THF | None | 43300 |
2.5 | 84 |
| LiHMDS | THF | TEMPO | 49400 |
3.5 | 82 |
Via the above described methodology, the general mechanisms of precursor polymerizations were elucidated. Moreover, for the sulfinyl route, a choice can be made on the reaction mode based on the solvent and base employed, giving access to much better control over the polymerizations.
000 g mol−1 to 500 g mol−1 when going from 0 to 8 equiv. of control agent relative to the monomer, respectively. The accompanying chain transfer constant (ratio of kinetic rate coefficient of chain transfer over the propagation rate coefficient) was determined as 0.46, a value lower than typical for CBr4 in vinyl polymerizations but still in the range of other CTAs.97 Controlling the molecular weight of the sulfinyl route, however, appears to be much more difficult, which is reflected in a 100-times lower value for the chain transfer constant; see Table 3. As a result, the molecular weight is not easily controlled, as Mn values showed a decrease from 89000 g mol−1 to roughly only 10000 g mol−1 upon the addition of 0 to 12 equiv. of control agent, respectively. Differences in value indicate that the use of CBr4 is less favoured in the sulfinyl route or that the rate of propagation of the sulfinyl route is much higher compared to the DTC route. Still, good control over the molecular weight for both routes was established, creating a controlled – yet non-living – radical pathway for the synthesis of PPVs via the sulfinyl and DTC routes.
| Conjugated PPVs | Chain transfer constant Ctr |
|---|---|
| MDMO-PPV (DTC route) | 0.46 |
| MDMO-PPV (sulfinyl route) | 0.0038 |
Interestingly, polymers produced via the CBr4 chain-transfer route feature bromine end-groups in the omega-position; see Fig. 6.97,101 At the other chain end, a CBr3 should be found; to date, however, this could not be confirmed. Nevertheless, the omega end-group bromine is of high interest as this functionality can serve as a macroinitiator in an ATRP chain extension. Based on CBr4-derived PPV with a molecular weight of roughly 10000 g mol−1, several block copolymers were derived via reactivation of the chains in the presence of tert-butyl acrylate (t-BuA) or styrene. In addition, copper-catalyzed azide–alkyne conjugation (CuAAC) (aka click reactions)102–106 were also tested, since a terminal bromine is easily converted into an azide.97,100
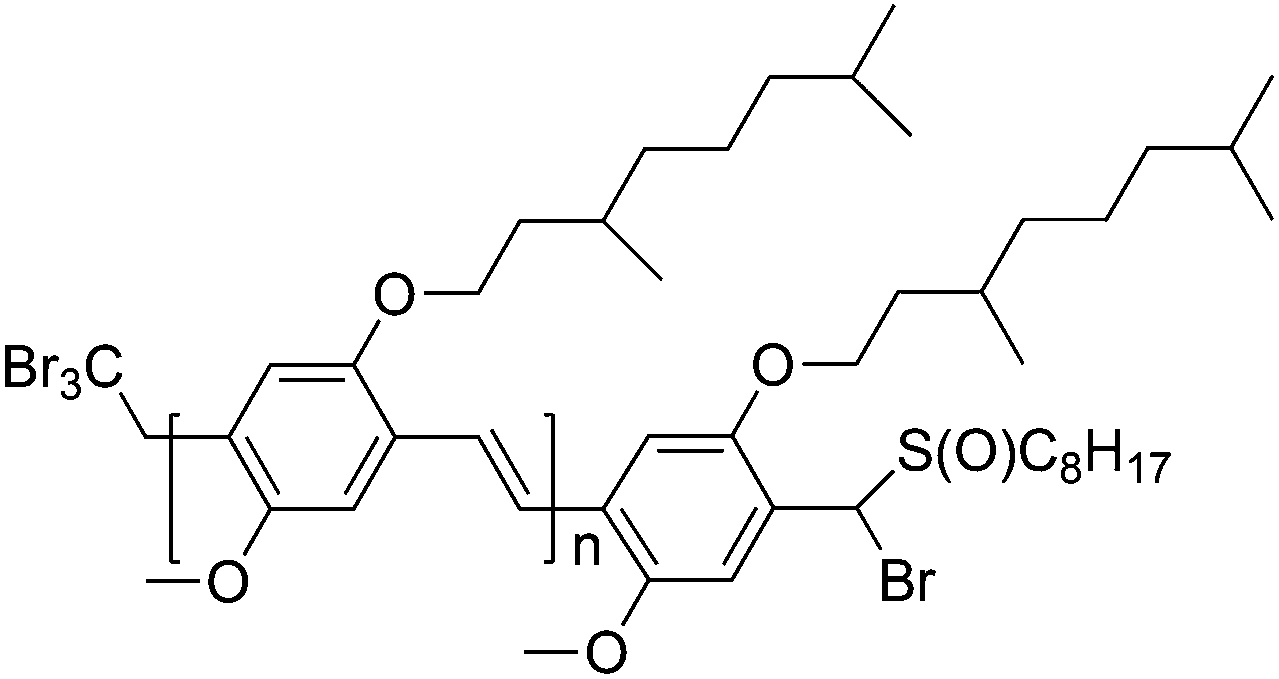 | ||
| Fig. 6 Suggested chemical structure of PPV synthesized via the radical precursor route using CBr4 as CTA. | ||
First, chain extension of the PPVs with either t-BuA or styrene was employed, leading to a PPV-b-PtBuA or PPV-b-PS block copolymer, respectively. A PPV homopolymer with Mn = 6200 g mol−1 led after chain extension with 100 equiv. of styrene up to Mn values of 10200 g mol−1 and a dispersity of 1.9 for reaction times of up to 2 h. A similar increase in molecular weight after chain extension was obtained when t-BuA was used as monomer. Mn values of up to 10900 g mol−1 and Đ = 1.8 were achieved for reaction times of up to 2 h when starting from a PPV homopolymer with Mn = 5300 g mol−1 and Đ = 1.9. In the next step, the bromine end of the PPV-b-PtBuA block copolymer (Mp = 10500 g mol−1) was substituted by an azide. CuAAC conjugation with an alkyne-functionalized PEG (Mp = 6500 g mol−1) led to successful PPV-b-PtBuA-b-PEG triblock copolymer synthesis with a peak molecular weight (Mp) of 19200 g mol−1. The major drawback of this synthetic route towards PPV block copolymers is the use of preparative recycling methods to remove the excess CBr4 after polymerization, as CBr4 may act as a co-initiator during ATRP. Still, chain-length control and especially the ability to form more complex macromolecular structures in both sequential and modular design approaches allow PPV segments to be built into any polymer architecture based on this concept (Fig. 7).
