
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhanced catalytic activity of oxovanadium complexes in oxidative polymerization of diphenyl disulfide†
Fuyuki
Aida
 ab,
Yohei
Takatori
a,
Daichi
Kiyokawa
a,
Kentaro
Nagamatsu
a,
Kenichi
Oyaizu
*a and
Hiroyuki
Nishide
*a
ab,
Yohei
Takatori
a,
Daichi
Kiyokawa
a,
Kentaro
Nagamatsu
a,
Kenichi
Oyaizu
*a and
Hiroyuki
Nishide
*a
aDepartment of Applied Chemistry, Waseda University, Tokyo 169-8555, Japan. E-mail: nishide@waseda.jp; oyaizu@waseda.jp
bJX Nippon Oil & Energy Corporation, Kanagawa, 231-0815, Japan
First published on 16th February 2016
Abstract
Oxygen-oxidative polymerization of diphenyl disulfide gives poly(1,4-phenylene sulfide) (PPS), catalyzed by a vanadyl complex and a protic acid. N,N′-Ethylenebis(salicylideneaminato)oxovanadium(IV) (VO(salen)) which has been reported to be catalytically inactive at ambient temperature because of the insufficient reactivity of its oxidized state, an oxovanadium(V) complex, for the oxidation of the monomer turned out to exhibit an excellent catalytic activity on raising the reaction temperature above 100 °C. The enhanced reactivity of VO(salen)-acid or bis(2,4-pentanedionato)oxovanadium(IV) (VO(acac)2)-acid catalytic systems at elevated temperatures gave insight into the catalytic mechanism where oxidation of the monomer with the oxovanadium(V) complex dominated the reactivity, which was also accelerated by a second redox shuttle molecule, such as quinones with a potential near that of the [VO(salen)]0/+ couple.
Introduction
In polymer synthesis, organic reaction of sulfur-containing molecules has been paid much attention, because of their unique properties. In addition to utilizing various kinds of sulfur-containing monomers represented by thiophenes, C–S bond formation is quite useful to yield polymers. Thiolates (RS−) as nucleophiles and sulfenyl cations (RS+) as electrophiles are used for this purpose. The nucleophilic reaction of RS− is typically represented by bis-hydrothiolation of thiols to alkynes to give thioacetals using calcium catalysts1 and thia-Michael addition of RS− to cyclohexenone followed by Baeyer–Villiger oxidation and finally giving sulfone-containing polyesters via ring-opening polymerization.2 Sulfonium electrophiles (RS+) have been employed under dehydrated conditions, such as the addition of electrochemically generated bis(arylthio)aryl sulfonium cation (ArS(ArSSAr)+) to olefins to give 1,2-bis(arylthio)-substituted compounds via the cationic chain reaction mechanism.3 Palladium-catalyzed intermolecular C–H chalcogenation of arenes with disulfides or diselenides has also been reported.4 Using phenylsulfenophthalimide as the PhS+ source, a Brønsted acid-chiral binaphthyl ligand system catalyzed carbosulfenylation of olefins enantioselectively and intramolecularly.5 The sulfonium electrophile, derived from the condensation of methyl(4-phenylthio)phenyl sulfoxide, was also used for the synthesis of poly(methylsulfonio-1,4-phenylenethio-1,4-phenylene trifluoromethanesulfonate).6a The condensation was applied in many kinds of polysulfonium syntheses including those for heteropolyacenes.6 Recently, as represented by copper acetate–dimethyl sulfoxide (DMSO) promoted methylthiolation of arenes,7 β-hydroxysulfide synthesis via air oxidative hydroxysulfurization8 and dithioether synthesis from 1,3-dithianes via ring-opening and palladium catalyzed C–S bond formation have been reported.9 Sulfur chemistry is thus one of the hot topics in current organic chemistry relevant to polymer synthesis.10 However, catalytic systems to produce the synthetically useful sulfonium electrophiles have been limited to those