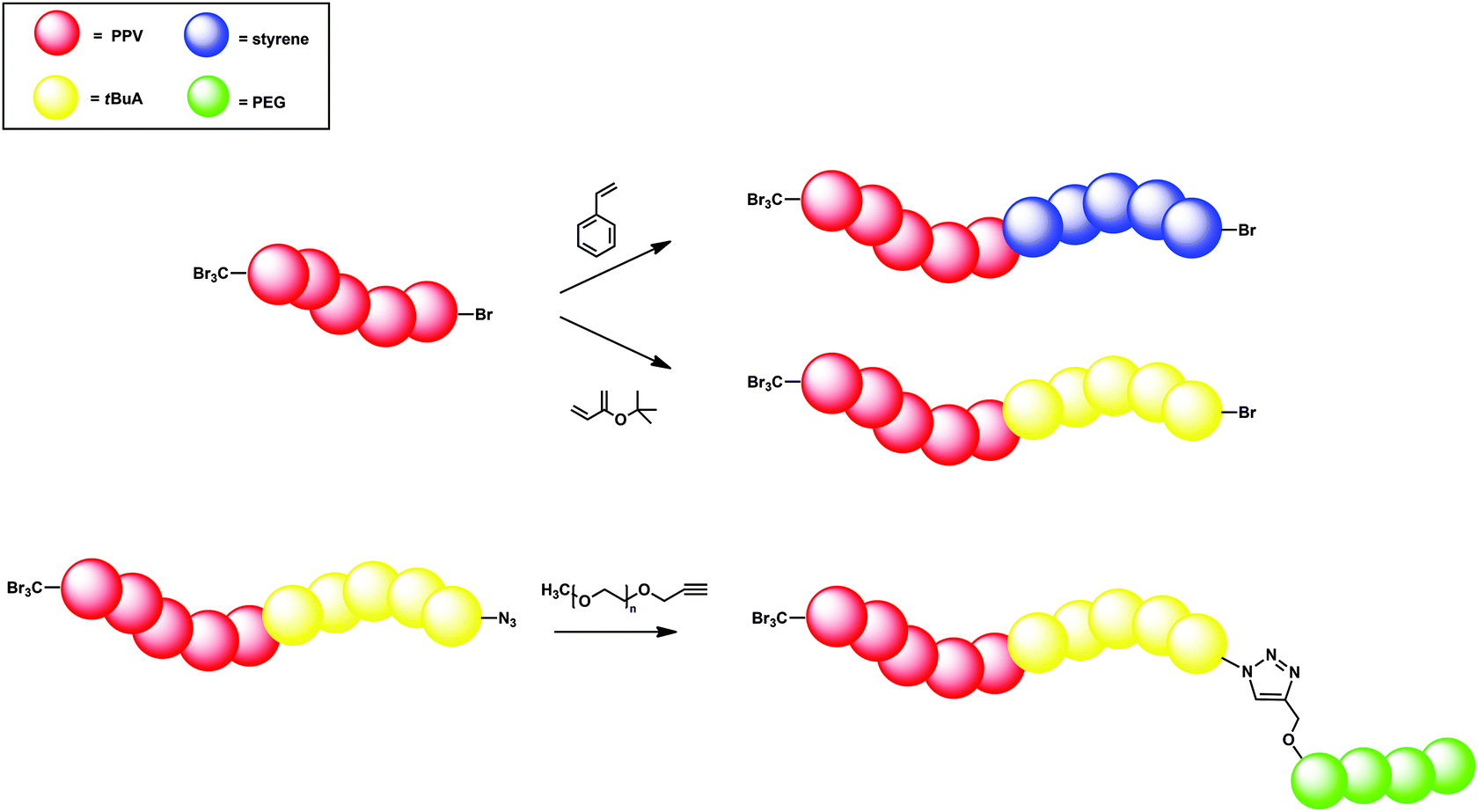 | ||
| Fig. 7 Schematic of (tri)block copolymers synthesized via the controlled radical sulfinyl precursor route in combination with ATRP (top) and ATRP/click (bottom) reaction conditions. | ||
700 g mol−1 to 51300 g mol−1 when 2.0 mol% of initiator was used. However, it was demonstrated by others108 that 4-methoxyphenol rather acts as a radical inhibitor for the Gilch polymerization route. The latter typically undergoes radical polymerization as indicated above. Recently the concept of anionic polymerization was further investigated for the sulfinyl route, using a different set of anionic initiators. For a variety of monomers and specifically designed anionic initiators – see Fig. 8 for chemical structures – the improvement that can be obtained from using anionic initiators was tested, for which the results are given in Table 4. Polymerizations were carried out in THF as solvent, using LiHMDS as base and a monomer concentration of 0.05 M at 0 °C for 5 minutes.