used in our previous research on the oxygen-oxidative polymerization of diphenyl disulfide catalyzed by VO(acac)2.Our research group has been focusing on the chemistry of the ArS(ArSSAr)+ electrophile, especially its formation and reactivity by catalytic oxidation of the corresponding ArSSAr using a vanadyl catalyst.11 The chemical structure of the sulfonium cation, having a three-membered sulfur ring, was determined by 1H and 13C NMR of bis(methylthio)methyl sulfonium11e and in situ Raman spectroscopy of ArS(ArSSAr)+ (Ar = p-FC6H4).3c This kind of sulfonium cation was applied to poly(1,4-arylene sulfide) (PAS) synthesis, under oxidative conditions with a vanadyl catalyst and oxygen. In previous studies, it was reported that the bis(phenylthio)phenyl sulfonium (PhS(PhSSPh)+) ion was identified to be the active species of the oxygen-oxidative polymerization of PhSSPh, propagating the “PhS” unit via Friedel–Crafts (FC) type electrophilic aromatic substitution (Fig. 1).12 Vanadyl complexes, such as VO(acac)2 (acac− is the acetylacetonato ligand), V(acac)3, bis(1-phenyl-2,4-pentanedionato)oxovanadium (VO(dbm)2), and (meso-tetraphenylporphyrinato)oxovanadium (VO(TPP)) catalyzed the oxygen-oxidative polymerization.11d However, the inherent lability of the β-diketonato ligand such as acac− has impeded to unravel the catalytic mechanism. The lack of the catalytic activity of VO(salen) (where salen2− is an inert four-coordinate N,N′-ethylenebis(salicylideneaminato) ligand) with a more negative redox potential suggested that the overall activity of the catalyst was dominated by the oxidation of the monomer with the catalyst (Fig. 1). We report here that the VO(salen)-catalyzed oxygen-oxidative polymerization of PhSSPh proceeds at elevated temperatures, which revealed that the complex was sufficiently robust as a result of the inertness of the ligand and yet the reactivity of VO(salen) is significantly enhanced.
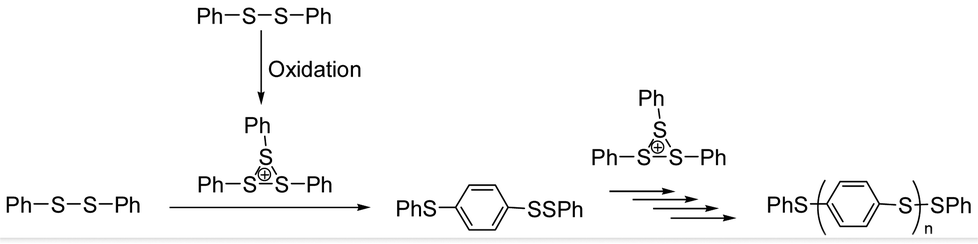 | ||
| Fig. 1 Oxidative polymerization of PhSSPh via FC electrophilic reaction of the sulfonium cation. | ||
H2salen and its derivatives are quite stable compounds and are useful as inert tetradentate ligands for various metal ions. Ruthenium(III)-, iron(II), copper(II)- and oxovanadium(IV)-salen complexes are typically classified into highly stable and robust complexes, which can be used as catalysts for oxidative reactions with peroxides and other oxidants.13 Oxygen or air is the ultimate oxidant in biological systems and the most economical reagent for oxidation in organic synthesis. To reduce oxygen, four-electron and four-proton transfers are essential according to O2 + 4H+ + 4e− → 2H2O. The two-electron and two-proton transfers give rise to a hydroxide according to O2 + 2H+ + 2e− → H2O2 which is often undesirable in the respiratory systems and organic synthesis. So far, many kinds of organometallic complexes have been reported, showing various types of the multi-electron transfer processes.14,15 The above mentioned oxygen-oxidative polymerization of PhSSPh is one of the four-electron-transfer preferred reactions to give H2O as the byproduct, because the hydroxide instantly oxygenates, rather than oxidating the disulfide to yield the undesired sulfoxides and sulfones.11,12,15,16 Although VO(salen) did not show the catalytic activity for polymerization under ambient conditions, the enhanced chemical robustness of VO(salen) and its oxidized [VVO(salen)]+, suggested by the electrochemically almost totally reversible nature of the couple which is in sharp contrast to the quasi-reversible response obtained for [VO(acac)2]0/+, clearly indicated the possibility of exploring more severe conditions with VO(salen) to undergo the four-electron transfer reaction.11d,16b
The mediated oxidation of PhSSPh catalyzed by the vanadyl complexes requires that the redox potentials of the vanadyl complexes be located in-between the potentials for the oxidation of PhSSPh near 1.7 V vs. Ag/AgCl and that for the four-electron reduction of oxygen near 0.5 V at room temperature. We previously reported that the potential range of 0.8–1.5 V for the vanadyl complex was appropriate to catalyze the polymerization.11d VO(acac)2 catalyzed the polymerization, because its redox potential was estimated to be near 1.1 V.11b The lack of the catalytic activity of VO(salen) was ascribed to the negative redox potential for the [VO(salen)]0/+ couple at a half-wave potential of E1/2 = 0.5 V.11d However, one could assume that the reactivity of those of the less oxidizing vanadyl complexes should be enhanced at elevated temperatures,17 according to Nernst's equation which suggests that the redox potential shifts positively with temperature. We report here our successful attempt to enhance the catalytic activity at moderately elevated temperatures by applying a pressurized condition (ca. 0.45 MPa) to partially maintain the oxygen concentration. Another effective means to enhance the catalytic activity is to combine an additional redox cycle with the oxovanadium(IV/V) couple. The effect of applying such multiple redox cycles formed in-between the monomer and oxygen has been examined in our previous work on the polymerization of bis(3,5-dimethylphenyl)disulfide using VO(acac)2 and 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) in the presence of trifluoroacetic acid (CF3CO2H) under ambient conditions with (CF3CO)2O in a halogenated hydrocarbon as a solvent.16a This method of enhancing the catalytic activity proved to have an inherent advantage over just raising the reaction temperature, because of the capability of undergoing the polymerization under atmospheric pressure. In search of quinones that match the conditions to undergo the polymerization of the disulfide at moderately elevated temperatures, we have found that 2,3,5,6-tetrabromo-p-benzoquinone(bromanil) significantly enhances the catalytic activity of the vanadyl complexes to produce polymers with larger molecular weights.
Experimental section
Materials
PhSSPh, VO(acac)2, bromanil, TrB(C6F5)4 (Tr+ = the triphenylmethylinium cation), TfOH, and CHF2CF2SO3H were obtained from Tokyo Kasei Co., and were used without further purification. VO(salen) was prepared as previously reported.18Measurements
Molecular weight measurements were done by gel permeation chromatography using an AGILENT PL-220 and 1-chloronaphthalene as the eluent (1 ml min−1) maintained at 210 °C. Calibration was done with polystyrene standards. Melting points were recorded using a differential scanning calorimeter (DSC, Seiko Instruments Inc. DSC-220), by scanning from 20 to 310 °C at a rate of 20 °C min−1 with alpha-alumina as the reference. Cyclic voltammetry was performed in 0.1 M solution of tetrabutylammonium tetrafluoroborate (TBABF4) in 1,2-dichloroethane with Pt working electrodes. The electrode potential of the working electrode was set versus the commercial Ag/AgCl reference electrode, and was scanned at a speed of 100 mV s−1 using an ALS/CH Instruments Electrochemical Analyzer (Model 660Dx) at −10, 20, and 70 °C. All potentials are shown with respect to this reference electrode.General procedure of oxygen-oxidative polymerization