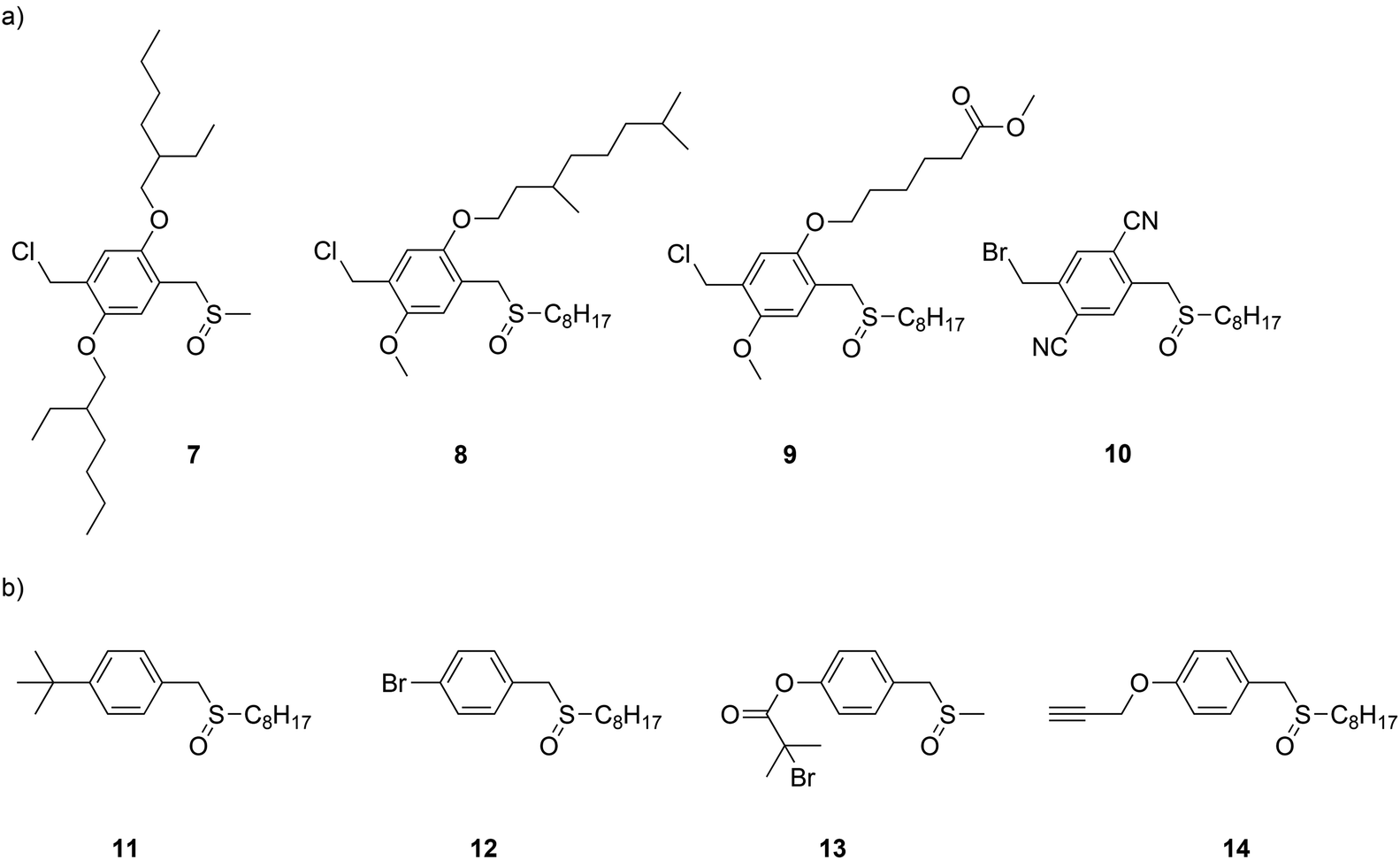 | ||
| Fig. 8 Chemical structures of monomers (a) and initiators (b) used to date in anionic sulfinyl route polymerizations. | ||
The anionic polymerization pathway for the sulfinyl route was explored using 1-chloromethyl-2,5-bis(2-ethylhexyloxy)-4-[(methylsulfinyl)methyl] benzene 7 (BEH-monomer),96 1-chloromethyl-2-methoxy-5-(3,7-dimethyloctyloxy)-4-[(octylsulfinyl)methyl] benzene 8 (MDMO-monomer),109 6-(2-chloromethoxy-5-[(octylsulfinyl)methyl]phenoxy) hexanoic acid methyl ester 9 (CPM-monomer)108 as monomers and 1-(tert-butyl)-4-[(n-octylsulfinyl)methyl] benzene 11 as initiator. In addition, 1-bromomethyl-2,5-dicyano-4-[(octylsulfinyl)methyl] benzene 10 (CN-monomer), in combination with initiator 14 was also tested.108 Results for the precursor polymer clearly indicate that an anionic pathway is followed, as low molecular weights (Mn values of roughly 4500 g mol−1) were obtained for MDMO-, CPM- and CN-PPV without the addition of an initiator (Table 4).96 BEH-PPV showed a somewhat higher molecular weight value (Mn = 20000 g mol−1), which could be related to the chemical structure of the monomer (e.g. bulky side chains) or small monomer impurities still present. The use of 25 mmol L−1 initiator indicated the profound effect of the initiator on the molecular weight and dispersity of all PPVs tested. Mn and Đ values of BEH-PPV decreased from 20000 g mol−1 to 1400 g mol−1 and 3.2 to 1.8, respectively. Results concerning MDMO- and CPM-PPV, on the other hand, showed a decrease from 4100 g mol−1 to 1300 g mol−1 and 4700 g mol−1 to 1300 g mol−1 in molecular weight, respectively. The latter effect was also shown for CN-PPV – however less pronounced for lower initiator concentrations – as molecular weights could be tuned between 4900 g mol−1 and 1300 g mol−1. In addition, lower dispersity values (<2.0) were obtained when adding an initiator. Generally linear relationships between initiator concentration and Mn are found, again underpinning the living nature of the reactions.