To a 10 ml flask, 5 mmol of PhSSPh, vanadyl compound, acid or borate compound (CHF2CF2SO3H or TrB(C6F5)4), and bromanil were introduced. The mixture was heated and stirred under oxygen flow for 20–80 h. After cooling down to room temperature, the reaction mixture was dispersed into 50 ml of CH2Cl2 and the dispersion was poured into 300 ml of MeOH–HCl mixture (MeOH/HCl = 95/5, in v/v). The precipitation was collected with a glass filter. The solid was washed with MeOH and H2O, and dried under vacuum. The purified polymer yields, melting points and molecular weights were determined according to the methods described above. See the ESI† for experimental details.Procedure of pressurized oxygen-oxidative polymerization
To a 50 ml autoclave with a Teflon® inner tube, 31 mmol of PhSSPh, 1 mol% of VO(salen) and 1 mol% of 1,1,2,2-tetrafluoroethanesulfonic acid were introduced. The mixture was heated and stirred for 20 hours at 160 °C under discontinuously pressurized oxygen (depressurized to ambient pressure and pressurized to 0.45 MPa every 1 h). After the reaction, oxygen was depressurized and purged with nitrogen. Then the autoclave was cooled down to room temperature. The reaction mixture was dispersed into 50 ml of CH2Cl2 and the dispersion was poured into 300 ml of MeOH–HCl mixture (MeOH/HCl = 95/5, in v/v). The precipitation was collected with a glass filter. The solid was washed with MeOH and H2O, and dried under vacuum to give purified PPS in 82% yield. Melting points and molecular weights were determined according to the methods described above.Results and discussion
Oxygen-oxidative polymerization of PhSSPh catalysed by VO(salen) at elevated temperatures
Cyclic voltammograms of VO(salen) were recorded at −10, 20, and 70 °C, which revealed the redox potentials (E1/2) at 0.44, 0.47, and 0.56 V, respectively. Assuming a linearity of the formal potential with temperature, E1/2 was extrapolated to be 0.60, 0.64, and 0.69 V at 100, 130, and 160 °C, respectively. The positive shift in the redox potential that came into the region of the suitable range for catalysis suggested that the oxygen-oxidative polymerization should proceed above 100 °C when VO(salen) is used as the catalyst (Fig. 2).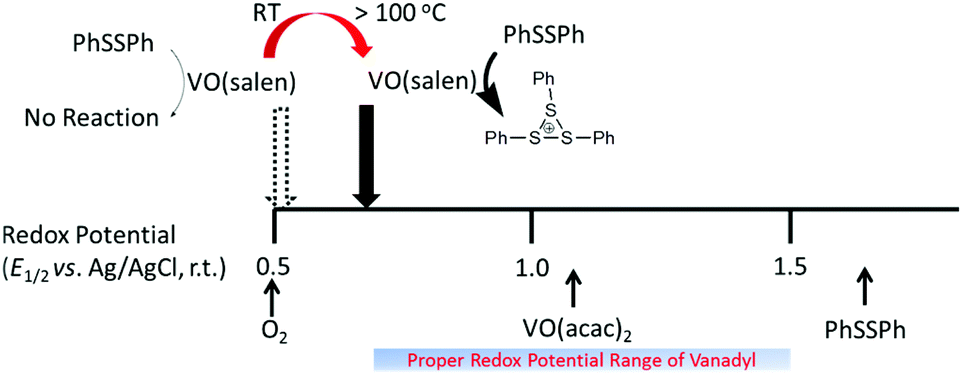 | ||
| Fig. 2 Potential diagram of the catalyst at elevated temperatures. | ||
Table 1 shows the results of the oxygen-oxidative polymerization of the disulfide with the VO(salen) catalyst, which was inactive at room temperature (entry 1) but gained activity at 100 °C (entry 2). Comparison between entries 2 and 4 suggested that higher reaction temperatures led to higher yields and melting points (Tm) of the products. Also, comparison between entries 3 and 6 indicated that the higher reaction temperatures resulted in higher yields and Tm of the products even in a shorter reaction time. Both the strong acid and the borate gave rise to the polymerization activity with VO(salen) (entries 4 and 5). Considering that the reactivity was limited by the equilibrated oxygen concentration which should be lowered at elevated temperatures, we adopted a pressurized condition (entry 7) to further enhance the reactivity. As expected, a higher molecular weight and Tm for the obtained PPS was accomplished in an even shorter time of the polymerization, compared with the results in entry 6. These results demonstrated the first catalysis of the oxidative polymerization of the disulfide by VO(salen) which has so far been regarded as inactive, suggesting that the electrochemical reversibility persisted at elevated temperatures as a result of the inertness of the salen2− ligand to give rise to the positive shift of the formal potential for the [VO(salen)]0/+ couple. The polymer obtained by this method was generally linear without branching or crosslinking as evidenced by the