Still, small deviations from the ideal anionic behaviour were found and the initiation efficiency of the anionic initiators was investigated by varying the type of initiator – tert-butyl functionalized initiator (11) and bromine-functionality (12) – and studying the polymer end-group by means of electronspray ionization mass spectrometry (ESI-MS).110,111 BEH-PPV with molecular weights of 1200 g mol−1 and 2800 g mol−1 for the precursor and conjugated polymer, respectively, was investigated. The elimination could be directly followed, showing that the endgroup structure is not affected by establishing the conjugated chain system. All chains contained the initiator moiety, which could be confirmed by comparing spectra with the tert-butyl terminal group compared to the bromine. Bromine features a distinct isotopic pattern, which clearly indicated that all peaks did indeed contain the anionic initiator, confirming the high initiation efficiency. At the ω-chain-end, mainly sulfinyl groups have been identified, showing that polymers could in principle be chain-extended in sequential monomer addition approaches.
This hypothesis was further tested, in order to see if direct block copolymer synthesis would be possible. Therefore, PPV was first synthesized using standard conditions as described above in combination with initiator 11. The mixture was allowed to react at 0 °C for 15 minutes, after which tert-butyl acrylate was added and allowed to react for an additional 15 minutes. In this way, partly successful PPV block copolymers were obtained, as indicated by the bimodality in the GPC chromatograms. Formation of PPV-b-PtBuA block copolymers did occur but PPV homopolymer (Mn = 5200 g mol−1 and Đ = 1.7) was still present in the mixture, indicating a somewhat hindered reinitiation of chains. Isolation of the block copolymer using preparative recycling GPC did lead nevertheless to isolation of the block copolymer (Mn = 48300 g mol−1 and Đ = 1.2). Still, this method is not suitable to efficiently produce PPV block copolymers on a large scale. As a result, alternative methods were further elucidated (Fig. 9).
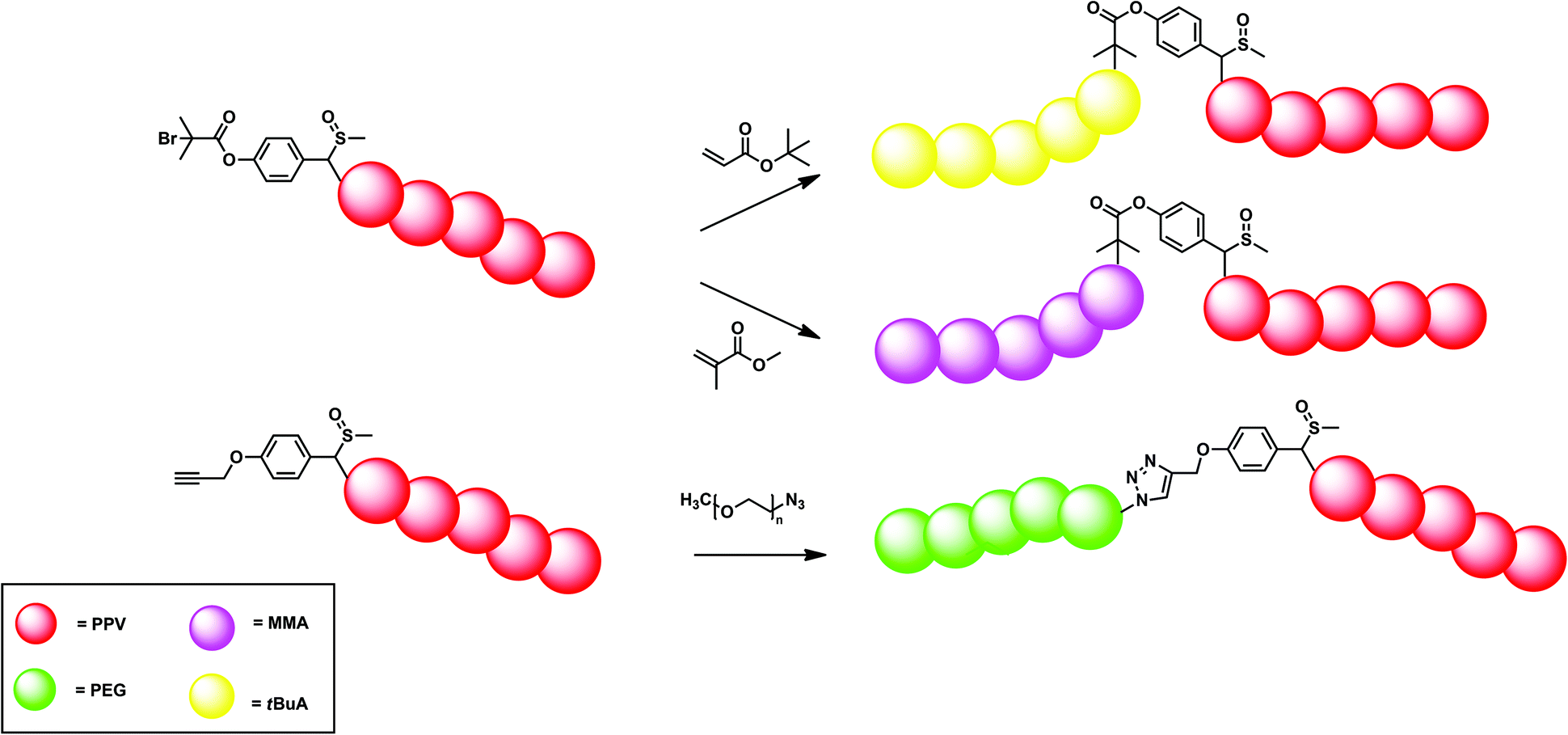 | ||
| Fig. 9 Schematic overview of the different block copolymers synthesized via the controlled anionic precursor route following the dual initiator approach in combination with either SET-LRP (top) or click (bottom) reaction conditions. | ||
As shown, the main advantage of the anionic polymerization approach is the ability to introduce specific functionalities into the polymer alpha chain end. Fig. 8 shows the structures of three functional initiators, two introducing a bromine end-group (12 and 13),110 of which only initiator 13 is suitable for ATRP re-initiation, and one for direct CuAAC conjugation (14).112Via the bromine-functional initiator, a dual initiator strategy could be followed, by employing single electron transfer living radical polymerization (SET-LRP).98,110,113–116 It is important to note here that chain extensions were performed at the precursor polymer level, thus the polymer still carried the sulfinyl groups on the backbone. In the case of the conjugated polymer being used, the conjugation could interfere with the copper species, leading to undesired oxidation reactions. t-BuA monomer in combination with copper species (Cu0) as metal and tris[2-(dimethylamino)ethylamine] (Me6TREN) as ligand were mixed together in DMF as solvent and used to chain-extend BEH-PPV homopolymers. Successful block copolymerization was achieved in this way. Molecular weights of the block copolymer showed an increase from 5300 g mol−1 for the homopolymer to 6800 g mol−1, 8900 g mol−1 and 24900 g mol−1 when adding 50, 100 and 200 equiv. of t-BuA monomer, respectively. Distributions were monomodal but featured a Đ of 1.8, due to the relatively high dispersity of 1.9 of the pure PPV block. Next to GPC analysis, infrared results also confirmed PPV-b-PtBA block copolymer formation. In addition, a blue shift in λmax in the UV-Vis as well as the emission of the fluorescence spectrum was observed after block copolymerization, indicating the quenching effect of the acrylate block on the photophysical properties of the PPV block.