presence of only two carbon signals in the CP-MAS spectrum corresponding to the structural symmetry and the IR peak at 820 cm−1 ascribed to the C–H out-of-plane vibration of adjacent two hydrogens in the 1,4-phenylene unit,10,17,19 suggesting the highly selective catalysis by VO(salen) (see the ESI†). The present study is the first achievement of catalytic activity of VO(salen) for the polymerization of the disulfide, while the catalytic mechanism of the vanadyl catalyst has so far been determined with the complexes bearing the inert salen2− ligand and yet the catalytic activity has so far been obtained only for those having labile acac− and related ligands.11d The results clearly demonstrated that the catalysis was accomplished by the incorporation of the multi-electron transfer process established for VO(salen).16e
| Entry | Acid or borate (mol%) | Temp. (°C) | Time (h) | Yield (%) |
T
m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (°C) b (°C) |
M
nc |
M
wc |
|---|---|---|---|---|---|---|---|
| a 5 mol% of VO(salen) was used. b Determined by DSC. c Measured by high temperature GPC. d Data from ref. 3a. e 1 mol% of VO(salen) was used. f 1 mol% of VO(salen) was used and the oxygen pressure was 0.45 MPa. | |||||||
| 1d | TfOH (10) | RT | 40 | 0 | |||
| 2 | TrB(C6F5)4 (10) | 100 | 20 | 52 | 149 | ||
| 3e | CHF2CF2SO3H (1) | 130 | 80 | 70 | 163 | 1200 | 1600 |
| 4 | TrB(C6F5)4 (10) | 160 | 20 | 69 | 254 | 880 | 1800 |
| 5 | CHF2CF2SO3H (10) | 160 | 20 | 69 | 198 | 1500 | 3400 |
| 6e | CHF2CF2SO3H (1) | 160 | 40 | 82 | 175 | 1100 | 1500 |
| 7f | CHF2CF2SO3H (1) | 160 | 20 | 82 | 205 | 1800 | 4400 |
Bromanil-assisted catalysis of oxygen-oxidative polymerization of PhSSPh by vanadyl complexes
Under ambient temperature conditions, quinones such as DDQ and bromanil are known to enhance the reactivity of the VO(acac)2-acid catalytic system for the oxygen-oxidative polymerization of PhSSPh or diaryl disulfide.16a Assuming that the quinones should also enhance the reactivity of VO(salen), we turned to examine the catalysis by VO(salen) under moderately elevated conditions in the presence of the quinones. These quinones are good chemical oxidants that undergo oxidative polymerization of PhSSPh when an equimolar amount of the oxidant is used.11a A catalytic amount of quinones gave only a trace amount of the polymerized product as typically shown for the case of 2.5 mol% bromanil and 5 mol% CHF2CF2SO3H with respect to the monomer after 4 h of the reaction (entry 1 in Table 2). It might be suggested that catalysis by bromanil was accomplished by placing the potential of bromanil near to an oxovanadium(IV/V) couple such as [VO(salen)]0/+. An interesting aspect of the bromanil was found for the combination with VO(acac)2 or VO(salen) at moderately elevated temperatures, which demonstrated the catalytic activity even higher than those found for the conventional catalysis by VO(acac)2 or VO(salen). When 2.5 mol% VO(acac)2 was combined with the bromanil-acid system, the polymerization proceeded rapidly to give PPS in a 51% yield within 1 h (entry 2). Under the same conditions as in entry 2, a higher molecular-weight PPS was obtained in 82% yield after 4 h (entry 3). In the case of the conventional oxidative polymerization without bromanil, 5 mol% VO(acac)2 and CHF2CF2SO3H gave PPS in yields of only 22% after 1 h (entry 4) and 74% after 4 h (entry 5), which were much lower than those for the entries 2 and 3. Strong acids or borate compounds, such as CHF2CF2SO3H and TrB(C6F5)4, were essential to activate the vanadyl compounds.19 Even for the oxidative polymerization with quinones, strong acids were found to be essential for