While the SET-LRP approach is very successful, it still limits the choice of the second block to vinyl-type monomers. As a result, development of a modular approach allowing any type of building block is likewise highly attractive. Therefore, CuAAC ligation was also approached, starting from an alkyne functional initiator (14).111 It is noteworthy to add that direct attachment of an alkyne to the phenyl ring does not lead to successful CuAAC reactions. With the introduction of a small spacer (see Fig. 8 for structure), however, click reactions were carried out with good success, even if preparative column chromatography was required to fully purify the residual block copolymers. Azide-functional poly(ethylene glycol) (PEG) was coupled using 5 equiv. of Cu(I)Br and N,N,N′,N′,N′′-pentamethyldiethylene triamine (PMDETA) as metal and ligand system, respectively, in dry DMF as solvent. GPC traces of the block copolymers confirmed the successful coupling of either a short PEG block (Mn = 2700 g mol−1) or a larger PEG block (Mn = 6200 g mol−1). Molecular weight distributions showed a clear shift from the PPV homopolymer (Mn = 4700 g mol−1) to higher molecular weights upon coupling with the large PEG-azide functionalized block (Mn = 14600 g mol−1). UV-Vis results for the PPV-b-PtBuA block synthesized via the dual-initiator approach indicated a quenching effect of the PPV optical properties upon coupling. PPV-b-PEG block copolymers, however, still show comparable λmax results compared to plain homopolymer PPVs. The nature and length of the PEG block seemingly did not influence the optical properties of the conjugated PPV block.
With the successful formation of both PPV block copolymers from materials stemming from the anionic precursor route, new application domains towards PPV materials with complex architectures were opened. Thus, first the acrylate block copolymers were converted to amphiphilic structures in order to test for self-assembly behavior. Consequently, PPV-b-PtBuA was treated with trifluoric acid, to yield PPV-b-PAA blocks. Both PPV-b-PtBuA and PPV-b-PEG blocks showed amphiphilic behavior and the ability to self-assemble in water. Preliminary dynamic light scattering (DLS) results indicated particle formation and hence provide a proof-of-concept for further studies into this domain.
3. Conclusions and future perspectives
Over the last decades, extensive research towards PPV materials via a living polymerization mechanism has led to a variety of synthesis routes. ROMP resulted in well-controlled PPV homopolymers or – coupled with norbornenes or MMA – block copolymers. However, synthesis is still accompanied by tedious monomer synthesis. As a result, more attention was drawn by the so called ‘precursor’ routes and more specifically the sulfinyl precursor route. Research towards the mechanism behind the sulfinyl route led to the discovery of a purely radical or anionic route, depending on the type of base (LiHMDS anionic; NatBuO radical) and solvent (THF anionic; s-BuOH radical) used during the polymerization. Although challenging, the radical polymerization route could be controlled with respect to molecular weight and dispersity when employing an excess amount of CBr4 as CTA. In the next step, reactivation of the bromine-endcapped PPV using ATRP reaction conditions, led to the formation of PPV diblock copolymer structures of a different nature. Taking this one step further, triblock copolymer synthesis using click reaction conditions was achieved. The anionic route, on the other hand leads, to a truly living polymerization and excellent molecular weight control when employing specific initiators during the polymerization. Upon functionalization of the initiator, the PPV alpha endgroup is easily functionalized, giving access to block copolymer formation via SET-LRP chain extension or click chemistry polymer ligation.The combination of both methods hence gives facile access to build PPVs into virtually any complex polymer architectures for the first time. PPVs of various structures can in this way be added, for example, to polymers dedicated to biomedical applications. In this case, PPV could, for example, serve as a fluorescence tag. Also, nanoparticles as inherently fluorescing nanocarriers for concomitant payload delivery and cell imaging could be foreseen. In any case, by implementing PPVs into other materials, pathways to yet unexplored applications are opened, giving access to new fields of application and hence a significant revival of PPV research. With the development of the techniques described herein, practically any polymer segment nowadays used in biologically relevant applications can be replaced by hydrophilic or hydrophobic PPVs, adding substantial functionality to materials. Without doubt, future applications will focus on the development of even more advanced structures as described herein.