stabilizing the key intermediate, the sulfonium cation (PhS(PhSSPh)+).19 In the case of utilizing a VO(salen)-acid catalytic system, bromanil similarly accelerated the oxidative polymerization (entries 6 and 7). These results gave insight into the nature of the catalysis which was dominated by the oxidation of the monomer and much improved by placing a second redox couple to enhance the overall reactivity under moderately elevated temperature conditions to yield PPS with a higher molecular weight than those with the conventional catalyst system. It was reported that the redox potentials of quinones were positively shifted under acidic conditions.20 Actually, the redox potential of bromanil (E1/2 = −0.71 and 0.11 V vs. Ag/AgCl, r.t.) was shifted to 0.1 and 0.5 V to show the irreversible reduction to yield the protonated product under acidified conditions (see the ESI†). During the polymerization, the anionic species, such as the reduced forms of the quinones or the conjugated bases of strong acids, were reported to stabilize the cationic species, typically the sulfonium cations.19 Bromanil turns out to be an anionic species by reduction, so the reduced bromanil might be able to form complexes with the cationic vanadyl compounds. It may be suggested that the effective enhanced catalytic systems of the vanadyl–acid–bromanil triad was ascribed to the re-oxidation of the possibly newly formed complex between the vanadyl catalyst and the reduced bromanil. Further investigation of the mechanism is the topic of our continuous research.| Entry | Vanadyl (mol%) | Bromanil (mol%) | Time (h) | Yield (%) |
M
nb |
M
wb |
|---|---|---|---|---|---|---|
| a [CHF2CF2SO3H]/[vanadyl] = 2, oxygen atmosphere, polymerized at 160 °C. b Determined by high temperature GPC. c 5 mol% of CHF2CF2SO3H was used. d VO(acac)2 was used. e VO(salen) was used. | ||||||
| 1c | 0 | 2.5 | 4 | Trace | ||
| 2 | 2.5d | 2.5 | 1 | 51 | 900 | 1100 |
| 3 | 2.5d | 2.5 | 4 | 82 | 1600 | 2400 |
| 4 | 5d | 0 | 1 | 22 | 680 | 850 |
| 5 | 5d | 0 | 4 | 74 | 1300 | 1600 |
| 6 | 2.5e | 2.5 | 4 | 56 | 960 | 1300 |
| 7 | 5e | 0 | 4 | 24 | ||
Conclusions
The VO(salen)-catalyzed oxygen-oxidative polymerization of PhSSPh was successfully established above 100 °C. Pressurized oxygen further accelerated the oxidative polymerization and gave higher molecular weight products in high yield. The results demonstrated that catalysis was dominated by the oxidation of the monomer rather than re-oxidation of the catalyst by oxygen, which was also supported by the enhanced reactivity of the vanadyl catalyst with bromanil. Further modification of the catalyst with a view to yield a higher molecular weight PPS is the topic of our continuous research.Acknowledgements
This work was partially supported by Grants-in-Aid for Scientific Research (No. 24225003, 25288056, 26620108) from MEXT, Japan.Notes and references
- M. Hut'ka, T. Tsubogo and S. Kobayashi, Organometallics, 2014, 33, 5626 CrossRef.
- M. Winkler, Y. Raupp, L. Koehl, H. Wagner and M. Meier, Macromolecules, 2014, 47, 2842 CrossRef CAS.
- (a) K. Matsumoto, T. Sanada, H. Shimazaki, K. Shimada, S. Hagiwara, S. Fujie, Y. Ashikari, S. Suga, S. Kashimura and J. Yoshida, Asian J. Org. Chem., 2013, 2, 325 CrossRef CAS; (b) K. Matsumoto, S. Fujie, S. Suga, T. Nokami and J. Yoshida, Chem. Commun., 2009, 5448 RSC; (c) K. Matsumoto, Y. Miyamoto, K. Shimada, Y. Morisawa, H. Zipse, S. Suga, J. Yoshida, S. Kashimura and T. Wakabayashi, Chem. Commun., 2015, 51, 13106 RSC.
- R. Qiu, V. P. Reddy, T. Iwasaki and N. Kambe, J. Org. Chem., 2015, 80, 367 CrossRef CAS PubMed.
- S. Denmark and H. Chi, J. Am. Chem. Soc., 2014, 136, 3655 CrossRef CAS PubMed.