Acknowledgements
All authors are grateful for funding from the Belgian Science Policy (BELSPO) in the framework of the Inter University Attraction Pole program P7/05 – Functional Supramolecular Systems (FS2). N. Z. is grateful for funding from the ‘Agency for Innovation by Science and Technology’ in Flanders (IWT), T. J. is grateful for funding from the ‘Fund for Scientific Research’ Flanders (FWO) in the framework of the Odysseus scheme.Notes and references
- C. K. Chang, C. R. Fincher, Y. W. Park, A. J. Heeger, H. Shirakawa, E. J. Louis, S. C. Gau and A. G. MacDiarmid, Phys. Rev. Lett., 1977, 39, 1098 CrossRef.
- A. J. Berresheim, M. Müller and K. Müllen, Chem. Rev., 1999, 99, 1747 CrossRef CAS PubMed.
- J. Roncali, Chem. Rev., 1992, 92, 711 CrossRef CAS.
- U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477 CrossRef CAS.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisch and A. B. Holmes, Chem. Rev., 2009, 109, 897 CrossRef CAS PubMed.
- E. D. Gomez, S. S. Lee, C. S. Kim and Y.-L. Loo, Mol. Org. Electron. Devices, 2010, 109 CAS.
- H. Spanggaard and F. C. Sariciftci, Sol. Energy Mater. Sol. Cells, 2004, 83, 125 CrossRef CAS.
- B. C. Thompson and J. M. J. Frechet, Angew. Chem., Int. Ed., 2008, 47, 58 CrossRef CAS PubMed.
- S. Gunes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324 CrossRef PubMed.
- S. A. Gevorgyan and F. C. Krebs, Mol. Org. Electron. Devices, 2010, 291 CAS.
- T. Kietzke, Mol. Org. Electron. Devices, 2010, 253 CAS.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868 CrossRef CAS PubMed.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539 CrossRef CAS.
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. Dos Santos, J. N. Bredas, M. Logdlund and W. R. Salaneck, Nature, 1999, 397, 121 CrossRef CAS.
- T. A. Skotheim and J. R. Reynolds, Handbook of conducting polymers: Conjugated polymers: processing and applications, CRC Press, 3rd edn, 2007, ch. 5 Search PubMed.
- A. P. Kulkarni, C. J. Tonzola, A. Babel and S. A. Jenekhe, Chem. Mater., 2004, 16, 4556 CrossRef CAS.
- K. H. Hendriks, W. Li, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2014, 136, 12130 CrossRef CAS PubMed.
- T. Junkers, J. Vandenbergh, P. Adriaensens, L. Lutsen and D. Vanderzande, Polym. Chem., 2012, 3, 275 RSC.
- J. F. Morin, N. Drolet, Y. Tao and M. Leclerc, Chem. Mater., 2004, 16, 4619 CrossRef CAS.
- L. Akcelrud, Prog. Polym. Sci., 2003, 28, 875 CrossRef CAS.
- F. Babudri, S. R. Cicco, G. M. Farinola, F. Naso, A. Bolognesi and W. Porzio, Macromol. Rapid Commun., 1996, 17, 905 CrossRef CAS.
- U. Scherf and K. Müllen, Synthesis, 1992, 23 CrossRef CAS.
- G. Odian, Principles of polymerization, Wiley–Interscience, 4th edn, 2004, ch. 3 Search PubMed.
- R. A. Wessling and R. G. Zimmerman, US Patent, 3401152, 1968 Search PubMed.
- F. Louwet, D. Vanderzande, J. Gelan and J. Mullens, Macromolecules, 1995, 28, 1330 CrossRef CAS.
- G. Trimmel, S. Riegler, G. Fuchs, C. Slugovc and F. Stelzer, Adv. Polym. Sci., 2005, 176, 43 CrossRef CAS.
- J. Wiesecke and M. Rehanh, Angew. Chem., Int. Ed., 2003, 42, 567 CrossRef CAS PubMed.
- E. Kesters, S. Gilissen, F. Motmans, L. Lutsen and D. Vanderzande, Macromolecules, 2002, 35, 7902 CrossRef CAS.
- A. Issaris, D. Vanderzande, P. Adriaensens and J. Gelan, Macromolecules, 1998, 31, 4426 CrossRef CAS.
- M. Kuik, G.-J. A. H. Wetzelaer, H. T. Nicolai, N. I. Craciun, D. M. De Leeuw and P. W. M. Blom, Adv. Mater., 2014, 26, 512–531 CrossRef CAS PubMed.
- D. Dini, Chem. Mater., 2005, 17, 1933 CrossRef CAS.
- B. J. Schwartz, Annu. Rev. Phys. Chem., 2003, 54, 141 CrossRef CAS PubMed.
- A. J. Heeger, Chem. Soc. Rev., 2010, 39, 2354 RSC.
- C. Wu, C. Szymanski, Z. Cain and J. McNeill, J. Am. Chem. Soc., 2007, 129, 12904 CrossRef CAS PubMed.
- W. Zhang, H. Sun, S. Yin, J. Chang, Y. Li, X. Guo and Z. Yuan, J. Mater. Sci., 2015, 50, 5571 CrossRef CAS.
- M. Doshi, M. Krienke, S. Khederzadeh, H. Sanchez, A. Copik, J. Oyer and A. J. Gesquiere, RSC Adv., 2015, 5, 37943 RSC.
- C. Wu, B. Bull, C. Szymanski, K. Christensen and J. McNeill, ACS Nano, 2008, 2, 2415 CrossRef CAS PubMed.
- J. Xu, Y. Zhou, G. Cheng, S. Liu, M. Dong and C. Huang, Luminescence, 2015, 30, 451 CrossRef CAS PubMed.
- M. Green, P. Howes, C. Berry, O. Argyros and M. Thanou, Proc. R. Soc. London, Ser. A, 2009, 465, 2751 CrossRef CAS.
- S. Kim, C.-K. Lim, J. Na, Y.-D. Lee, K. Kim, K. Choi, J. Leary and I. Kwon, Chem. Commun., 2010, 46, 1617 RSC.
- N. Zaquen, P. H. M. Van Steenberge, D. R. D'Hooge, M.-F. Reyniers, G. B. Marin, J. Vandenergh, L. Lutsen, D. J. M. Vanderzande and T. Junkers, Macromolecules, 2015, 48, 8294 CrossRef CAS.
- M. C. Stefan, M. P. Bhatt, P. Sista and H. D. Magurudeniya, Polym. Chem., 2012, 3, 1693 RSC.
- H. S. Eleuterio, J. Mol. Catal., 1991, 65, 55 CrossRef CAS.
- N. Calderon, Acc. Chem. Res., 1972, 5, 127 CrossRef CAS.
- N. Calderon, H.-Y. Chen and K. W. Scott, Tetrahedron Lett., 1967, 34, 3328 Search PubMed.
- J. L. Herisson and Y. Chauvin, Makromol. Chem., 1971, 141, 161 CrossRef CAS.
- C. P. Casey, J. Chem. Educ., 2006, 83, 192 CrossRef CAS.
- M. Siaj and P. H. McBreen, Science, 2005, 309, 588 CrossRef CAS PubMed.
- M. B. Herbert and R. H. Grubbs, Angew. Chem., Int. Ed., 2015, 54, 5018 CrossRef CAS PubMed.
- T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 19 CrossRef.
- R. M. Johnson and C. L. Fraser, Macromolecules, 2004, 37, 2718 CrossRef CAS.
- O. Ikkala and G. ten Brinke, Science, 2002, 295, 2407 CrossRef CAS PubMed.
- F. L. Klavetter and R. H. Grubbs, J. Am. Chem. Soc., 1988, 110, 7807 CrossRef CAS.
- A. Kumar and B. E. Eichinger, Makromol. Chem., Rapid Commun., 1992, 13, 311 CrossRef CAS.
- L. K. Johnson, S. C. Virgil and R. H. Grubbs, J. Am. Chem. Soc., 1990, 112, 5384 CrossRef CAS.
- H. H. Fox, R. R. Schrock and R. O'Dell, Organometallics, 1994, 13, 635 CrossRef CAS.
- V. P. Conticello, D. L. Gin and R. H. Grubbs, J. Am. Chem. Soc., 1992, 114, 9708 CrossRef CAS.
- Y.-J. Miao and G. C. Bazan, J. Am. Chem. Soc., 1994, 116, 9379 CrossRef CAS.
- S. Son, A. Dodabalapur, A. J. Lovinger and M. E. Glavin, Science, 1995, 269, 376 CAS.
- A. M. Spring, C.-Y. Yu, M. Horie and M. L. Turner, Chem. Commun., 2009, 2676 RSC.
- G. C. Bazan, M. L. Renak and B. J. Sun, Macromolecules, 1996, 29, 1085 CrossRef CAS.
- M. Porz, D. Mäker, K. Brödner and U. W. F. Bunz, Macromol. Rapid Commun., 2013, 34, 873 CrossRef CAS PubMed.
- C.-Y. Yu, M. Horie, A. M. Spring, K. Tremel and M. L. Turner, Macromolecules, 2010, 42, 222 CrossRef.
- B. J. Lidster, J. M. Behrendt and M. L. Turner, Chem. Commun., 2014, 50, 11867 RSC.
- R. N. McDonald and T. W. Campbell, J. Am. Chem. Soc., 1960, 82, 4669–4671 CrossRef CAS.
- G. Kossmehl, J. Phys. Chem., 1979, 83, 417–426 CAS.
- A. P. Davey, A. Drury, S. Maier, H. J. Byrne and W. J. Blau, Synth. Met., 1999, 103, 2478–2479 CrossRef CAS.
- S. Pfeiffer and H.-H. Hörhold, Macromol. Chem. Phys., 1999, 200, 1870 CrossRef CAS.
- W. J. Feast, I. S. Millichamp, R. H. Friend, M. E. Horton, D. Phillips, S. D. D. V. Rughooputh and G. Rumbles, Synth. Met., 1985, 10, 181 CrossRef CAS.
- M. Rehahn and A. D. Schlüter, Makromol. Chem., Rapid Commun., 1990, 11, 375 CrossRef CAS.
- N. C. Greenham, S. C. Moratti, D. D. C. Bradley, R. H. Friend and A. B. Holmes, Nature, 1993, 365, 628–630 CrossRef CAS.
- S. C. Moratti, R. Cervini, A. B. Holmes, D. R. Baigent, R. H. Friend, N. C. Greenham, J. Grüner and P. J. Hamer, Synth. Met., 1995, 71, 2117 CrossRef CAS.
- H. Kretzschmann and H. Meier, Tetrahedron Lett., 1991, 32, 5059 CrossRef CAS.
- Z. Bao, Y. Chen, R. Cai and L. Yu, Macromolecules, 1993, 26, 5281 CrossRef CAS.
- M. Pan, Z. Bao and L. Yu, Macromolecules, 1995, 28, 5151 CrossRef CAS.
- F. Babudri, S. R. Cicco, G. M. Farinola, F. Naso, A. Bolognesi and W. Porzio, Macromol. Rapid Commun., 1996, 17, 905 CrossRef CAS.
- F. Koch and W. Heitz, Macromol. Chem. Phys., 1997, 198, 1531 CrossRef CAS.
- L. Hermosilla, S. Catak, V. Van Speybroeck, M. Waroquier, J. Vandenbergh, F. Motmans, P. Adriaensens, L. Lutsen, T. Cleij and D. Vanderzande, Macromolecules, 2010, 43, 7424 CrossRef CAS.