- (a) E. Tsuchida, K. Yamamoto and E. Shouji, Macromolecules, 1993, 26, 7389 CrossRef CAS; (b) K. Miyatake, K. Oyaizu, Y. Nishimura and E. Tsuchida, Macromolecules, 2001, 34, 1172 CrossRef CAS; (c) K. Oyaizu, T. Mikami, F. Mitsuhashi and E. Tsuchida, Macromolecules, 2002, 35, 67 CrossRef CAS; (d) K. Oyaizu, F. Mitsuhashi and E. Tsuchida, Macromol. Chem. Phys., 2002, 203, 1328 CrossRef CAS; (e) K. Oyaizu, T. Iwasaki, Y. Tsukahara and E. Tsuchida, Macromolecules, 2004, 37, 1257 CrossRef CAS; (f) K. Oyaizu, T. Mikami and E. Tsuchida, Macromolecules, 2004, 37, 2325 CrossRef CAS.
- P. Sharma, S. Rohilla and N. Jain, J. Org. Chem., 2015, 80, 4116 CrossRef CAS PubMed.
- S. Zhou, X. Pan, Z. Zhou, A. Shoberu and J. Zou, J. Org. Chem., 2015, 80, 3682 CrossRef CAS PubMed.
- N. Abidi and J. Schmink, J. Org. Chem., 2015, 80, 4123 CrossRef CAS PubMed.
- F. Aida and K. Oyaizu, Chem. Lett., 2016, 45, 201 CrossRef.
- (a) E. Tsuchida, K. Yamamoto, M. Jikei, E. Shouji and H. Nishide, J. Macromol. Sci., Part A: Pure Appl. Chem., 1991, 28, 1285 CrossRef; (b) K. Yamamoto, M. Jikei, J. Katoh, H. Nishide and E. Tsuchida, Macromolecules, 1992, 25, 2698 CrossRef CAS; (c) E. Shouji, K. Yamamoto, J. Katoh, H. Nishide and E. Tsuchida, Polym. Adv. Technol., 1991, 2, 149 CrossRef CAS; (d) K. Yamamoto, E. Tsuchida, H. Nishide, M. Jikei and K. Oyaizu, Macromolecules, 1993, 26, 3432 CrossRef CAS; (e) E. Tsuchida, K. Yamamoto and E. Shouji, J. Macromol. Sci., Part A: Pure Appl. Chem., 1994, 31, 1579 CrossRef.
- E. Tsuchida and K. Oyaizu, Bull. Chem. Soc. Jpn., 2003, 76, 15 CrossRef CAS.
- (a) Y. Fukunaga, T. Uchida, Y. Ito, K. Matsumoto and T. Katsuki, Org. Lett., 2012, 12, 4658 CrossRef PubMed; (b) S. Koya, Y. Nishioka, H. Mizoguchi, T. Uchida and T. Katsuki, Angew. Chem., Int. Ed., 2012, 51, 8243 CrossRef CAS PubMed; (c) H. Mizoguchi, T. Uchida and T. Katsuki, Angew. Chem., Int. Ed., 2014, 53, 3178 CrossRef CAS PubMed; (d) Y. Nishioka, T. Uchida and T. Katsuki, Angew. Chem., Int. Ed., 2013, 52, 1739 CrossRef CAS PubMed; (e) K. Matsumoto, H. Egami, T. Oguma and T. Katsuki, Chem. Commun., 2012, 48, 5823 RSC; (f) T. Oguma and T. Katsuki, Chem. Commun., 2014, 50, 5053 RSC; (g) M. Nikoorazm, A. G-Choghamarani, F. Ghorbani, H. Mahdavi and Z. Karamshahi, J. Porous Mater., 2015, 22, 261 CrossRef CAS; (h) P. Du, H. Li, Y. Wang, J. Cheng and X. Wan, Org. Lett., 2014, 16, 6350 CrossRef CAS PubMed; (i) A. Mathavan, A. Ramdass and S. Rajagopal, Transition Met. Chem., 2015, 40, 355 CrossRef CAS; (j) A. Berkessel, T. Günther, T. Wang and J.-M. Neudröft, Angew. Chem., Int. Ed., 2013, 52, 8467 CrossRef CAS PubMed.
- (a) S. Fukuzumi, L. Tahsini, Y. M. Lee, K. Ohkubo, W. Nam and K. D. Karlin, J. Am. Chem. Soc., 2012, 134, 7025 CrossRef CAS PubMed; (b) S. Kakuda, R. L. Peterson, K. Ohkubo, K. D. Karlin and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 6513 CrossRef CAS PubMed; (c) J. Y. Lee, R. L. Peterson, K. Ohkubo, I. Garcia-Bosh, R. A. Himes, J. Woertink, C. D. Moore, E. I. Solomon, S. Fukuzumi and K. D. Karlin, J. Am. Chem. Soc., 2014, 136, 9925 CrossRef CAS PubMed; (d) A. Yokoyama, J. E. Han, K. D. Karlin and W. Nam, Chem. Commun., 2014, 50, 1742 RSC; (e) I. Garcia-Bosh, S. M. Adam, A. W. Schaefer, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2015, 137, 1032 CrossRef PubMed; (f) S. Kim, J. Y. Lee, R. E. Cowley, J. W. Ginsbach, M. A. Siegler, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2015, 137, 2769 CrossRef PubMed; (g) A. Hay, US Patent, 3306874, 1967 Search PubMed.
- (a) K. Oyaizu, Y. Kumaki, K. Saito and E. Tsuchida, Macromolecules, 2000, 33, 5766 CrossRef CAS; (b) K. Oyaizu, K. Saito and E. Tsuchida, Chem. Lett., 2000, 1318 CrossRef CAS; (c) K. Saito, T. Masuyama, K. Oyaizu and H. Nishide, Chem. – Eur. J., 2003, 9, 4240 CrossRef CAS PubMed; (d) E. Tsuchida, K. Yamamoto and K. Oyaizu, Multi-electron Transfer and Catalytic Mechanisms in Oxidative Polymerization, Marcel Dekker, New York, 1999, p. 535 Search PubMed.
- (a) K. Yamamoto, M. Jikei, K. Oyaizu, F. Suzuki, H. Nishide and E. Tsuchida, Bull. Chem. Soc. Jpn., 1994, 67, 251 CrossRef CAS; (b) K. Yamamoto, K. Oyaizu and E. Tsuchida, Polym. Adv. Technol., 1995, 6, 155 CrossRef CAS; (c) K. Oyaizu, K. Yamamoto, K. Yoneda and E. Tsuchida, Inorg. Chem., 1996, 35, 6634 CrossRef CAS PubMed; (d) E. Tsuchida, K. Yamamoto, K. Oyaizu, N. Iwasaki and F. Anson, Inorg. Chem., 1994, 33, 1056 CrossRef CAS; (e) E. Tsuchida and K. Oyaizu, Coord. Chem. Rev., 2003, 237, 213 CrossRef CAS; (f) E. Tsuchida, K. Yamamoto and K. Oyaizu, Molecular Conversion Based on Multi-electron Transfer System, Kodansha, Tokyo, 1995, p. 41 Search PubMed; (g) E. Tsuchida, K. Yamamoto and K. Oyaizu, Multi-electron Transfer Process of a Vanadium Dinuclear Complex for Molecular Conversions, Plenum Press, New York, 1996, p. 139 Search PubMed; (h) M. Jikei, J. Katoh, N. Sato, K. Yamamoto, H. Nishide and E. Tsuchida, Bull. Chem. Soc. Jpn., 1992, 65, 2029 CrossRef CAS.
- F. Aida, Y. Takatori, D. Kiyokawa, K. Nagamatsu, H. Nishide and K. Oyaizu, Chem. Lett., 2015, 44, 767 CrossRef CAS.
- J. A. Bonadies and C. J. Carrano, J. Am. Chem. Soc., 1986, 108, 4088 CrossRef CAS.
- F. Aida, S. Yamaguchi, Y. Takatori, K. Nagamatsu, D. Kiyokawa, K. Oyaizu and H. Nishide, Macromol. Chem. Phys., 2015, 216, 1850 CrossRef CAS.
- K. Oyaizu, N. Iwasaki, K. Yamamoto, H. Nishide and E. Tsuchida, Bull. Chem. Soc. Jpn., 1994, 67, 1456 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5py01980j |
| This journal is © The Royal Society of Chemistry 2016 |