- E. Kesters, L. Lutsen, D. Vanderzande and J. Gelan, Synth. Met., 2001, 119, 311 CrossRef CAS.
- A. J. J. M. van Breemen, A. D. J. Issaris, M. M. de Kok, M. J. A. N. Van Der Borght, P. J. Adriaensens, J. Gelan and D. Vanderzande, Macromolecules, 1999, 32, 5728 CrossRef CAS.
- H. G. Gilch and W. L. Weelwright, J. Polym. Sci., Polym. Chem. Ed., 1966, 4, 1337 CrossRef CAS.
- R. A. Wessling and R. G. Zimmerman, US Patent, 3401152, 1968 Search PubMed.
- R. A. Wessling and R. G. Zimmerman, US Patent, 3706677, 1972 Search PubMed.
- F. R. Denton, A. Serker, P. M. Lathi, R. O. Garay and F. E. Karasz, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 2233 CrossRef CAS.
- S. Son, A. Dodabalapur, A. J. Lovinger and M. E. Galvin, Science, 1995, 269, 376 CAS.
- A. Henckens, I. Duyssens, L. Lutsen, D. Vanderzande and T. Cleij, Polymer, 2006, 47, 123 CrossRef CAS.
- F. Louwet, D. Vanderzande and J. Gelan, Synth. Met., 1995, 69, 509 CrossRef CAS.
- A. Van Breemen, D. Vanderzande, P. Adriaensens and J. Gelan, J. Org. Chem., 1999, 64, 3106 CrossRef CAS PubMed.
- L. Lutsen, A. Van Breemen, W. Kreuder, D. Vanderzande and J. Gelan, Helv. Chim. Acta, 2000, 83, 3113 CrossRef CAS.
- M. Van Der Borght, D. Vanderzande, P. Adriaensens and J. Gelan, J. Org. Chem., 2000, 65, 284 CrossRef CAS PubMed.
- H. Roex, P. Adriaensens, D. Vanderzande and J. Gelan, Macromolecules, 2003, 36, 5613 CrossRef CAS.
- E. Kesters, L. Lutsen, D. Vanderzande, J. Gelan, T. P. Nguyen and P. Molinié, Thin Solid Films, 2002, 403–404, 120 CrossRef CAS.
- L. Hontis, M. Van Der Borght, D. Vanderzande and J. Gelan, Polymer, 1999, 40, 6615 CrossRef CAS.
- M. Vanderboght, P. Adriaensens, D. Vanderzande and J. Gelan, Polymer, 2000, 41, 2743 CrossRef.
- I. Cosemans, L. Hontis, D. Van Den Berghe, A. Palmaerts, J. Wouters, T. Cleij, L. Lutsen, W. Maes, T. Junkers and D. Vanderzande, Macromolecules, 2011, 44, 7610 CrossRef CAS.
- D. Vanderzande, L. Hontis, A. Palmaerts, D. Van Den Berghe, J. Wouters, L. Lutsen and T. Cleij, Proc. SPIE, 2005, 5937 Search PubMed.
- I. Cosemans, J. Wouters, T. Cleij, L. Lutsen, W. Maes, T. Junkers and D. Vanderzande, Macromol. Rapid Commun., 2012, 33, 242 CrossRef CAS PubMed.
- N. Zaquen, J. Vandenbergh, M. Schneider-Baumann, L. Lutsen, D. Vanderzande and T. Junkers, Polymers, 2015, 7, 418 CrossRef CAS.
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069 CrossRef CAS PubMed.
- J. Vandenbergh, PhD Thesis, Universiteit Hasselt, 2011 Search PubMed.
- J. Vandenbergh, I. Cosemans, L. Lutsen, D. Vanderzande and T. Junkers, Polym. Chem., 2012, 3, 1722 RSC.
- L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60 CrossRef CAS PubMed.
- S. Chakraborty, A. Keightley, V. Dusevich, Y. Wang and Z. Peng, Chem. Mater., 2010, 22, 3995 CrossRef CAS.
- C.-C. Ho, Y.-H. Lee, C.-A. Dai, R. A. Segalman and W.-F. Su, Macromolecules, 2009, 42, 4208 CrossRef CAS.
- C. J. Neef and J. P. Ferraris, Macromolecules, 2000, 33, 2311 CrossRef CAS.
- L. Hontis, V. Vrindts, D. Vanderzande and L. Lutsen, Macromolecules, 2003, 36, 3035 CrossRef CAS.
- I. Cosemans, PhD Thesis, Universiteit Hasselt, 2014 Search PubMed.
- I. Cosemans, J. Vandenbergh, V. S. D. Voet, K. Loos, L. Lutsen, D. Vanderzande and T. Junkers, Polymer, 2013, 54, 1298 CrossRef CAS.
- I. Cosemans, J. Vandenbergh, L. Lutsen, D. Vanderzande and T. Junkers, Polym. Chem., 2013, 4, 3471 RSC.
- I. Cosemans, J. Vandenbergh, L. Lutsen, D. Vanderzande and T. Junkers, Eur. Polym. J., 2014, 55, 114 CrossRef CAS.
- V. Percec, T. Guliashvili, J. Ladislaw, A. Wistrand, A. Stjerndahl, M. Sienkowska, M. Montiero and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156 CrossRef CAS PubMed.
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5109 CrossRef CAS.
- N. H. Nguyen, B. M. Rosen, G. Ligadas and V. Percec, Macromolecules, 2009, 42, 2379 CrossRef CAS.
- A. Anastasaki, C. Waldron, P. Wilson, R. McHale and D. M. Haddleton, Polym. Chem., 2013, 4, 2672 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py01987g |
| This journal is © The Royal Society of Chemistry 2016 |